
Senescent Cells: Dual Implications on the Retinal Vascular System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Cellular Senescence
1.1. Cell Senescence and Inducers
1.2. Cell Cycle Arrest
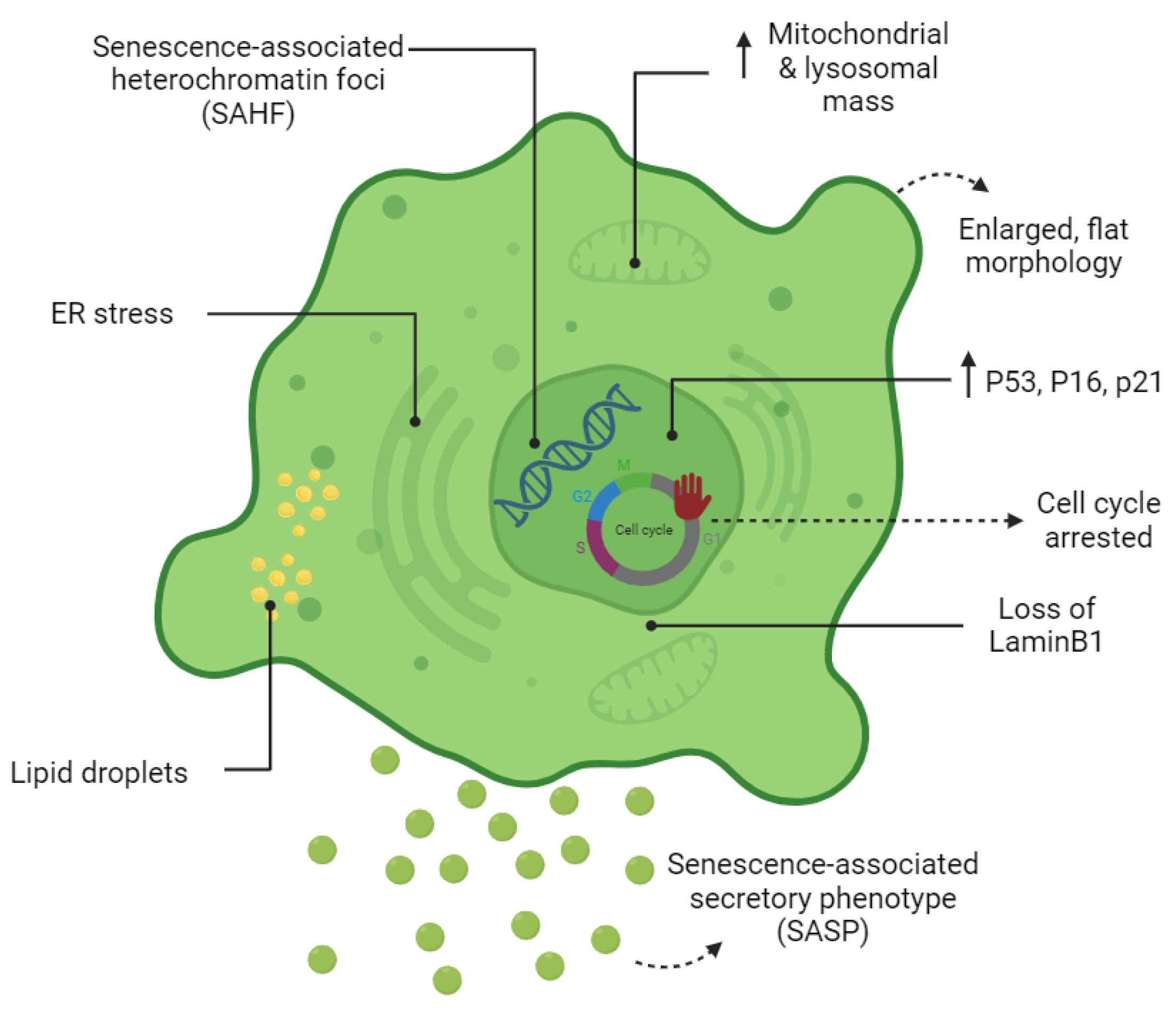
1.3. The Senescent Phenotype
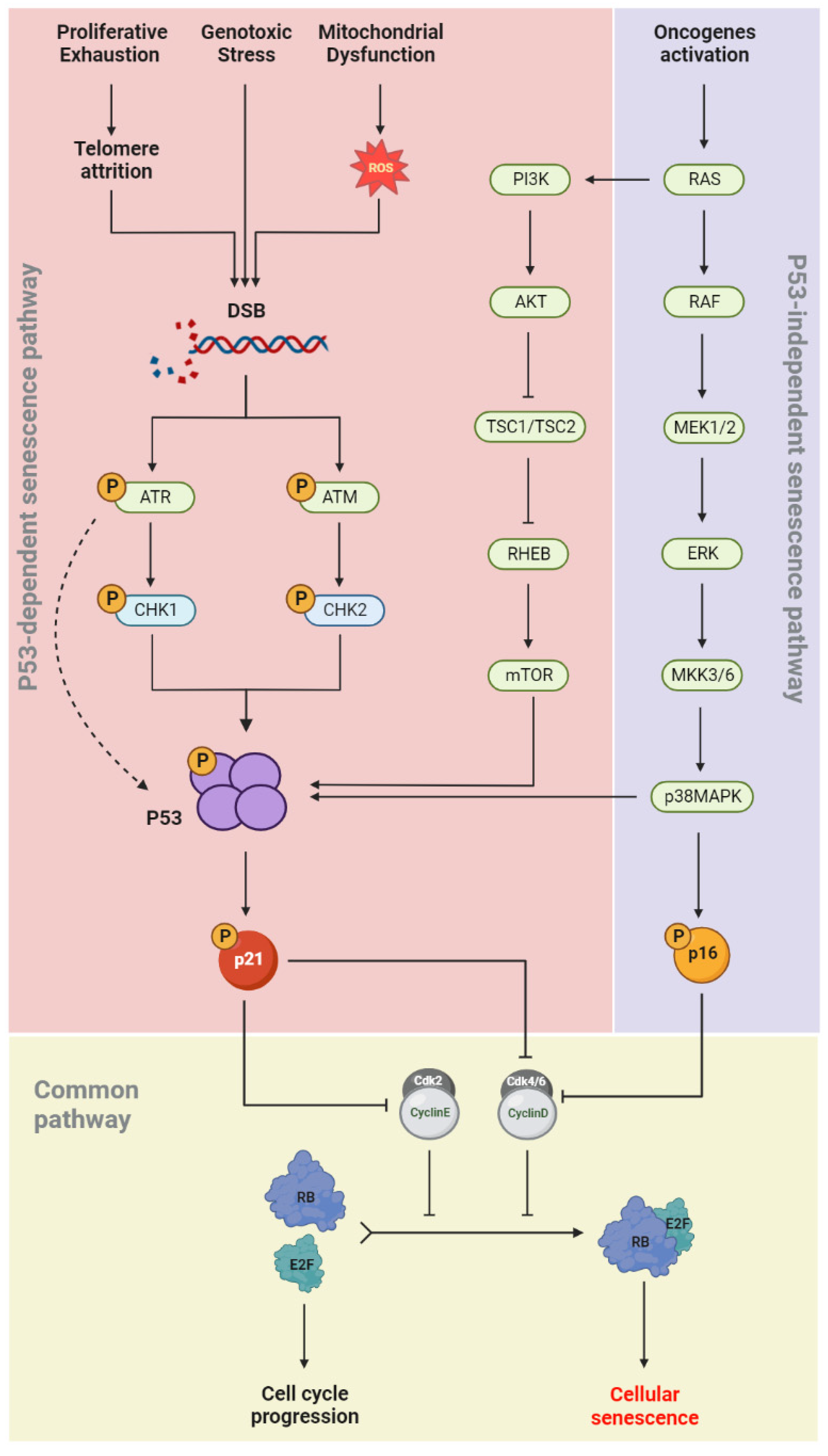
1.4. Senescence Molecular Pathways
1.5. Autophagy: A Pro-Senescence or an Anti-Senescence Mechanism?
2. Regulatory Mechanisms of the SASP
2.1. NF-κB Signaling
2.2. cGAS/STING Signaling
2.3. mTOR Signaling
2.4. C/EBPβ Signaling
2.5. NOTCH1 Signaling
2.6. JAK-STAT Signaling
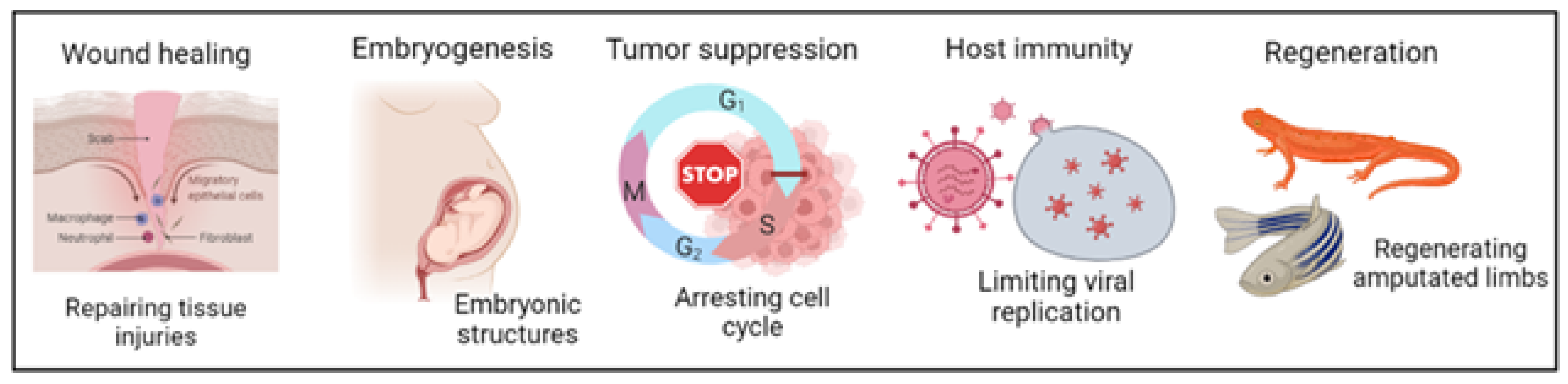
3. Physiological Roles of Cellular Senescence
3.1. Embryogenesis
3.2. Regeneration
3.3. Wound Healing
4. Retinal Vascular System
4.1. Retina
4.2. Retinal Blood Vessel Development
4.3. Vasculogenesis
4.4. Angiogenesis
4.5. Retinal Vasculature in Eye Diseases
4.5.1. Retinopathy of Prematurity (ROP)
4.5.2. Diabetic Retinopathy (DR)
4.5.3. Age-Related Macular Degeneration (AMD)
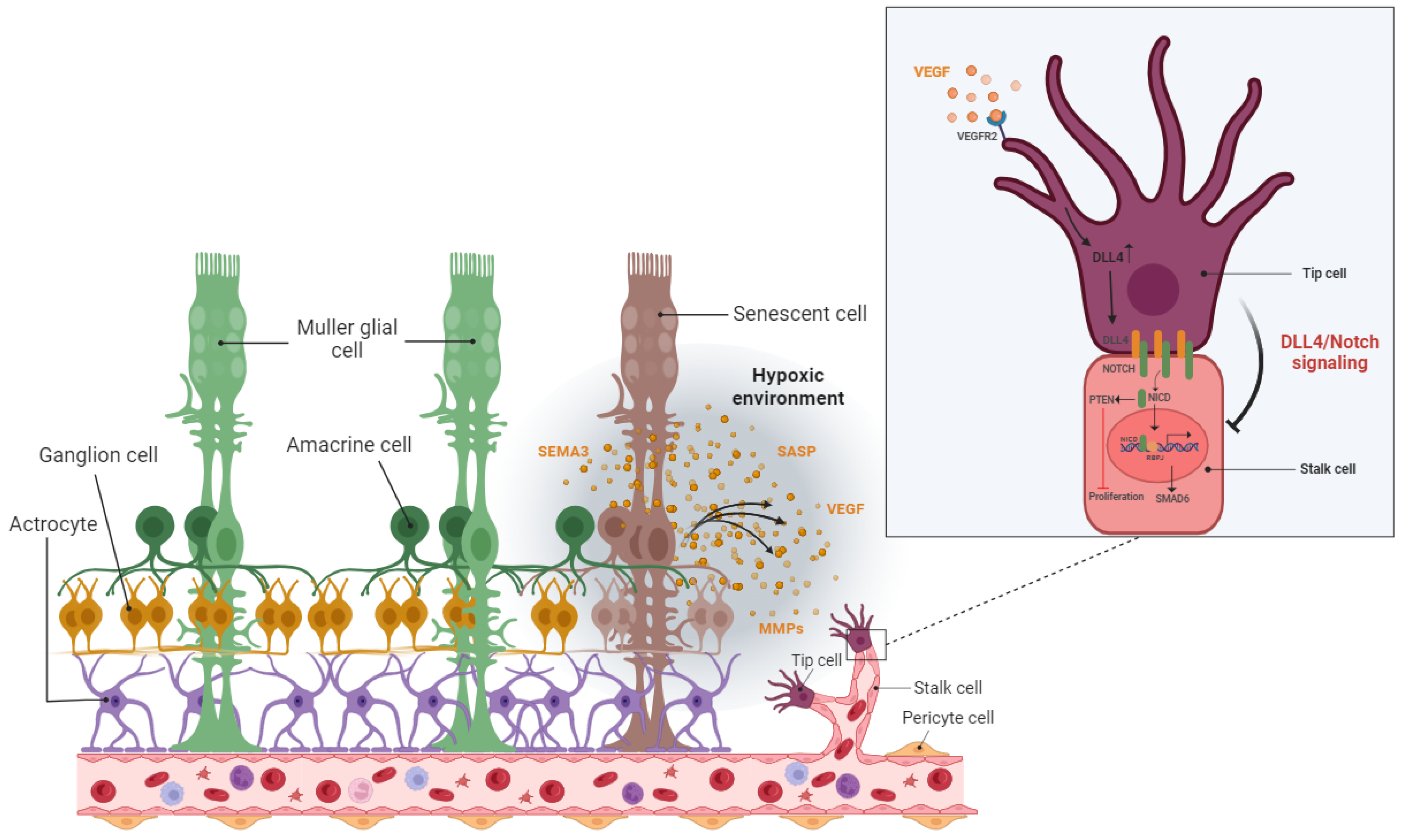
5. Implications of SCs on the Retinal Vascular System
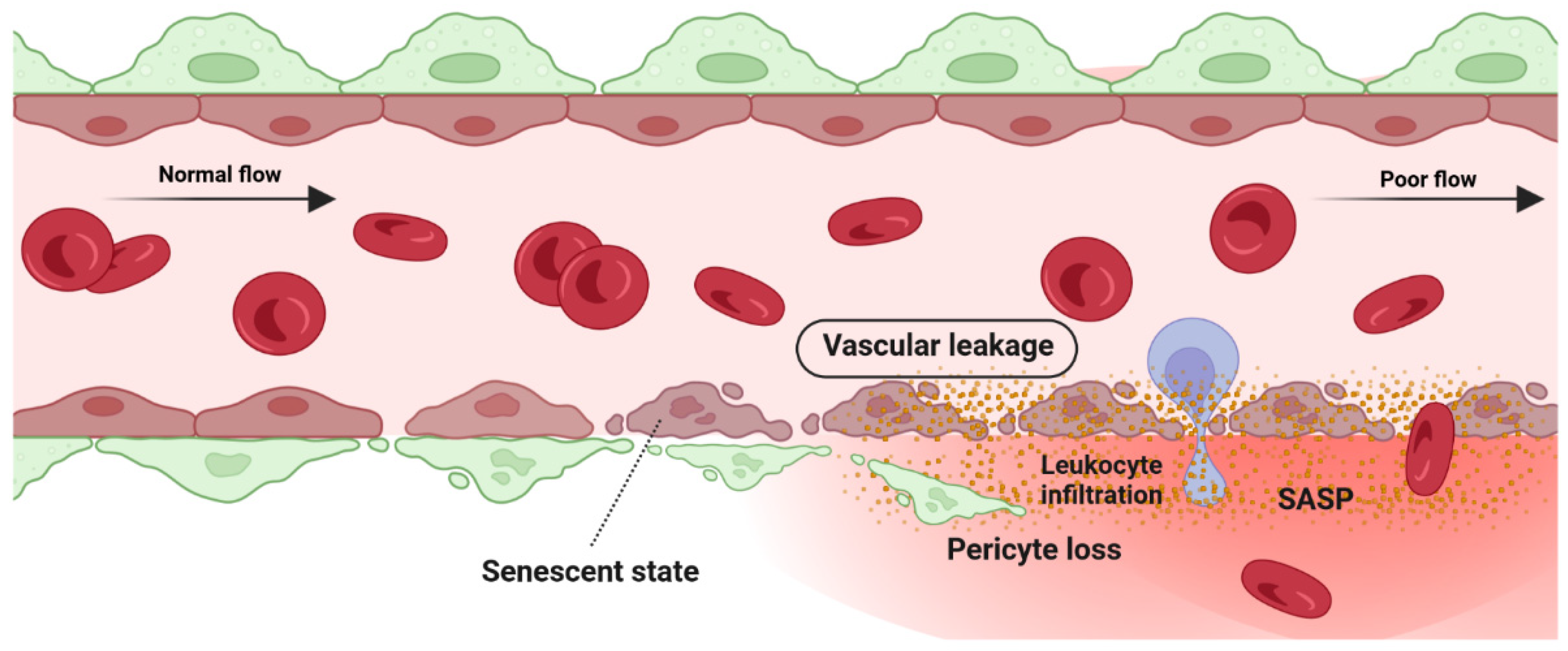
5.1. Detrimental Effect: Vascular Degeneration
5.2. Detrimental Effect: Pathological Angiogenesis
5.3. Beneficial Effect: Vascular Remodeling
6. Therapeutic Targeting of Senescent Cells
6.1. Senolytics
6.2. Senomorphics
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Rabiee, A.; Kovary, K.M.; Bahrami-Nejad, Z.; Taylor, B.; Teruel, M.N. Molecular Competition in G1 Controls When Cells Simultaneously Commit to Terminally Differentiate and Exit the Cell Cycle. Cell Rep. 2020, 31, 107769. [Google Scholar] [CrossRef]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef]
- Rumman, M.; Dhawan, J.; Kassem, M. Concise Review: Quiescence in Adult Stem Cells: Biological Significance and Relevance to Tissue Regeneration. Stem. Cells 2015, 33, 2903–2912. [Google Scholar] [CrossRef]
- Maciel-Baron, L.A.; Moreno-Blas, D.; Morales-Rosales, S.L.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Castro-Obregon, S.; Konigsberg, M. Cellular Senescence, Neurological Function, and Redox State. Antioxid Redox. Signal 2018, 28, 1704–1723. [Google Scholar] [CrossRef]
- Cho, I.J.; Lui, P.P.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.L.; Spagnoli, F.; Watt, F.M. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Rep. 2019, 12, 1190–1200. [Google Scholar] [CrossRef]
- De Morree, A.; Rando, T.A. Regulation of adult stem cell quiescence and its functions in the maintenance of tissue integrity. Nat. Rev. Mol. Cell Biol. 2023, 24, 334–354. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Vergel, M.; Marin, J.J.; Estevez, P.; Carnero, A. Cellular senescence as a target in cancer control. J. Aging Res. 2010, 2011, 725365. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene 2008, 27, 7070–7082. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.; Pacios-Bras, C.; Choi, J.; Yenjerla, M.; Sussman, M.A.; Fotedar, R.; Margolis, R.L. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol. Biol. Cell 2014, 25, 3105–3118. [Google Scholar] [CrossRef]
- Walen, K.H. Human diploid fibroblast cells in senescence; cycling through polyploidy to mitotic cells. Vitr. Cell Dev. Biol. Anim. 2006, 42, 216–224. [Google Scholar] [CrossRef]
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496. [Google Scholar] [CrossRef]
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Diaz, A.; Garcia, F.; Munoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. The aging myocardium: Roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. 2005, 14, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis Rev. 2021, 40, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Zwerschke, W.; Mazurek, S.; Stockl, P.; Hutter, E.; Eigenbrodt, E.; Jansen-Durr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Invest. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef]
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Cormenier, J.; Martin, N.; Desle, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6alpha arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10 (Suppl 1), S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Cassidy, L.D.; Narita, M. Autophagy and senescence, converging roles in pathophysiology as seen through mouse models. Adv. Cancer Res. 2021, 150, 113–145. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol. Cells 2017, 40, 607–612. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy 2016, 12, 1158373. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef]
- Ozcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 2016, 8, 1316–1329. [Google Scholar] [CrossRef]
- Severino, V.; Alessio, N.; Farina, A.; Sandomenico, A.; Cipollaro, M.; Peluso, G.; Galderisi, U.; Chambery, A. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013, 4, e911. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Baron, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martinez, F.; Galvan-Arzate, S.; Luna-Lopez, A.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Konigsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 2016, 38, 26. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- Wu, Z.H.; Wong, E.T.; Shi, Y.; Niu, J.; Chen, Z.; Miyamoto, S.; Tergaonkar, V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell 2010, 40, 75–86. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Prasnikar, E.; Borisek, J.; Perdih, A. Senescent cells as promising targets to tackle age-related diseases. Ageing Res. Rev. 2021, 66, 101251. [Google Scholar] [CrossRef]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.Y.; Londono, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Sun, Y.; Coppe, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Zullo, J.M.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e745. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Serrano, M. The InflammTORy Powers of Senescence. Trends Cell Biol. 2015, 25, 634–636. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e2046. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, X.; Mao, L.; Wang, S. The CCAAT/Enhancer-Binding Protein Family: Its Roles in MDSC Expansion and Function. Front. Immunol. 2019, 10, 1804. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Sebastian, T.; Malik, R.; Thomas, S.; Sage, J.; Johnson, P.F. C/EBPbeta cooperates with RB:E2F to implement Ras(V12)-induced cellular senescence. EMBO J. 2005, 24, 3301–3312. [Google Scholar] [CrossRef]
- Huggins, C.J.; Malik, R.; Lee, S.; Salotti, J.; Thomas, S.; Martin, N.; Quinones, O.A.; Alvord, W.G.; Olanich, M.E.; Keller, J.R.; et al. C/EBPgamma suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPbeta. Mol. Cell Biol. 2013, 33, 3242–3258. [Google Scholar] [CrossRef]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct Target Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Kirkland, J.L. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 2016, 111, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xiao, L.; Dai, G.; Lu, P.; Zhang, Y.; Li, Y.; Ni, M.; Rui, Y. Inhibition of JAK-STAT Signaling Pathway Alleviates Age-Related Phenotypes in Tendon Stem/Progenitor Cells. Front. Cell Dev. Biol. 2021, 9, 650250. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.X.; Jiang, L.J.; Chen, Q.; Wang, T.; Ye, D.W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; David, G. Pro- and anti-tumorigenic functions of the senescence-associated secretory phenotype. Expert. Opin. Ther. Targets 2019, 23, 1041–1051. [Google Scholar] [CrossRef]
- Davaapil, H.; Brockes, J.P.; Yun, M.H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 2017, 144, 106–114. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Villiard, E.; Denis, J.F.; Hashemi, F.S.; Igelmann, S.; Ferbeyre, G.; Roy, S. Senescence gives insights into the morphogenetic evolution of anamniotes. Biol. Open 2017, 6, 891–896. [Google Scholar] [CrossRef]
- Nacher, V.; Carretero, A.; Navarro, M.; Armengol, C.; Llombart, C.; Rodriguez, A.; Herrero-Fresneda, I.; Ayuso, E.; Ruberte, J. The quail mesonephros: A new model for renal senescence? J. Vasc. Res. 2006, 43, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bautista, J.A.; Acevo-Rodriguez, P.S.; Castro-Obregon, S. Programmed Cell Senescence in the Mouse Developing Spinal Cord and Notochord. Front. Cell Dev. Biol. 2021, 9, 587096. [Google Scholar] [CrossRef]
- Antelo-Iglesias, L.; Picallos-Rabina, P.; Estevez-Souto, V.; Da Silva-Alvarez, S.; Collado, M. The role of cellular senescence in tissue repair and regeneration. Mech. Ageing Dev. 2021, 198, 111528. [Google Scholar] [CrossRef]
- Eguchi, G.; Eguchi, Y.; Nakamura, K.; Yadav, M.C.; Millan, J.L.; Tsonis, P.A. Regenerative capacity in newts is not altered by repeated regeneration and ageing. Nat. Commun. 2011, 2, 384. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 2015, 4, e05505. [Google Scholar] [CrossRef]
- Elder, S.S.; Emmerson, E. Senescent cells and macrophages: Key players for regeneration? Open Biol. 2020, 10, 200309. [Google Scholar] [CrossRef]
- Azevedo, A.S.; Grotek, B.; Jacinto, A.; Weidinger, G.; Saude, L. The regenerative capacity of the zebrafish caudal fin is not affected by repeated amputations. PLoS ONE 2011, 6, e22820. [Google Scholar] [CrossRef]
- Itou, J.; Kawakami, H.; Burgoyne, T.; Kawakami, Y. Life-long preservation of the regenerative capacity in the fin and heart in zebrafish. Biol. Open 2012, 1, 739–746. [Google Scholar] [CrossRef]
- Da Silva-Alvarez, S.; Guerra-Varela, J.; Sobrido-Camean, D.; Quelle, A.; Barreiro-Iglesias, A.; Sanchez, L.; Collado, M. Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell 2020, 19, e13052. [Google Scholar] [CrossRef]
- Bednarek, D.; Gonzalez-Rosa, J.M.; Guzman-Martinez, G.; Gutierrez-Gutierrez, O.; Aguado, T.; Sanchez-Ferrer, C.; Marques, I.J.; Galardi-Castilla, M.; de Diego, I.; Gomez, M.J.; et al. Telomerase Is Essential for Zebrafish Heart Regeneration. Cell Rep. 2015, 12, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Meng, J.; Kou, S.; Jiang, Z.; Huang, X.; Lu, Z.; Zhao, H.; Lau, L.F.; Zhou, B.; Zhang, H. CCN1-Induced Cellular Senescence Promotes Heart Regeneration. Circulation 2019, 139, 2495–2498. [Google Scholar] [CrossRef]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef]
- Chen, J.; Liu, C.-H.; Sapieha, P. Retinal Vascular Development; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Sun, Y.; Smith, L.E.H. Retinal Vasculature in Development and Diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; McClung, J.A.; Aronow, W.S. Translational Research in Coronary Artery Disease, 1 ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar] [CrossRef]
- Uhrin, P. Cellular and Molecular Mechanisms of Vasculogenesis, Angiogenesis, and Lymphangiogenesis; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Asahara, T.; Kawamoto, A. Endothelial progenitor cells for postnatal vasculogenesis. Am. J. Physiol. Cell Physiol. 2004, 287, C572–C579. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Hantusch, B. Morphological and Functional Characteristics of Blood and Lymphatic Vessels; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–43. [Google Scholar] [CrossRef]
- McLeod, D.S.; Hasegawa, T.; Prow, T.; Merges, C.; Lutty, G. The initial fetal human retinal vasculature develops by vasculogenesis. Dev. Dyn. 2006, 235, 3336–3347. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Keramidas, M.; Lamande, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef]
- Larrivee, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Mahmoud, M.; Allinson, K.R.; Redgrave, R.E.; Zhai, Z.; Oh, S.P.; Fruttiger, M.; Arthur, H.M. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE 2014, 9, e98646. [Google Scholar] [CrossRef] [PubMed]
- Bourne, R.; Steinmetz, J.D.; Flaxman, S.; Briant, P.S.; Taylor, H.R.; Resnikoff, S.; Casson, R.J.; Abdoli, A.; Abu-Gharbieh, E.; Afshin, A.; et al. Trends in prevalence of blindness and distance and near vision impairment over 30 years: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e130–e143. [Google Scholar] [CrossRef] [PubMed]
- Lad, E.M.; Hernandez-Boussard, T.; Morton, J.M.; Moshfeghi, D.M. Incidence of retinopathy of prematurity in the United States: 1997 through 2005. Am. J. Ophthalmol. 2009, 148, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Good, W.V.; Early Treatment for Retinopathy of Prematurity Cooperative Group. Final results of the Early Treatment for Retinopathy of Prematurity (ETROP) randomized trial. Trans. Am. Ophthalmol. Soc. 2004, 102, 233–248; discussion 248–250. [Google Scholar]
- Forrester, J.; Gardner, T.W.; Kirchhof, B.; Ryan, S.J. Retinal Vascular Disease; Springer: Berlin/Heidelberg, Germany, 2010; Volume 247, p. 816. [Google Scholar]
- Forrester, J. Retinal vascular disease by A.M. Joussen, T.W. Gardner, B. Kirchhof, S.J. Ryan. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 143–144. [Google Scholar] [CrossRef]
- Olivares, A.M.; Althoff, K.; Chen, G.F.; Wu, S.; Morrisson, M.A.; DeAngelis, M.M.; Haider, N. Animal Models of Diabetic Retinopathy. Curr. Diab. Rep. 2017, 17, 93. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the Epidemiology of Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.A.; Williams, H.; George, S.J. Evidence for the Involvement of Matrix-Degrading Metalloproteinases (MMPs) in Atherosclerosis. Prog. Mol. Biol. Transl. Sci. 2017, 147, 197–237. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, P.M.; Pedrini, E.; Hughes, D.; McDonnell, S.; Pathak, V.; Peixoto, E.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. Long term high glucose exposure induces premature senescence in retinal endothelial cells. Front. Physiol. 2022, 13, 929118. [Google Scholar] [CrossRef]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [CrossRef]
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef]
- Del Cuore, A.; Pacinella, G.; Riolo, R.; Tuttolomondo, A. The Role of Immunosenescence in Cerebral Small Vessel Disease: A Review. Int. J. Mol. Sci. 2022, 23, 7136. [Google Scholar] [CrossRef]
- Cheung, T.M.; Ganatra, M.P.; Peters, E.B.; Truskey, G.A. Effect of cellular senescence on the albumin permeability of blood-derived endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1374–H1383. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Yamada, T.; Tai-Nagara, I.; Okabe, K.; Kitagawa, Y.; Ema, M.; Kubota, Y. Developmental regression of hyaloid vasculature is triggered by neurons. J. Exp. Med. 2016, 213, 1175–1183. [Google Scholar] [CrossRef]
- Hamrick, S.E.G.; Sallmon, H.; Rose, A.T.; Porras, D.; Shelton, E.L.; Reese, J.; Hansmann, G. Patent Ductus Arteriosus of the Preterm Infant. Pediatrics 2020, 146, e20201209. [Google Scholar] [CrossRef]
- Girling, J.E.; Rogers, P.A. Regulation of endometrial vascular remodelling: Role of the vascular endothelial growth factor family and the angiopoietin-TIE signalling system. Reproduction 2009, 138, 883–893. [Google Scholar] [CrossRef]
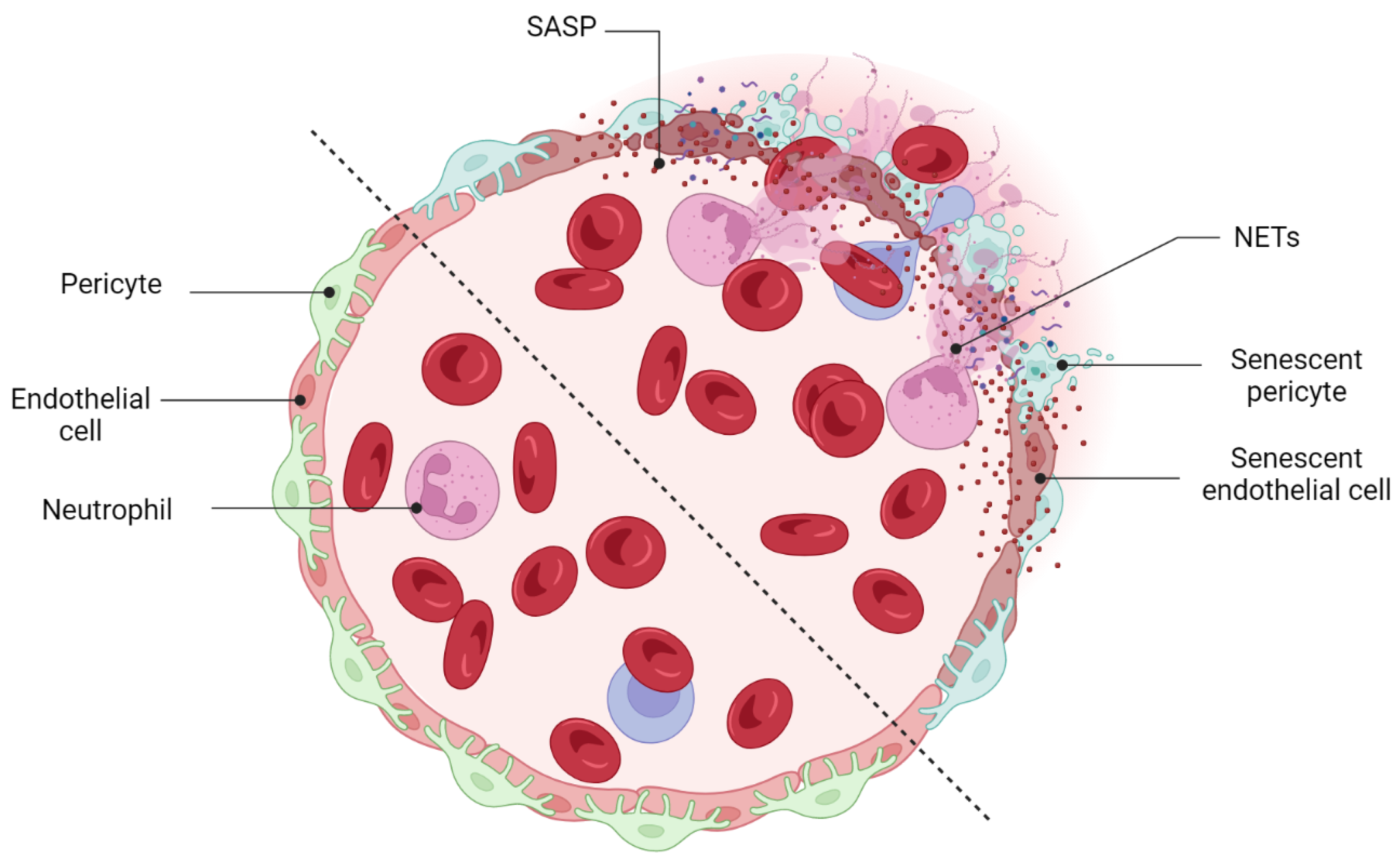
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E.; Penn, J.S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Klotzsche-von Ameln, A.; Sprott, D. Harnessing retinal phagocytes to combat pathological neovascularization in ischemic retinopathies? Pflug. Arch. 2022, 474, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Ju, R.H.; Zhang, J.Q.; Ke, X.Y.; Lu, X.H.; Liang, L.F.; Wang, W.J. Spontaneous regression of retinopathy of prematurity: Incidence and predictive factors. Int. J. Ophthalmol. 2013, 6, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Han, J.R.; Ju, W.K.; Park, I.W. Spontaneous regression of neovascularization at the disc in diabetic retinopathy. Korean J. Ophthalmol. 2004, 18, 41–46. [Google Scholar] [CrossRef]
- Bandello, F.; Gass, J.D.; Lattanzio, R.; Brancato, R. Spontaneous regression of neovascularization at the disk and elsewhere in diabetic retinopathy. Am. J. Ophthalmol. 1996, 122, 494–501. [Google Scholar] [CrossRef]
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 179–185. [Google Scholar] [CrossRef]
- Melo, L.G.; Gnecchi, M.; Ward, C.A.; Dzau, V.J. Vascular Remodeling in Health and Disease. J. Cardiovasc. Med. 2007, 1541–1565. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Egashira, M.; Hirota, Y.; Shimizu-Hirota, R.; Saito-Fujita, T.; Haraguchi, H.; Matsumoto, L.; Matsuo, M.; Hiraoka, T.; Tanaka, T.; Akaeda, S.; et al. F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinology 2017, 158, 2344–2353. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Talia, D.M.; Zhu, T.; Maxwell, M.J.; Agrotis, A.; Jerome, J.R.; Hargreaves, E.M.; Gerondakis, S.; Hibbs, M.L.; Mackay, F.; et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat. Commun. 2017, 8, 748. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Hernandez, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Rhoades, W.R.; Hanout, M.; Soliman, M.K.; Sarwar, S.; Sadiq, M.A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Management of neovascular age-related macular degeneration: Current state-of-the-art care for optimizing visual outcomes and therapies in development. Clin. Ophthalmol. 2015, 9, 1001–1015. [Google Scholar] [CrossRef]
- Sapieha, P.; Joyal, J.S.; Rivera, J.C.; Kermorvant-Duchemin, E.; Sennlaub, F.; Hardy, P.; Lachapelle, P.; Chemtob, S. Retinopathy of prematurity: Understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 2010, 120, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 582060. [Google Scholar] [CrossRef]
- Arroba, A.I.; Alcalde-Estevez, E.; Garcia-Ramirez, M.; Cazzoni, D.; de la Villa, P.; Sanchez-Fernandez, E.M.; Mellet, C.O.; Garcia Fernandez, J.M.; Hernandez, C.; Simo, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. Biochim. Biophys. Acta 2016, 1862, 1663–1674. [Google Scholar] [CrossRef]
- Zeng, H.Y.; Green, W.R.; Tso, M.O. Microglial activation in human diabetic retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef]
- Li, J.; Yu, S.; Lu, X.; Cui, K.; Tang, X.; Xu, Y.; Liang, X. The phase changes of M1/M2 phenotype of microglia/macrophage following oxygen-induced retinopathy in mice. Inflamm. Res. 2021, 70, 183–192. [Google Scholar] [CrossRef]
- Zhou, Y.; Yoshida, S.; Nakao, S.; Yoshimura, T.; Kobayashi, Y.; Nakama, T.; Kubo, Y.; Miyawaki, K.; Yamaguchi, M.; Ishikawa, K.; et al. M2 Macrophages Enhance Pathological Neovascularization in the Mouse Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4767–4777. [Google Scholar] [CrossRef] [PubMed]
- Korovina, I.; Neuwirth, A.; Sprott, D.; Troullinaki, M.; Poitz, D.M.; Deussen, A.; Klotzsche-von Ameln, A. Myeloid SOCS3 Deficiency Regulates Angiogenesis via Enhanced Apoptotic Endothelial Cell Engulfment. J. Innate Immun. 2020, 12, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Korovina, I.; Neuwirth, A.; Sprott, D.; Weber, S.; Sardar Pasha, S.P.B.; Gercken, B.; Breier, G.; El-Armouche, A.; Deussen, A.; Karl, M.O.; et al. Hematopoietic hypoxia-inducible factor 2alpha deficiency ameliorates pathological retinal neovascularization via modulation of endothelial cell apoptosis. FASEB J. 2019, 33, 1758–1770. [Google Scholar] [CrossRef]
- He, P. Leucocyte/endothelium interactions and microvessel permeability: Coupled or uncoupled? Cardiovasc. Res. 2010, 87, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Cai, W.Y.; Suzuma, I.; Pak, J.; Ju, S.T.; Rook, S.L.; Esser, P.; Mitsiades, C.S.; et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. 2003, 17, 76–78. [Google Scholar] [CrossRef]
- Davies, M.H.; Stempel, A.J.; Powers, M.R. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Colman, R.J. Non-human primates as a model for aging. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2733–2741. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989, 5, 155–171; discussion 172. [Google Scholar] [PubMed]
- Martel, J.; Ojcius, D.M.; Ko, Y.F.; Chang, C.J.; Young, J.D. Antiaging effects of bioactive molecules isolated from plants and fungi. Med. Res. Rev. 2019, 39, 1515–1552. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535. [Google Scholar] [CrossRef]
- Tulipano, G. Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area. Int. J. Mol. Sci. 2021, 22, 3068. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Dowling, R.J.; Goodwin, P.J.; Stambolic, V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011, 9, 33. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Park, S.K.; Shin, O.S. Metformin alleviates ageing cellular phenotypes in Hutchinson-Gilford progeria syndrome dermal fibroblasts. Exp. Dermatol. 2017, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Halicka, H.D.; Zhao, H.; Li, J.; Traganos, F.; Zhang, S.; Lee, M.; Darzynkiewicz, Z. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging 2011, 3, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016, 5, e16351. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Vautier, M.; Allenbach, Y.; Zahr, N.; Benveniste, O.; Funck-Brentano, C.; Salem, J.E. Sirolimus and mTOR Inhibitors: A Review of Side Effects and Specific Management in Solid Organ Transplantation. Drug Saf. 2019, 42, 813–825. [Google Scholar] [CrossRef]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Wu, C.M.; Zheng, L.; Wang, Q.; Hu, Y.W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. 2020, 59, 27–38. [Google Scholar] [CrossRef]
- Muhlberger, D.; Zumholz, A.K.; Brenner, E.; Mumme, A.; Stucker, M.; Falkenstein, T.; Hummel, T. Cellular senescence at the saphenofemoral junction in patients with healthy, primary varicose and recurrent varicose veins—A pilot study. Vascular 2022, 30, 559–567. [Google Scholar] [CrossRef]
- Molnar, A.A.; Nadasy, G.L.; Dornyei, G.; Patai, B.B.; Delfavero, J.; Fulop, G.A.; Kirkpatrick, A.C.; Ungvari, Z.; Merkely, B. The aging venous system: From varicosities to vascular cognitive impairment. Geroscience 2021, 43, 2761–2784. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habibi-Kavashkohie, M.R.; Scorza, T.; Oubaha, M. Senescent Cells: Dual Implications on the Retinal Vascular System. Cells 2023, 12, 2341. https://doi.org/10.3390/cells12192341
Habibi-Kavashkohie MR, Scorza T, Oubaha M. Senescent Cells: Dual Implications on the Retinal Vascular System. Cells. 2023; 12(19):2341. https://doi.org/10.3390/cells12192341
Chicago/Turabian StyleHabibi-Kavashkohie, Mohammad Reza, Tatiana Scorza, and Malika Oubaha. 2023. "Senescent Cells: Dual Implications on the Retinal Vascular System" Cells 12, no. 19: 2341. https://doi.org/10.3390/cells12192341