
Kinetic Mathematical Modeling of Oxidative Phosphorylation in Cardiomyocyte Mitochondria


Abstract
:1. Introduction
2. Description of Enzyme Reaction Rates
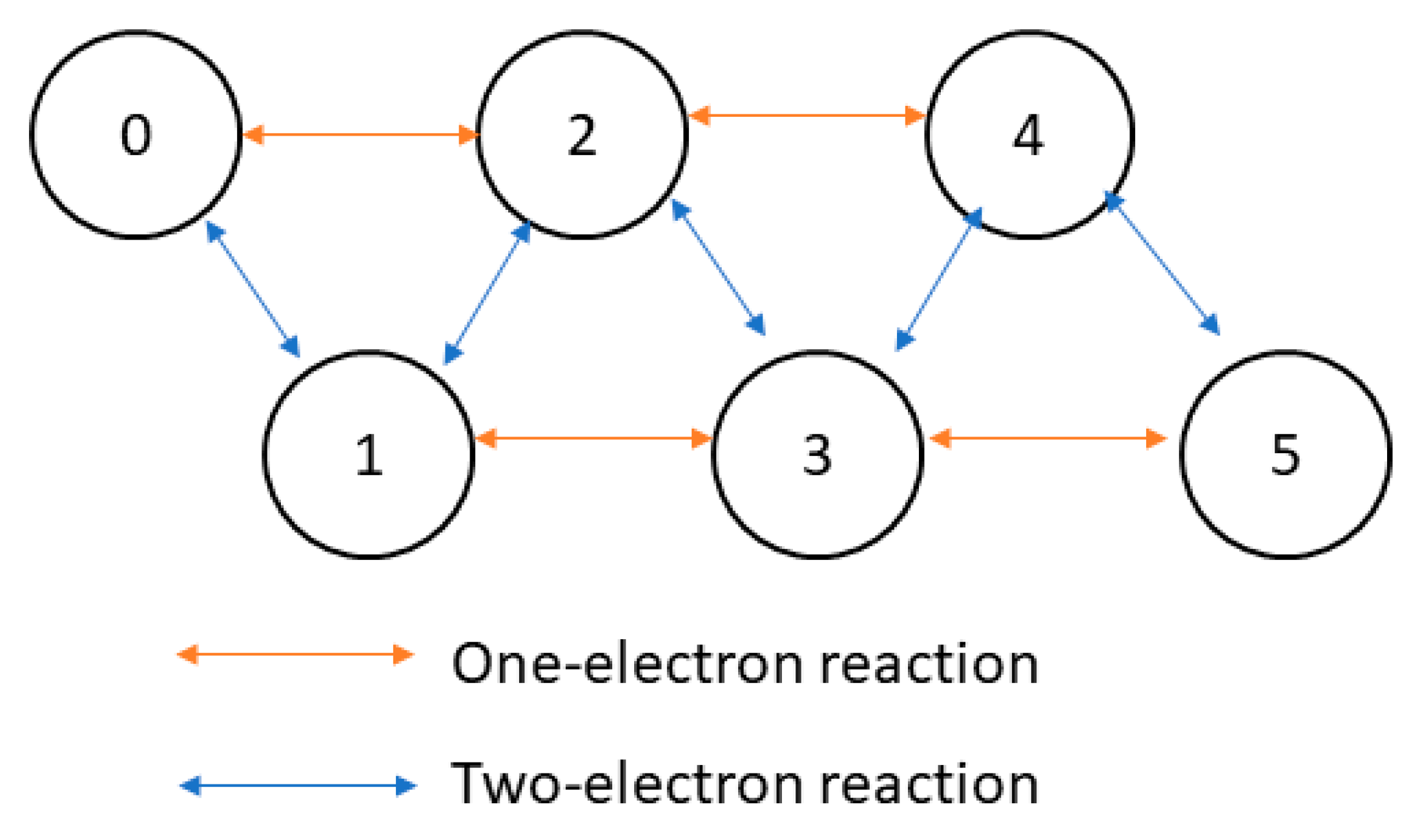
2.1. Law of Mass Action
2.2. Quasi-Steady State Enzyme Kinetics
3. Lumped Models of the Electron Transport Chain
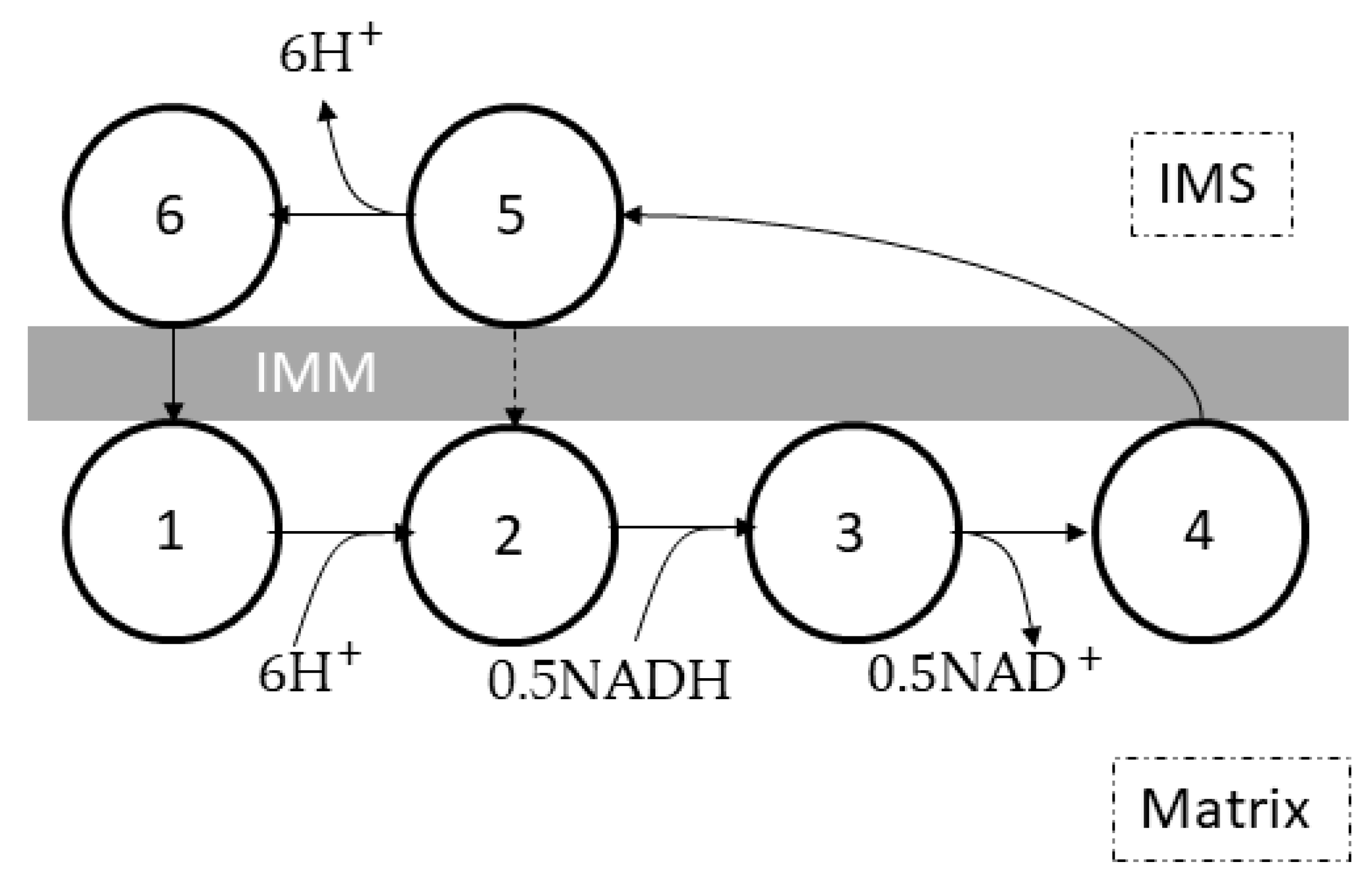
4. Complex I
4.1. The Molecular Structure and Electron Transport in Complex I
4.2. Reactive Oxygen Species (ROS) Generation Mechanisms
4.3. Kinetic Models of Complex I
5. Complex II
5.1. Molecular Structure and Reaction Mechanism
5.2. Kinetic Models of Complex II
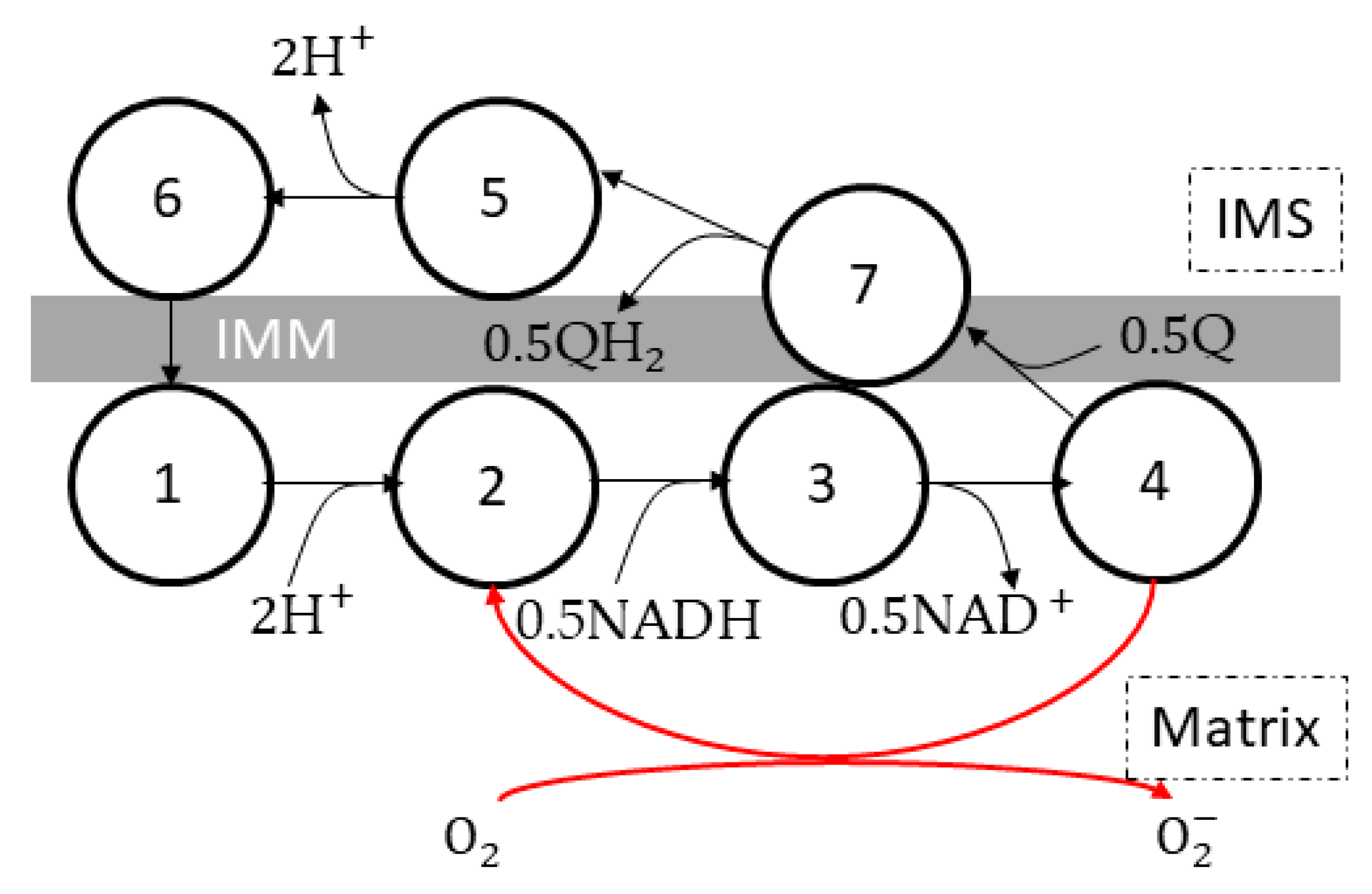
6. Complex III
6.1. Molecular Structure and Reaction Mechanism
6.2. Reactive Oxygen Species Generation Mechanisms
6.3. Kinetic Models of Complex III
7. Complex IV
7.1. Molecular Structure and Reaction Mechanism
7.2. Kinetic Models of Complex IV
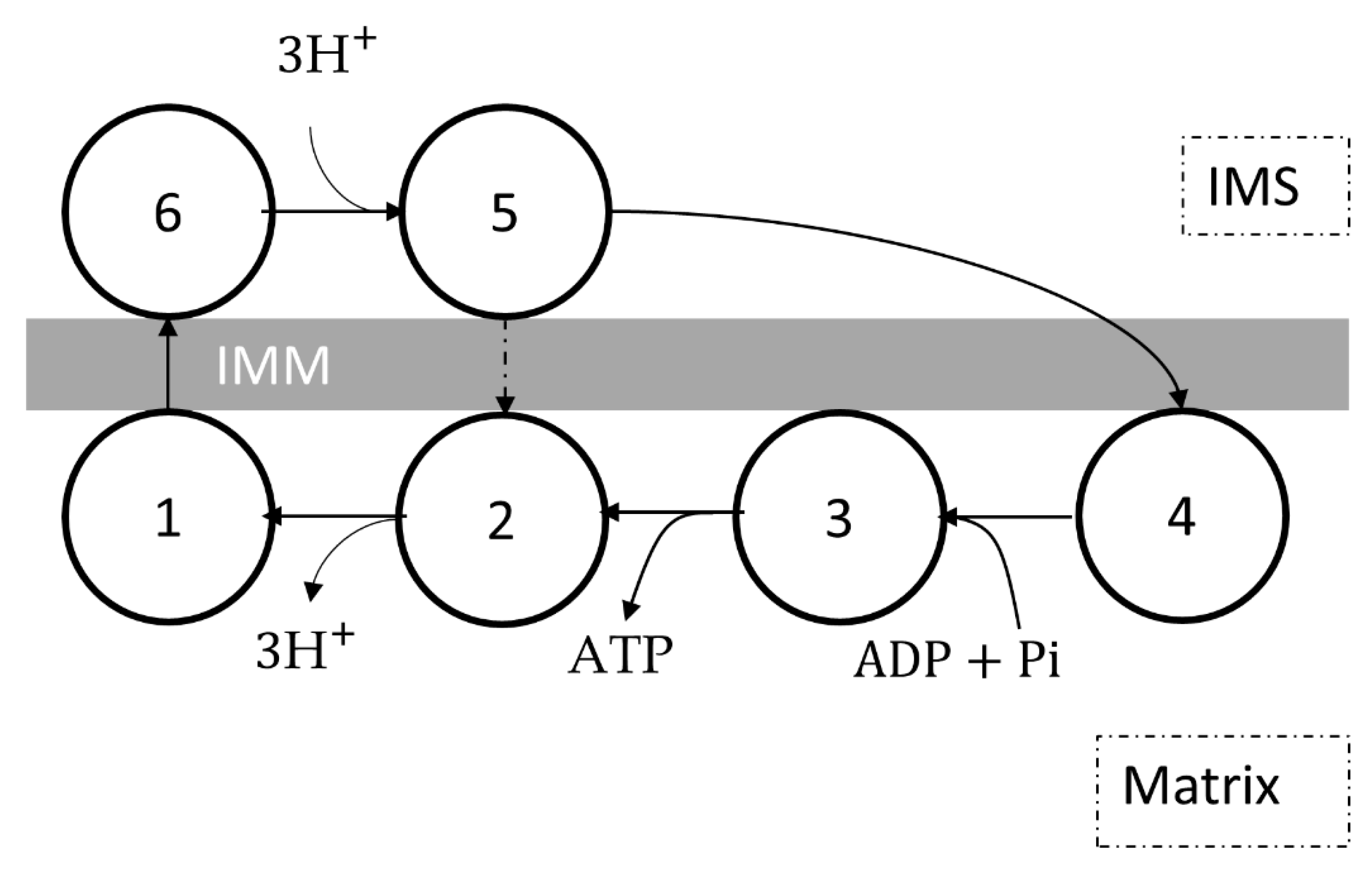
8. Complex V
8.1. Molecular Structure and Reaction Mechanism
8.2. Kinetic Models of Complex V
9. Summaries of the OXPHOS Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Respiratory Complex | Cell Type | Method | Internal State Variables |
|---|---|---|---|---|
| Korzeniewski, 1991 [52] | I-III IV V | Rat hepatocytes | TS MAK TS | 0 2 0 |
| Korzeniewski, 1996 [114] | IV | Rat hepatocytes | MAK | 0 |
| Korzeniewski, 1996 [53] | I, III | Rat skeletal muscle cells | TS | 0 |
| Magnus and Keizer, 1997 [54] | I-III-IV V | Mouse pancreatic beta-cells | KAH KAH | 0 0 |
| Cortassa, 2003 [56] | I-III-IV II-III-IV V | Guinea pig ventricular cardiomyocytes | KAH KAH KAH | 0 0 0 |
| Saa, 2013 [159] | I-III-IV V | Not specified | Phenomenal Phenomenal | 0 0 |
| Beard, 2005 [69] | I, III, IV, V | Porcine heart and skeletal muscle cells | MAK | 0 |
| Wu, 2007 [82] | II | Porcine heart and skeletal muscle cells and rat cardiomyocytes | Theorell–Chance Bi-Bi mechanism | 0 |
| Heiske, 2017 [71] | I, III, IV, V | Bovine cardiomyocytes | MM | 0 |
| Markevich and Hoek, 2015 [63] | I III | Bovine cardiomyocytes | MAK | 12 14 |
| Gauthier, 2013 [62] | I III | Guinea pig ventricular cardiomyocytes | KAH MAK | 0 10 |
| Bazil et al., 2014 [61] | I | Bovine cardiomyocytes and rat cardiomyocytes | HKA | 0 |
| Manhas et al., 2020 [84] | II | Bovine/Pig/Guinea pig cardiomyocytes | KAH | 0 |
| Markevich et al., 2020 [85] | II | Bovine and rat cardiomyocytes | MAK | 35 |
| Demin et al., 1998 [79] | III | Not specified | MAK | 9 (minimal)/12 (channeled) |
| Demin et al., 2001 [78] | III IV | Hepatocyte | MAK MAK | 12 4 |
| Selivanov et al., 2009 [96] | III | Rat brain mitochondria | MAK | 400 |
| Guillaud et al., 2014 [97] | III | Rat skeletal muscle, heart, liver, kidney, and brain cells | MAK | 16 |
| Bazil et al., 2013 [98] | III | Bovine cardiomyocytes | KAH | 0 |
| Krab et al., 2011 [115] | IV | Bovine cardiomyocytes | KAH | 0 |
| Wilson et al., 2014 [169] | IV | Rat hepatocytes | Quasi-steady state from MAK | 0 |
| Pannala et al., 2016 [117] | IV | Rat hepatocytes and bovine cardiomyocytes | KAH | 0 |
| Nguyen et al., 2007 [141] | V | Guinea pig ventricular cardiomyocytes | Hill kinetics for MMP, Random Bi-Uni kinetics for substrate | 0 |
| Cortassa et al., 2022 [140] | V | Guinea pig ventricular cardiomyocytes | KAH | 0 |
10. Other OXPHOS Aspects and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Junge, W.; Nelson, N. ATP synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef] [Green Version]
- Boyer, P.D.; Chance, B.; Ernster, L.; Mitchell, P.; Racker, E.; Slater, E.C. Oxidative phosphorylation and photophosphorylation. Annu. Rev. Biochem. 1977, 46, 955–966. [Google Scholar] [CrossRef]
- Mitchell, P. Keilin’s respiratory chain concept and its chemiosmotic consequences. Science 1979, 206, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Siekevitz, P. Powerhouse of the cell. Sci. Am. 1957, 197, 131–144. [Google Scholar] [CrossRef]
- Wood, P.M. The potential diagram for oxygen at pH 7. Biochem. J. 1988, 253, 287–289. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Scialò, F.; Sriram, A.; Fernández-Ayala, D.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Kembro, J.M.; Aon, M.A.; Winslow, R.L.; O’Rourke, B.; Cortassa, S. Integrating mitochondrial energetics, redox and ROS metabolic networks: A two-compartment model. Biophys. J. 2013, 104, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef] [Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [Green Version]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spégel, P.; Mulder, H. The pathogenetic role of β-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef] [Green Version]
- Mihajlovic, M.; Vinken, M. Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. Int. J. Mol. Sci. 2022, 23, 3315. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Will, Y. Current concepts in drug-induced mitochondrial toxicity. Curr. Protoc. Toxicol. 2009, 40, 2–15. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Distelmaier, F.; Smeitink, J.A.M.; Willems, P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metab. Clin. Exp. 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr. 2009, 14, 8–13; discussion 16. [Google Scholar] [CrossRef]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [Green Version]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, and Council on Functional Genomics and Translational Biology Mitochondrial function, biology, and role in disease: A scientific statement from the american heart association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.; Loiselle, D.S.; Crampin, E.J. Regulation of cardiac cellular bioenergetics: Mechanisms and consequences. Physiol. Rep. 2015, 3, e12464. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The role of oxidative stress in cardiac disease: From physiological response to injury factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Sollott, S.J.; Aon, M.A. Computational Modeling of Mitochondrial Function from a Systems Biology Perspective. Methods Mol. Biol. 2018, 1782, 249–265. [Google Scholar] [CrossRef]
- Takeuchi, A.; Matsuoka, S. Integration of mitochondrial energetics in heart with mathematical modelling. J. Physiol. 2020, 598, 1443–1457. [Google Scholar] [CrossRef]
- Winslow, R.L.; Cortassa, S.; Greenstein, J.L. Using models of the myocyte for functional interpretation of cardiac proteomic data. J. Physiol. 2005, 563, 73–81. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A. Computational modeling of mitochondrial function. Methods Mol. Biol. 2012, 810, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Imperial, S.; Centelles, J.J. Enzyme kinetic equations of irreversible and reversible reactions in metabolism. JBM 2014, 2, 24–29. [Google Scholar] [CrossRef]
- Alberty, R.A. Relations between biochemical thermodynamics and biochemical kinetics. Biophys. Chem. 2006, 124, 11–17. [Google Scholar] [CrossRef]
- Gunawardena, J. A linear framework for time-scale separation in nonlinear biochemical systems. PLoS ONE 2012, 7, e36321. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B. A guide to the Michaelis-Menten equation: Steady state and beyond. FEBS J. 2021, 289, 6086–6098. [Google Scholar] [CrossRef]
- Einav, T.; Mazutis, L.; Phillips, R. Statistical mechanics of allosteric enzymes. J. Phys. Chem. B 2016, 120, 6021–6037. [Google Scholar] [CrossRef] [Green Version]
- Leskovac, V.; Trivić, S.; Pericin, D.; Kandrac, J. Deriving the rate equations for product inhibition patterns in bisubstrate enzyme reactions. J. Enzyme Inhib. Med. Chem. 2006, 21, 617–634. [Google Scholar] [CrossRef]
- Cornish-Bowden, A.; Hofmeyr, J.-H.S. Enzymes in context: Kinetic characterization of enzymes for systems biology. Biochemist 2005, 27, 11–14. [Google Scholar] [CrossRef] [Green Version]
- King, E.L.; Altman, C. A Schematic Method of Deriving the Rate Laws for Enzyme-Catalyzed Reactions. J. Phys. Chem. 1956, 60, 1375–1378. [Google Scholar] [CrossRef]
- Qi, F.; Dash, R.K.; Han, Y.; Beard, D.A. Generating rate equations for complex enzyme systems by a computer-assisted systematic method. BMC Bioinform. 2009, 10, 238. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, B.; Froncisz, W. An extended dynamic model of oxidative phosphorylation. Biochim. Biophys. Acta BBA Bioenerg. 1991, 1060, 210–223. [Google Scholar] [CrossRef]
- Korzeniewski, B.; Mazat, J.P. Theoretical studies on the control of oxidative phosphorylation in muscle mitochondria: Application to mitochondrial deficiencies. Biochem. J. 1996, 319 Pt 1, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnus, G.; Keizer, J. Minimal model of beta-cell mitochondrial Ca2+ handling. Am. J. Physiol. 1997, 273, C717–C733. [Google Scholar] [CrossRef]
- Pietrobon, D.; Caplan, S.R. Flow-force relationships for a six-state proton pump model: Intrinsic uncoupling, kinetic equivalence of input and output forces, and domain of approximate linearity. Biochemistry 1985, 24, 5764–5776. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Marbán, E.; Winslow, R.L.; O’Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef] [Green Version]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 863–871. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef]
- Bazil, J.N.; Pannala, V.R.; Dash, R.K.; Beard, D.A. Determining the origins of superoxide and hydrogen peroxide in the mammalian NADH:ubiquinone oxidoreductase. Free Radic. Biol. Med. 2014, 77, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O’Rourke, B.; Winslow, R.L. A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys. J. 2013, 105, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markevich, N.I.; Hoek, J.B. Computational modeling analysis of mitochondrial superoxide production under varying substrate conditions and upon inhibition of different segments of the electron transport chain. Biochim. Biophys. Acta 2015, 1847, 656–679. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransac, S.; Arnarez, C.; Mazat, J.-P. The flitting of electrons in complex I: A stochastic approach. Biochim. Biophys. Acta 2010, 1797, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzeniewski, B. Regulation of ATP supply during muscle contraction: Theoretical studies. Biochem. J. 1998, 330 Pt 3, 1189–1195. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, B.; Zoladz, J.A. A model of oxidative phosphorylation in mammalian skeletal muscle. Biophys. Chem. 2001, 92, 17–34. [Google Scholar] [CrossRef]
- Korzeniewski, B.; Noma, A.; Matsuoka, S. Regulation of oxidative phosphorylation in intact mammalian heart in vivo. Biophys. Chem. 2005, 116, 145–157. [Google Scholar] [CrossRef]
- Beard, D.A. A biophysical model of the mitochondrial respiratory system and oxidative phosphorylation. PLoS Comput. Biol. 2005, 1, e36. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; French, S.; Evans, F.J.; Joubert, F.; Balaban, R.S. Metabolic network control of oxidative phosphorylation: Multiple roles of inorganic phosphate. J. Biol. Chem. 2003, 278, 39155–39165. [Google Scholar] [CrossRef]
- Heiske, M.; Letellier, T.; Klipp, E. Comprehensive mathematical model of oxidative phosphorylation valid for physiological and pathological conditions. FEBS J. 2017, 284, 2802–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.D.; Greenstein, J.L.; O’Rourke, B.; Winslow, R.L. An integrated mitochondrial ROS production and scavenging model: Implications for heart failure. Biophys. J. 2013, 105, 2832–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, R.; Kirshenbaum, L.A. Succinate dehydrogenase/complex II activity obligatorily links mitochondrial reserve respiratory capacity to cell survival in cardiac myocytes. Cell Death Dis. 2015, 6, e1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, A.; Shurubor, Y.; Valsecchi, F.; Manfredi, G.; Galkin, A. Differential susceptibility of mitochondrial complex II to inhibition by oxaloacetate in brain and heart. Biochim. Biophys. Acta 2016, 1857, 1561–1568. [Google Scholar] [CrossRef] [Green Version]
- Wei, A.-C.; Aon, M.A.; O’Rourke, B.; Winslow, R.L.; Cortassa, S. Mitochondrial energetics, pH regulation, and ion dynamics: A computational-experimental approach. Biophys. J. 2011, 100, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Demin, O.V.; Gorianin, I.I.; Kholodenko, B.N.; Westerhoff, H.V. Kinetic modeling of energy metabolism and generation of active forms of oxygen in hepatocyte mitochondria. Mol. Biol. 2001, 35, 1095–1104. [Google Scholar] [CrossRef]
- Demin, O.V.; Kholodenko, B.N.; Skulachev, V.P. A model of O2.-generation in the complex III of the electron transport chain. Mol. Cell. Biochem. 1998, 184, 21–33. [Google Scholar] [CrossRef]
- Mogilevskaya, E.; Demin, O.; Goryanin, I. Kinetic model of mitochondrial Krebs cycle: Unraveling the mechanism of salicylate hepatotoxic effects. J. Biol. Phys. 2006, 32, 245–271. [Google Scholar] [CrossRef]
- Bazil, J.N.; Beard, D.A.; Vinnakota, K.C. Catalytic coupling of oxidative phosphorylation, ATP demand, and reactive oxygen species generation. Biophys. J. 2016, 110, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Yang, F.; Vinnakota, K.C.; Beard, D.A. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 2007, 282, 24525–24537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazil, J.N.; Buzzard, G.T.; Rundell, A.E. Modeling mitochondrial bioenergetics with integrated volume dynamics. PLoS Comput. Biol. 2010, 6, e1000632. [Google Scholar] [CrossRef] [Green Version]
- Manhas, N.; Duong, Q.V.; Lee, P.; Richardson, J.D.; Robertson, J.D.; Moxley, M.A.; Bazil, J.N. Computationally modeling mammalian succinate dehydrogenase kinetics identifies the origins and primary determinants of ROS production. J. Biol. Chem. 2020, 295, 15262–15279. [Google Scholar] [CrossRef] [PubMed]
- Markevich, N.I.; Markevich, L.N.; Hoek, J.B. Computational modeling analysis of generation of reactive oxygen species by mitochondrial assembled and disintegrated complex II. Front. Physiol. 2020, 11, 557721. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Protonmotive redox mechanism of the cytochrome b-c 1 complex in the respiratory chain: Protonmotive ubiquinone cycle. FEBS Lett. 1975, 56, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Crofts, A.R.; Holland, J.T.; Victoria, D.; Kolling, D.R.J.; Dikanov, S.A.; Gilbreth, R.; Lhee, S.; Kuras, R.; Kuras, M.G. The Q-cycle reviewed: How well does a monomeric mechanism of the bc(1) complex account for the function of a dimeric complex? Biochim. Biophys. Acta 2008, 1777, 1001–1019. [Google Scholar] [CrossRef] [Green Version]
- Crofts, A.R.; Hong, S.; Wilson, C.; Burton, R.; Victoria, D.; Harrison, C.; Schulten, K. The mechanism of ubihydroquinone oxidation at the Qo-site of the cytochrome bc1 complex. Biochim. Biophys. Acta 2013, 1827, 1362–1377. [Google Scholar] [CrossRef] [Green Version]
- Ransac, S.; Parisey, N.; Mazat, J.-P. The loneliness of the electrons in the bc1 complex. Biochim. Biophys. Acta 2008, 1777, 1053–1059. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Truong, D.; Shangari, N.; O’Brien, P.J. Drug-induced mitochondrial toxicity. Expert Opin. Drug Metab. Toxicol. 2005, 1, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Vinnakota, K.C.; Bazil, J.N.; Van den Bergh, F.; Wiseman, R.W.; Beard, D.A. Feedback regulation and time hierarchy of oxidative phosphorylation in cardiac mitochondria. Biophys. J. 2016, 110, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Selivanov, V.A.; Votyakova, T.V.; Zeak, J.A.; Trucco, M.; Roca, J.; Cascante, M. Bistability of mitochondrial respiration underlies paradoxical reactive oxygen species generation induced by anoxia. PLoS Comput. Biol. 2009, 5, e1000619. [Google Scholar] [CrossRef] [Green Version]
- Guillaud, F.; Dröse, S.; Kowald, A.; Brandt, U.; Klipp, E. Superoxide production by cytochrome bc1 complex: A mathematical model. Biochim. Biophys. Acta 2014, 1837, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Bazil, J.N.; Vinnakota, K.C.; Wu, F.; Beard, D.A. Analysis of the kinetics and bistability of ubiquinol:cytochrome c oxidoreductase. Biophys. J. 2013, 105, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazil, J.N. Analysis of a functional dimer model of ubiquinol cytochrome c oxidoreductase. Biophys. J. 2017, 113, 1599–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinov, A.A. Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 2012, 586, 630–639. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [Green Version]
- Bloch, D.; Belevich, I.; Jasaitis, A.; Ribacka, C.; Puustinen, A.; Verkhovsky, M.I.; Wikström, M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc. Natl. Acad. Sci. USA 2004, 101, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Muramoto, K.; Ohta, K.; Shinzawa-Itoh, K.; Kanda, K.; Taniguchi, M.; Nabekura, H.; Yamashita, E.; Tsukihara, T.; Yoshikawa, S. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. USA 2010, 107, 7740–7745. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.G.; Nicholls, P.; Wilson, M.T.; Cooper, C.E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [Google Scholar] [CrossRef] [Green Version]
- Sarti, P.; Forte, E.; Giuffrè, A.; Mastronicola, D.; Magnifico, M.C.; Arese, M. The Chemical Interplay between Nitric Oxide and Mitochondrial Cytochrome c Oxidase: Reactions, Effectors and Pathophysiology. Int. J. Cell Biol. 2012, 2012, 571067. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.; Wilson, M.T.; Aasa, R.; Malmström, B.G. Cyanide inhibition of cytochrome c oxidase. A rapid-freeze e.p.r. investigation. Biochem. J. 1984, 224, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Alonso, J.-R.; Cardellach, F.; López, S.; Casademont, J.; Miró, O. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol. Toxicol. 2003, 93, 142–146. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Impact of hydrogen sulfide on mitochondrial and bacterial bioenergetics. Int. J. Mol. Sci. 2021, 22, 12688. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Caughey, W.S. Infrared evidence of azide binding to iron, copper, and non-metal sites in heart cytochrome c oxidase. J. Biol. Chem. 1992, 267, 9757–9766. [Google Scholar] [CrossRef]
- Liesivuori, J.; Savolainen, H. Methanol and formic acid toxicity: Biochemical mechanisms. Pharmacol. Toxicol. 1991, 69, 157–163. [Google Scholar] [CrossRef]
- Hashmi, J.T.; Huang, Y.-Y.; Osmani, B.Z.; Sharma, S.K.; Naeser, M.A.; Hamblin, M.R. Role of low-level laser therapy in neurorehabilitation. PM R 2010, 2, S292–S305. [Google Scholar] [CrossRef] [Green Version]
- Antonini, E.; Brunori, M.; Colosimo, A.; Greenwood, C.; Wilson, M.T. Oxygen “pulsed” cytochrome c oxidase: Functional properties and catalytic relevance. Proc. Natl. Acad. Sci. USA 1977, 74, 3128–3132. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, B. Simulation of oxidative phosphorylation in hepatocytes. Biophys. Chem. 1996, 58, 215–224. [Google Scholar] [CrossRef]
- Krab, K.; Kempe, H.; Wikström, M. Explaining the enigmatic K(M) for oxygen in cytochrome c oxidase: A kinetic model. Biochim. Biophys. Acta 2011, 1807, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.F.; Harrison, D.K.; Vinogradov, A. Mitochondrial cytochrome c oxidase and control of energy metabolism: Measurements in suspensions of isolated mitochondria. J. Appl. Physiol. 2014, 117, 1424–1430. [Google Scholar] [CrossRef] [Green Version]
- Pannala, V.R.; Camara, A.K.S.; Dash, R.K. Modeling the detailed kinetics of mitochondrial cytochrome c oxidase: Catalytic mechanism and nitric oxide inhibition. J. Appl. Physiol. 2016, 121, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Senior, A.E. ATP synthase: What we know about ATP hydrolysis and what we do not know about ATP synthesis. Biochim. Biophys. Acta BBA Bioenerg. 2000, 1458, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Gout, E.; Rébeillé, F.; Douce, R.; Bligny, R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: Unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. USA 2014, 111, E4560-7. [Google Scholar] [CrossRef] [Green Version]
- Senior, A.E.; Nadanaciva, S.; Weber, J. The molecular mechanism of ATP synthesis by F1F0-ATP synthase. Biochim. Biophys. Acta BBA Bioenerg. 2002, 1553, 188–211. [Google Scholar] [CrossRef]
- Petersen, J.; Förster, K.; Turina, P.; Gräber, P. Comparison of the H+/ATP ratios of the H+-ATP synthases from yeast and from chloroplast. Proc. Natl. Acad. Sci. USA 2012, 109, 11150–11155. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, A.; Reynafarje, B.; Lehninger, A.L. Stoichiometry of vectorial H+ movements coupled to electron transport and to ATP synthesis in mitochondria. Proc. Natl. Acad. Sci. USA 1978, 75, 5296–5300. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Pedersen, P.L. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol. Mol. Biol. Rev. 2008, 72, 590–641. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef]
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kühlbrandt, W. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc. Natl. Acad. Sci. USA 2019, 116, 4250–4255. [Google Scholar] [CrossRef] [Green Version]
- Campanella, M.; Parker, N.; Tan, C.H.; Hall, A.M.; Duchen, M.R. IF(1): Setting the pace of the F(1)F(o)-ATP synthase. Trends Biochem. Sci. 2009, 34, 343–350. [Google Scholar] [CrossRef]
- García-Bermúdez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta 2016, 1857, 1167–1182. [Google Scholar] [CrossRef]
- Chinopoulos, C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011, 585, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP Synthase K+- and H+-Fluxes Drive ATP Synthesis and Enable Mitochondrial K+-”Uniporter” Function: I. Characterization of Ion Fluxes. Function 2022, 3, zqab065. [Google Scholar] [CrossRef]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP Synthase K+- and H+-fluxes Drive ATP Synthesis and Enable Mitochondrial K+-”Uniporter” Function: II. Ion and ATP Synthase Flux Regulation. Function 2022, 3, zqac001. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K+ cycle. Biochim. Biophys. Acta BBA Bioenerg. 2003, 1606, 23–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.; Moyle, J. Translocation of some anions cations and acids in rat liver mitochondria. Eur. J. Biochem. 1969, 9, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.-C.; Liu, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: Low and high affinity effects of cyclosporine A. Biochim. Biophys. Acta 2011, 1813, 1373–1381. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004, 87, 2060–2073. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; O’Rourke, B.; Jacques, R.; Tseng, H.-J.; Marbán, E.; Winslow, R.L. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 2006, 91, 1564–1589. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; O’Rourke, B. Cardiac mitochondrial network excitability: Insights from computational analysis. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2178–H2189. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Cortassa, S.; Wei, A.-C.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys. J. 2009, 97, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Solhjoo, S.; Millare, B.; Plank, G.; Abraham, M.R.; Cortassa, S.; Trayanova, N.; O’Rourke, B. Effects of regional mitochondrial depolarization on electrical propagation: Implications for arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2014, 7, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Sollott, S.J. Computational modeling of mitochondrial K+- and H+-driven ATP synthesis. J. Mol. Cell. Cardiol. 2022, 165, 9–18. [Google Scholar] [CrossRef]
- Nguyen, M.-H.T.; Dudycha, S.J.; Jafri, M.S. Effect of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am. J. Physiol. Cell Physiol. 2007, 292, C2004–C2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metelkin, E.; Demin, O.; Kovács, Z.; Chinopoulos, C. Modeling of ATP-ADP steady-state exchange rate mediated by the adenine nucleotide translocase in isolated mitochondria. FEBS J. 2009, 276, 6942–6955. [Google Scholar] [CrossRef] [PubMed]
- Chance, E.M. A computer simulation of oxidative phosphorylation. Comput. Biomed. Res. 1967, 1, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Bohnensack, R. Control of energy transformation of mitochondria. Analysis by a quantitative model. Biochim. Biophys. Acta 1981, 634, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Holzhütter, H.G.; Henke, W.; Dubiel, W.; Gerber, G. A mathematical model to study short-term regulation of mitochondrial energy transduction. Biochim. Biophys. Acta 1985, 810, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.P.J.; Vanlier, J.; van Riel, N.A.W.; Jeneson, J.A.L. Computational modeling of mitochondrial energy transduction. Crit. Rev. Biomed. Eng. 2011, 39, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Mazat, J.-P.; Ransac, S.; Heiske, M.; Devin, A.; Rigoulet, M. Mitochondrial energetic metabolism-some general principles. IUBMB Life 2013, 65, 171–179. [Google Scholar] [CrossRef]
- Korzeniewski, B.; Froncisz, W. Theoretical studies on the control of the oxidative phosphorylation system. Biochim. Biophys. Acta 1992, 1102, 67–75. [Google Scholar] [CrossRef]
- Korzeniewski, B. The modeling of oxidative phosphorylation in skeletal muscle. Jpn. J. Physiol. 2004, 54, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Takeuchi, A.; Himeno, Y.; Inagaki, N.; Matsuoka, S. A simulation study on the constancy of cardiac energy metabolites during workload transition. J. Physiol. 2016, 594, 6929–6945. [Google Scholar] [CrossRef]
- LaNoue, K.; Nicklas, W.J.; Williamson, J.R. Control of citric acid cycle activity in rat heart mitochondria. J. Biol. Chem. 1970, 245, 102–111. [Google Scholar] [CrossRef]
- Heiske, M.; Nazaret, C.; Mazat, J.-P. Modeling the respiratory chain complexes with biothermokinetic equations—The case of complex I. Biochim. Biophys. Acta 2014, 1837, 1707–1716. [Google Scholar] [CrossRef]
- Duong, Q.V.; Levitsky, Y.; Dessinger, M.J.; Strubbe-Rivera, J.O.; Bazil, J.N. Identifying Site-Specific Superoxide and Hydrogen Peroxide Production Rates from the Mitochondrial Electron Transport System Using a Computational Strategy. Function 2021, 2, zqab050. [Google Scholar] [CrossRef]
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef] [Green Version]
- Magnus, G.; Keizer, J. Model of β-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am. J. Physiol. Cell Physiol. 1998, 274, C1158–C1173. [Google Scholar] [CrossRef]
- Magnus, G.; Keizer, J. Model of β-cell mitochondrial calcium handling and electrical activity. II. Mitochondrial variables. Am. J. Physiol. Cell Physiol. 1998, 274, C1174–C1184. [Google Scholar] [CrossRef]
- Jafri, M.S.; Dudycha, S.J. Increases in mitochondrial Ca can result in increases in NADH production. Biophys. J. 2001, 80, 589. [Google Scholar]
- Bertram, R.; Gram Pedersen, M.; Luciani, D.S.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Saa, A.; Siqueira, K.M. Modeling the ATP production in mitochondria. Bull. Math. Biol. 2013, 75, 1636–1651. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.-H.T.; Jafri, M.S. Mitochondrial calcium signaling and energy metabolism. Ann. N. Y. Acad. Sci. 2005, 1047, 127–137. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Marbán, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010, 6, e1000657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Ernst, P.; Song, J.; Liu, X.M.; Huke, S.; Wang, S.; Zhang, J.J.; Zhou, L. Mitochondrial-Mediated Oxidative Ca2+/Calmodulin-Dependent Kinase II Activation Induces Early Afterdepolarizations in Guinea Pig Cardiomyocytes: An in Silico Study. J. Am. Heart Assoc. 2018, 7, e008939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Su, D.; O’Rourke, B.; Pogwizd, S.M.; Zhou, L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: A theoretical study. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H623–H636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortassa, S.; Sollott, S.J.; Aon, M.A. Mitochondrial respiration and ROS emission during β-oxidation in the heart: An experimental-computational study. PLoS Comput. Biol. 2017, 13, e1005588. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Ripple, M.O.; Springett, R. Measurement of the mitochondrial membrane potential and pH gradient from the redox poise of the hemes of the bc1 complex. Biophys. J. 2012, 102, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, B.L.; Niederer, S. A biophysical systems approach to identifying the pathways of acute and chronic doxorubicin mitochondrial cardiotoxicity. PLoS Comput. Biol. 2016, 12, e1005214. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.F.; Vinogradov, S.A. Mitochondrial cytochrome c oxidase: Mechanism of action and role in regulating oxidative phosphorylation. J. Appl. Physiol. 2014, 117, 1431–1439. [Google Scholar] [CrossRef] [Green Version]
- Pereira, E.J.; Smolko, C.M.; Janes, K.A. Computational Models of Reactive Oxygen Species as Metabolic Byproducts and Signal-Transduction Modulators. Front. Pharmacol. 2016, 7, 457. [Google Scholar] [CrossRef] [Green Version]
- Mazat, J.-P.; Devin, A.; Ransac, S. Modelling mitochondrial ROS production by the respiratory chain. Cell. Mol. Life Sci. 2020, 77, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Kembro, J.M.; Cortassa, S.; Aon, M.A. Complex oscillatory redox dynamics with signaling potential at the edge between normal and pathological mitochondrial function. Front. Physiol. 2014, 5, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metelkin, E.; Goryanin, I.; Demin, O. Mathematical modeling of mitochondrial adenine nucleotide translocase. Biophys. J. 2006, 90, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saks, V.A.; Kongas, O.; Vendelin, M.; Kay, L. Role of the creatine/phosphocreatine system in the regulation of mitochondrial respiration. Acta Physiol. Scand. 2000, 168, 635–641. [Google Scholar] [CrossRef]
- Meyer, L.E.; Machado, L.B.; Santiago, A.P.S.A.; da-Silva, W.S.; De Felice, F.G.; Holub, O.; Oliveira, M.F.; Galina, A. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: Antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J. Biol. Chem. 2006, 281, 37361–37371. [Google Scholar] [CrossRef] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Vendelin, M.; Lemba, M.; Saks, V.A. Analysis of functional coupling: Mitochondrial creatine kinase and adenine nucleotide translocase. Biophys. J. 2004, 87, 696–713. [Google Scholar] [CrossRef] [Green Version]
- Hettling, H.; van Beek, J.H.G.M. Analyzing the functional properties of the creatine kinase system with multiscale “sloppy” modeling. PLoS Comput. Biol. 2011, 7, e1002130. [Google Scholar] [CrossRef]
- Fridlyand, L.E.; Philipson, L.H. Glucose sensing in the pancreatic beta cell: A computational systems analysis. Theor. Biol. Med. Model. 2010, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Genova, M.L. Mobility and function of coenzyme Q (ubiquinone) in the mitochondrial respiratory chain. Biochim. Biophys. Acta 2009, 1787, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: Random collisions vs. solid state electron channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, A.; Okada, J.-I.; Washio, T.; Hisada, T.; Sugiura, S. Distinct functional roles of cardiac mitochondrial subpopulations revealed by a 3D simulation model. Biophys. J. 2015, 108, 2732–2739. [Google Scholar] [CrossRef] [Green Version]
- Occhipinti, R.; Somersalo, E.; Calvetti, D. Astrocytes as the glucose shunt for glutamatergic neurons at high activity: An in silico study. J. Neurophysiol. 2009, 101, 2528–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloutier, M.; Bolger, F.B.; Lowry, J.P.; Wellstead, P. An integrative dynamic model of brain energy metabolism using in vivo neurochemical measurements. J. Comput. Neurosci. 2009, 27, 391–414. [Google Scholar] [CrossRef] [Green Version]
- Cakir, T.; Alsan, S.; Saybaşili, H.; Akin, A.; Ulgen, K.O. Reconstruction and flux analysis of coupling between metabolic pathways of astrocytes and neurons: Application to cerebral hypoxia. Theor. Biol. Med. Model. 2007, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Nadanaciva, S.; Will, Y.; Woodhead, J.L.; Howell, B.A.; Watkins, P.B.; Siler, S.Q. Mitosym®: A mechanistic, mathematical model of hepatocellular respiration and bioenergetics. Pharm. Res. 2015, 32, 1975–1992. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lai, N.; Kirwan, J.P.; Saidel, G.M. Computational Model of Cellular Metabolic Dynamics in Skeletal Muscle Fibers during Moderate Intensity Exercise. Cell. Mol. Bioeng. 2012, 5, 92–112. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.K.; Dibella, J.A.; Cabrera, M.E. A computational model of skeletal muscle metabolism linking cellular adaptations induced by altered loading states to metabolic responses during exercise. Biomed. Eng. Online 2007, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Benard, G.; Faustin, B.; Passerieux, E.; Galinier, A.; Rocher, C.; Bellance, N.; Delage, J.P.; Casteilla, L.; Letellier, T.; Rossignol, R. Physiological diversity of mitochondrial oxidative phosphorylation. Am. J. Physiol. Cell Physiol. 2006, 291, C1172–C1182. [Google Scholar] [CrossRef] [Green Version]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Niederer, S.A.; Lumens, J.; Trayanova, N.A. Computational models in cardiology. Nat. Rev. Cardiol. 2019, 16, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Kernik, D.C.; Morotti, S.; Wu, H.; Garg, P.; Duff, H.J.; Kurokawa, J.; Jalife, J.; Wu, J.C.; Grandi, E.; Clancy, C.E. A computational model of induced pluripotent stem-cell derived cardiomyocytes incorporating experimental variability from multiple data sources. J. Physiol. 2019, 597, 4533–4564. [Google Scholar] [CrossRef] [Green Version]
- Moser, C.C.; Farid, T.A.; Chobot, S.E.; Dutton, P.L. Electron tunneling chains of mitochondria. Biochim. Biophys. Acta 2006, 1757, 1096–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Hummer, G. Proton-pumping mechanism of cytochrome c oxidase: A kinetic master-equation approach. Biochim. Biophys. Acta 2012, 1817, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maarleveld, T.R.; Khandelwal, R.A.; Olivier, B.G.; Teusink, B.; Bruggeman, F.J. Basic concepts and principles of stoichiometric modeling of metabolic networks. Biotechnol. J. 2013, 8, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.M.; Taha, F.; Rahman, J.; Rahman, S. Systems biology approaches toward understanding primary mitochondrial diseases. Front. Genet. 2019, 10, 19. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, W.-W.; Wei, A.-C. Kinetic Mathematical Modeling of Oxidative Phosphorylation in Cardiomyocyte Mitochondria. Cells 2022, 11, 4020. https://doi.org/10.3390/cells11244020
Tseng W-W, Wei A-C. Kinetic Mathematical Modeling of Oxidative Phosphorylation in Cardiomyocyte Mitochondria. Cells. 2022; 11(24):4020. https://doi.org/10.3390/cells11244020
Chicago/Turabian StyleTseng, Wen-Wei, and An-Chi Wei. 2022. "Kinetic Mathematical Modeling of Oxidative Phosphorylation in Cardiomyocyte Mitochondria" Cells 11, no. 24: 4020. https://doi.org/10.3390/cells11244020