

Altered Metabolism in Motor Neuron Diseases: Mechanism and Potential Therapeutic Target

Abstract
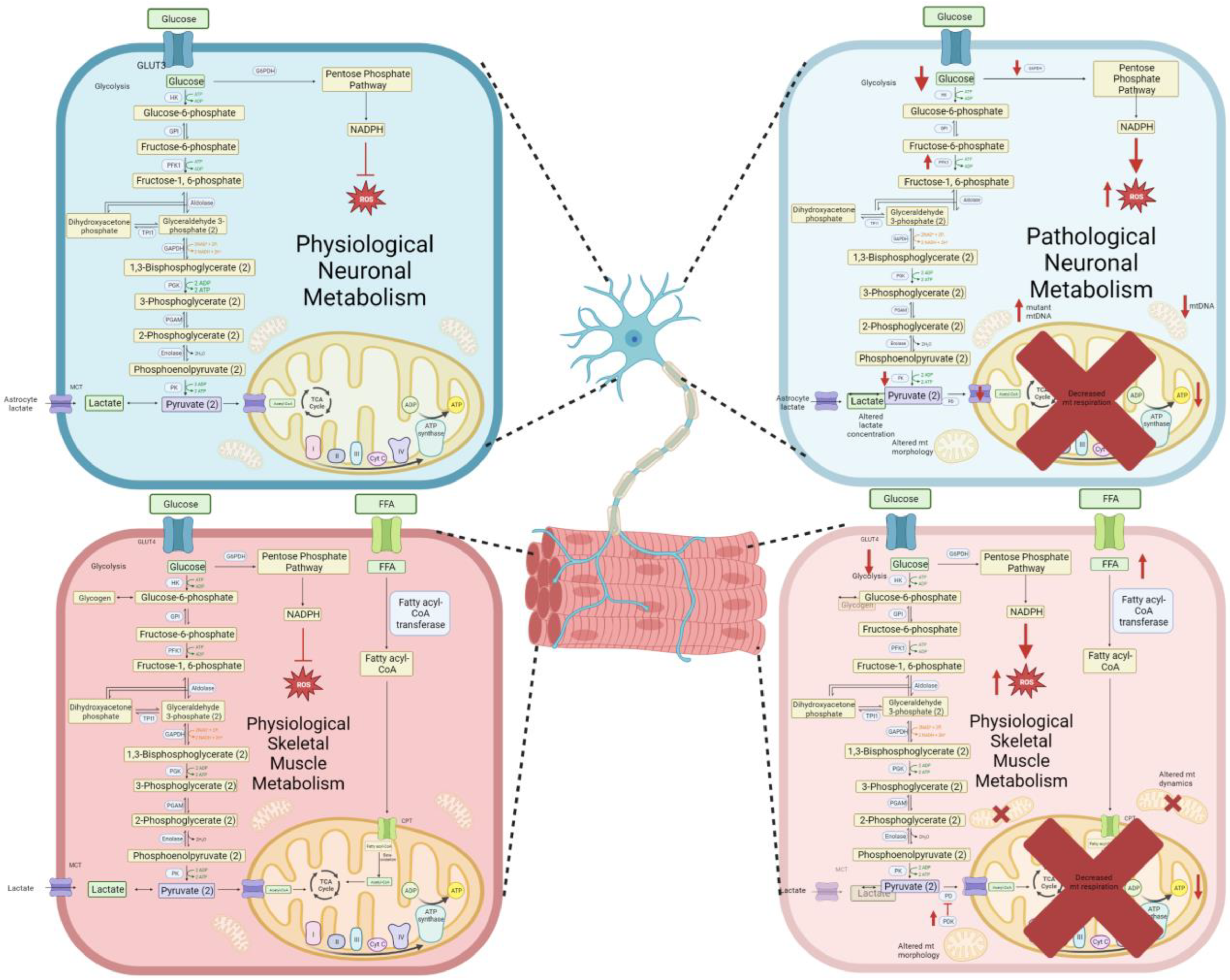
:1. Introduction
2. Types of MNDs
2.1. Amyotrophic Lateral Sclerosis
2.2. Primary Lateral Sclerosis
2.3. Spinal Muscular Atrophy
2.4. Kennedy’s Disease
2.5. Hereditary Spastic Paraplegia
3. Metabolism in Neuron-Muscle Communication
3.1. Neuronal
3.2. Neuromuscular Junction
3.3. Skeletal Muscle
4. Metabolic Dysfunction in Motor Neuron Diseases
4.1. CNS Metabolic Malfunction in ALS
4.2. Peripheral Metabolic Malfunction in ALS
5. Metabolic Malfunction in Other Motor Neuron Diseases
6. Strategies to Manipulate Metabolic Dysregulation in Motor Neuron Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meo, G.; Ferraro, P.M.; Cillerai, M.; Gemelli, C.; Cabona, C.; Zaottini, F.; Roccatagliata, L.; Villani, F.; Schenone, A.; Caponnetto, C. MND Phenotypes Differentiation: The Role of Multimodal Characterization at the Time of Diagnosis. Life 2022, 12, 1506. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef] [PubMed]
- Boido, M.; Vercelli, A. Neuromuscular Junctions as Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costamagna, D.; Casters, V.; Beltrà, M.; Sampaolesi, M.; Van Campenhout, A.; Ortibus, E.; Desloovere, K.; Duelen, R. Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia. Cells 2022, 11, 3351. [Google Scholar] [CrossRef] [PubMed]
- Molotsky, E.; Liu, Y.; Lieberman, A.P.; Merry, D.E. Neuromuscular junction pathology is correlated with differential motor unit vulnerability in spinal and bulbar muscular atrophy. Acta Neuropathol. Commun. 2022, 10, 97. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.R.; Barohn, R.J.; Corcia, P.; Fink, J.K.; Harms, M.B.; Kiernan, M.C.; Ravits, J.; Silani, V.; Simmons, Z.; Statland, J.; et al. Primary lateral sclerosis: Consensus diagnostic criteria. J. Neurol. Neurosurg. Psychiatry 2020, 91, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [Green Version]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Shellikeri, S.; Karthikeyan, V.; Martino, R.; Black, S.; Zinman, L.; Keith, J.; Yunusova, Y. The neuropathological signature of bulbar-onset ALS: A systematic review. Neurosci. Biobehav. Rev. 2017, 75, 378–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Lyall, R.A.; Donaldson, N.; Polkey, M.I.; Leigh, P.N.; Moxham, J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain 2001, 124, 14. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [Green Version]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.A.; Statland, J.M.; Wolfe, G.I.; Barohn, R.J. Primary lateral sclerosis. Muscle Nerve 2007, 35, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, F.M.; Moersch, F.P. Primary Lateral Sclerosis A Distinct Clinical Entity. J. Nerv. Ment. Dis. 1945, 102, 6. [Google Scholar] [CrossRef]
- Swank, R.L.; Putnam, T.J. Amyotrophic Lateral Sclerosis and Related Conditions A Clinical Analysis. Arch. Neurol. Psychiatry 1943, 49, 26. [Google Scholar] [CrossRef]
- Pringle, C.E.; Hudson, A.J.; Munoz, D.; Kiernan, J.; Brown, W.F.; Ebers, G. Primary lateral sclerosis: Clinical features, neuropathology and diagnostic criteria. Brain 1992, 115, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Mittal, S.O.; Hu, W.T.; Josephs, K.A.; Sorenson, E.J.; Ahlskog, J.E. Natural History of “Pure” Primary Lateral Sclerosis. Neurology 2021, 96, e2231–e2238. [Google Scholar] [CrossRef]
- Le Forestier, N.; Maisonobe, T.; Piquard, A.; Rivaud, S.; Crevier-Buchman, L.; Salachas, F.; Pradat, P.F.; Lacomblez, L.; Meininger, V. Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain 2001, 124, 11. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.M.; Barohn, R.J.; Dimachkie, M.M.; Floeter, M.K.; Mitsumoto, H. Primary Lateral Sclerosis. Neurol. Clin. 2015, 33, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Silani, V.; Corcia, P.; Harms, M.B.; Rouleau, G.; Siddique, T.; Ticozzi, N. Genetics of primary lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21 (Suppl. 1), 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hentati, A.; Deng, H.-X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.-Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Bohlega, S.; Al-Mohanna, F. Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann. Neurol. 2012, 72, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy-a literature review. Orphanet. J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 11. [Google Scholar] [CrossRef] [Green Version]
- Rochette, C.; Gilbert, N.; Simard, L. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef]
- Hahnen, E.; Forkert, R.; Marke, C.; Rudnik-Schöneborn, S.; Schönling, J.; Zerres, K.; Creavin, T.; Wirth, B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 1995, 4, 1927–1933. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal Muscular Atrophy: Clinical Classification and Disease Heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 16. [Google Scholar] [CrossRef] [Green Version]
- Macleod, M.J.; Taylor, J.E.; Lunt, P.W.; Mathew, C.G.; Robb, S.A. Prenatal onset spinal muscular atrophy. Eur. J. Paediatr. Neurol. 1999, 3, 8. [Google Scholar] [CrossRef]
- Alves, C.N.; Braga, T.K.K.; Somensi, D.N.; Nascimento, B.S.V.D.; De Lima, J.A.S.; Fujihara, S. X-linked spinal and bulbar muscular atrophy (Kennedy’s disease): The first case described in the Brazilian Amazon. Einstein 2018, 16, eRC4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradat, P.-F.; Bernard, E.; Corcia, P.; Couratier, P.; Jublanc, C.; Querin, G.; Panzini, C.M.; Salachas, F.; Vial, C.; on behalf of the French Kennedy’s Disease Writing Group. The French national protocol for Kennedy’s disease (SBMA): Consensus diagnostic and management recommendations. Orphanet J. Rare Dis. 2020, 15, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querin, G.; Bede, P.; Marchand-Pauvert, V.; Pradat, P.-F. Biomarkers of Spinal and Bulbar Muscle Atrophy (SBMA): A Comprehensive Review. Front. Neurol. 2018, 9, 844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, C.; White, K.; Bushby, K.; Shaw, P. Hereditary spastic paraparesis: A review of new developments. J. Neurol. Neurosurg. Psychiatry 2000, 69, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Gumeni, S.; Vantaggiato, C.; Montopoli, M.; Orso, G. Hereditary Spastic Paraplegia and Future Therapeutic Directions: Beneficial Effects of Small Compounds Acting on Cellular Stress. Front. Neurosci. 2021, 15, 660714. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, Z.; Shen, H.; Bing, Q.; Li, N.; Guo, X.; Hu, J. Genetic origin of patients having spastic paraplegia with or without other neurologic manifestations. BMC Neurol. 2022, 22, 180. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. [Google Scholar] [CrossRef]
- Elsayed, L.E.O.; Eltazi, I.Z.; Ahmed, A.E.; Stevanin, G. Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview. Front. Mol. Biosci. 2021, 8, 690899. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; Berg, L.H.V.D.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Quessada, C.; Bouscary, A.; René, F.; Valle, C.; Ferri, A.; Ngo, S.T.; Loeffler, J.P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells 2021, 10, 1449. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef] [Green Version]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997, 21, 2–21. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Castets, P.; Ham, D.J.; Ruegg, M.A. The TOR Pathway at the Neuromuscular Junction: More Than a Metabolic Player? Front. Mol. Neurosci. 2020, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.R.; Cossins, J.; Beeson, D.; Vincent, A. The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.J.; Rüegg, M.A. Causes and consequences of age-related changes at the neuromuscular junction. Curr. Opin. Physiol. 2018, 4, 32–39. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Altman, T.; Geller, D.; Kleeblatt, E.; Gradus-Perry, T.; Perlson, E. In vitro compartmental system underlines the contribution of mitochondrial immobility to the ATP supply in the NMJ. J. Cell Sci. 2019, 132, jcs234492. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Mika, A.; Macaluso, F.; Barone, R.; Di Felice, V.; Sledzinski, T. Effect of Exercise on Fatty Acid Metabolism and Adipokine Secretion in Adipose Tissue. Front. Physiol. 2019, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Ignatieva, E.; Smolina, N.; Kostareva, A.; Dmitrieva, R. Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype. Int. J. Mol. Sci. 2021, 22, 7349. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Powers, S.K. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Sun, X.-H.; Cai, Z.-Y.; Shen, D.-C.; Yang, X.-Z.; Liu, M.-S.; Cui, L.-Y. Correlation of weight and body composition with disease progression rate in patients with amyotrophic lateral sclerosis. Sci. Rep. 2022, 12, 13292. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Coccurello, R. What is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm 2017, 2017, 7821672. [Google Scholar] [CrossRef]
- Endo, H.; Sekiguchi, K.; Ueda, T.; Kowa, H.; Kanda, F.; Toda, T. Regional glucose hypometabolic spread within the primary motor cortex is associated with amyotrophic lateral sclerosis disease progression: A fluoro-deoxyglucose positron emission tomography study. Eneurologicalsci 2017, 6, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Pagani, M.; Chiò, A.; Valentini, M.C.; Öberg, J.; Nobili, F.; Calvo, A.; Moglia, C.; Bertuzzo, D.; Morbelli, S.; De Carli, F.; et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology 2014, 83, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis-a valid pharmacological target? Br. J. Pharm. 2014, 171, 2191–2205. [Google Scholar] [CrossRef] [Green Version]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Dhaliwal, G.K.; Grewal, R.P. Mitochondrial DNA deletion mutation levels are elevated in ALS brains. Neuroreport 2000, 11, 2507–2509. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Paitents With Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Iwata, M. Ultrastructual study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 4. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, S.E.; Yang, L.; DiMauro, J.-P.; Fuller, S.W.; Licata, S.C.; Beal, M.F. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 2006, 22, 599–610. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- McCombe, P.A.; Henderson, R.D. Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 2010, 7, 557–570. [Google Scholar] [CrossRef]
- Cacabelos, D.; Ramírez-Núñez, O.; Granado-Serrano, A.B.; Torres, P.; Ayala, V.; Moiseeva, V.; Povedano, M.; Ferrer, I.; Pamplona, R.; Portero-Otin, M.; et al. Early and gender-specific differences in spinal cord mitochondrial function and oxidative stress markers in a mouse model of ALS. Acta Neuropathol. Commun. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLOS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, M.; Gunay, A.; Chandel, N.S.; Ozdinler, P.H. Mitochondrial dysregulation occurs early in ALS motor cortex with TDP-43 pathology and suggests maintaining NAD(+) balance as a therapeutic strategy. Sci. Rep. 2022, 12, 4287. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 2019, 8, 101. [Google Scholar] [CrossRef]
- Loganathan, S.; Wilson, B.A.; Carey, S.B.; Manzo, E.; Joardar, A.; Ugur, B.; Zarnescu, D.C. TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS. Metabolites 2022, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.-H.; Santosa, M.M.; Lim, V.J.W.; Ho, B.X.; Taylor, A.; Khong, Z.J.; Ravits, J.; Fan, Y.; Liou, Y.-C.; Soh, B.-S.; et al. ALS motor neurons exhibit hallmark metabolic defects that are rescued by SIRT3 activation. Cell Death Differ. 2020, 28, 1379–1397. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.P.; Rajan, S.; Duffy, L.; Mortiboys, H.; Higginbottom, A.; Grierson, A.J.; Shaw, P.J. Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis. Neurobiol. Aging 2014, 35, 1499–1509. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [Green Version]
- Riechers, S.-P.; Mojsilovic-Petrovic, J.; Belton, T.B.; Chakrabarty, R.P.; Garjani, M.; Medvedeva, V.; Dalton, C.; Wong, Y.C.; Chandel, N.S.; Dienel, G.; et al. Neurons undergo pathogenic metabolic reprogramming in models of familial ALS. Mol. Metab. 2022, 60, 101468. [Google Scholar] [CrossRef]
- Tefera, T.W.; Bartlett, K.; Tran, S.S.; Hodson, M.P.; Borges, K. Impaired Pentose Phosphate Pathway in the Spinal Cord of the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 5844–5855. [Google Scholar] [CrossRef]
- Crugnola, V.; Lamperti, C.; Lucchini, V.; Ronchi, D.; Peverelli, L.; Prelle, A.; Sciacco, M.; Bordoni, A.; Fassone, E.; Fortunato, F.; et al. Mitochondrial Respiratory Chain Dysfunction in Muscle From Patients With Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Ríos, E.; Deng, H.-X. Hyperactive Intracellular Calcium Signaling Associated with Localized Mitochondrial Defects in Skeletal Muscle of an Animal Model of Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Claasse, D.O.; Josephs, K.A.; Peller, P.J. The Stripe of Primary Lateral Sclerosis Focal Primary Motor Cortex Hypometabolism Seen on Fluordeozyglucose F18 Positron Emission Tomography. Arch. Neurol. 2010, 67, 4. [Google Scholar]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.-C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [Green Version]
- Malik, B.; Devine, H.; Patani, R.; La Spada, A.R.; Hanna, M.G.; Greensmith, L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci. Rep. 2019, 9, 3539. [Google Scholar] [CrossRef] [Green Version]
- Mignarri, A.; Rubegni, A.; Tessa, A.; Stefanucci, S.; Malandrini, A.; Cardaioli, E.; Meschini, M.C.; Stromillo, M.L.; Doccini, S.; Federico, A.; et al. Mitochondrial dysfunction in hereditary spastic paraparesis with mutations in DDHD1/SPG28. J. Neurol. Sci. 2016, 362, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Wali, G.; Kumar, K.R.; Liyanage, E.; Davis, R.L.; Mackay-Sim, A.; Sue, C.M. Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 2020, 14, 820. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chai, E.; Mou, Y.; Roda, R.H.; Blackstone, C.; Li, X.-J. Inhibiting mitochondrial fission rescues degeneration in hereditary spastic paraplegia neurons. Brain 2022, 145, 4016–4031. [Google Scholar] [CrossRef]
- Simon, N.G.; Lee, M.; Bae, J.S.; Mioshi, E.; Lin, C.S.-Y.; Pfluger, C.M.; Henderson, R.D.; Vucic, S.; Swash, M.; Burke, D.; et al. Dissociated lower limb muscle involvement in amyotrophic lateral sclerosis. J. Neurol. 2015, 262, 1424–1432. [Google Scholar] [CrossRef]
- Hu, F.; Jin, J.; Chen, Q.; Kang, L.; Jia, R.; Qin, X.; Liu, X.; Dang, Y.; Dang, J. Dissociated lower limb muscle involvement in amyotrophic lateral sclerosis and its differential diagnosis value. Sci. Rep. 2019, 9, 17786. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, A.; Nicholson, G. Detection of preclinical motor neuron loss in SOD1 mutation carrier using motor unit number estimation. J. Neurol. Neurosurg. Psychiatry 2002, 73, 3. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef] [PubMed]
- Desseille, C.; Deforges, S.; Biondi, O.; Houdebine, L.; D’amico, D.; Lamazière, A.; Caradeuc, C.; Bertho, G.; Bruneteau, G.; Weill, L.; et al. Specific Physical Exercise Improves Energetic Metabolism in the Skeletal Muscle of Amyotrophic-Lateral-Sclerosis Mice. Front. Mol. Neurosci. 2017, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers-Upmeijer, J.; Jager, A.E.J.D.; Hew, J.M.; Snoek, J.W.; van Weerden, T.W. Primary lateral sclerosis: Clinical, neurophysiological, and magnetic resonance findings. J. Neurol. Neurosurg. Psychiatry 2001, 71, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilio, E.; Piano, V.; Wirth, B. Mitochondrial Dysfunction in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2022, 23, 10878. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Miao, D.; Yu, J.T.; Tan, L. Mitochondrial dysfunction in Kennedy’s disease: A new pharmacological target? Ann. Transl. Med. 2015, 3, 66. [Google Scholar]
- Finsterer, J.; Mishra, A.; Wakil, S.; Pennuto, M.; Soraru, G. Mitochondrial implications in bulbospinal muscular atrophy (Kennedy disease). Amyotroph. Lateral Scler. Front. Degener. 2015, 17, 112–118. [Google Scholar] [CrossRef]
- Tanaka, F.; Katsuno, M.; Banno, H.; Suzuki, K.; Adachi, H.; Sobue, G. Current Status of Treatment of Spinal and Bulbar Muscular Atrophy. Neural Plast. 2012, 2012, 369284. [Google Scholar] [CrossRef]
- Tesson, C.; Nawara, M.; Salih, M.A.; Rossignol, R.; Zaki, M.S.; Al Balwi, M.; Schule, R.; Mignot, C.; Obre, E.; Bouhouche, A.; et al. Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2012, 91, 1051–1064. [Google Scholar] [CrossRef] [Green Version]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, G.; Thomas, R.E.; Pallanck, L.J. Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis. 2018, 9, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, J.; Borner, G.H.H.; Edgar, J.; Hein, M.Y.; Mann, M.; Buchholz, F.; Antrobus, R.; Robinson, M.S. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell 2013, 24, 2558–2569. [Google Scholar] [CrossRef]
- Khamaysa, M.; Pradat, P.-F. Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas. J. Pers. Med. 2022, 12, 1601. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Amadio, S.; Nesci, V.; Torcinaro, A.; Giacovazzo, G.; Primiano, A.; Gloriani, M.; Candelise, N.; Pieroni, L.; et al. Repurposing of Trimetazidine for amyotrophic lateral sclerosis: A study in SOD1(G93A) mice. Br. J. Pharmacol. 2022, 179, 1732–1752. [Google Scholar] [CrossRef]


{kind=link}
| Disease | Model | Findings |
|---|---|---|
| ALS | Neuronal—Patients |
|
| Neuronal—in vivo | ||
| Neuronal—in vitro |
| |
| Skeletal—Patients |
| |
| Skeletal—in vivo | ||
| PLS | Neuronal—Patients | |
| SMA | Neuronal—in vivo |
|
| Kennedy’s Disease | Neuronal—in vitro |
|
| HSP | Skeletal—Patients |
|
| Neuronal—in vitro |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barone, C.; Qi, X. Altered Metabolism in Motor Neuron Diseases: Mechanism and Potential Therapeutic Target. Cells 2023, 12, 1536. https://doi.org/10.3390/cells12111536
Barone C, Qi X. Altered Metabolism in Motor Neuron Diseases: Mechanism and Potential Therapeutic Target. Cells. 2023; 12(11):1536. https://doi.org/10.3390/cells12111536
Chicago/Turabian StyleBarone, Cassandra, and Xin Qi. 2023. "Altered Metabolism in Motor Neuron Diseases: Mechanism and Potential Therapeutic Target" Cells 12, no. 11: 1536. https://doi.org/10.3390/cells12111536