
Role of Heat Shock Proteins in Atrial Fibrillation: From Molecular Mechanisms to Diagnostic and Therapeutic Opportunities

Abstract
:1. Introduction
2. Definition and Function of HSPs in AF
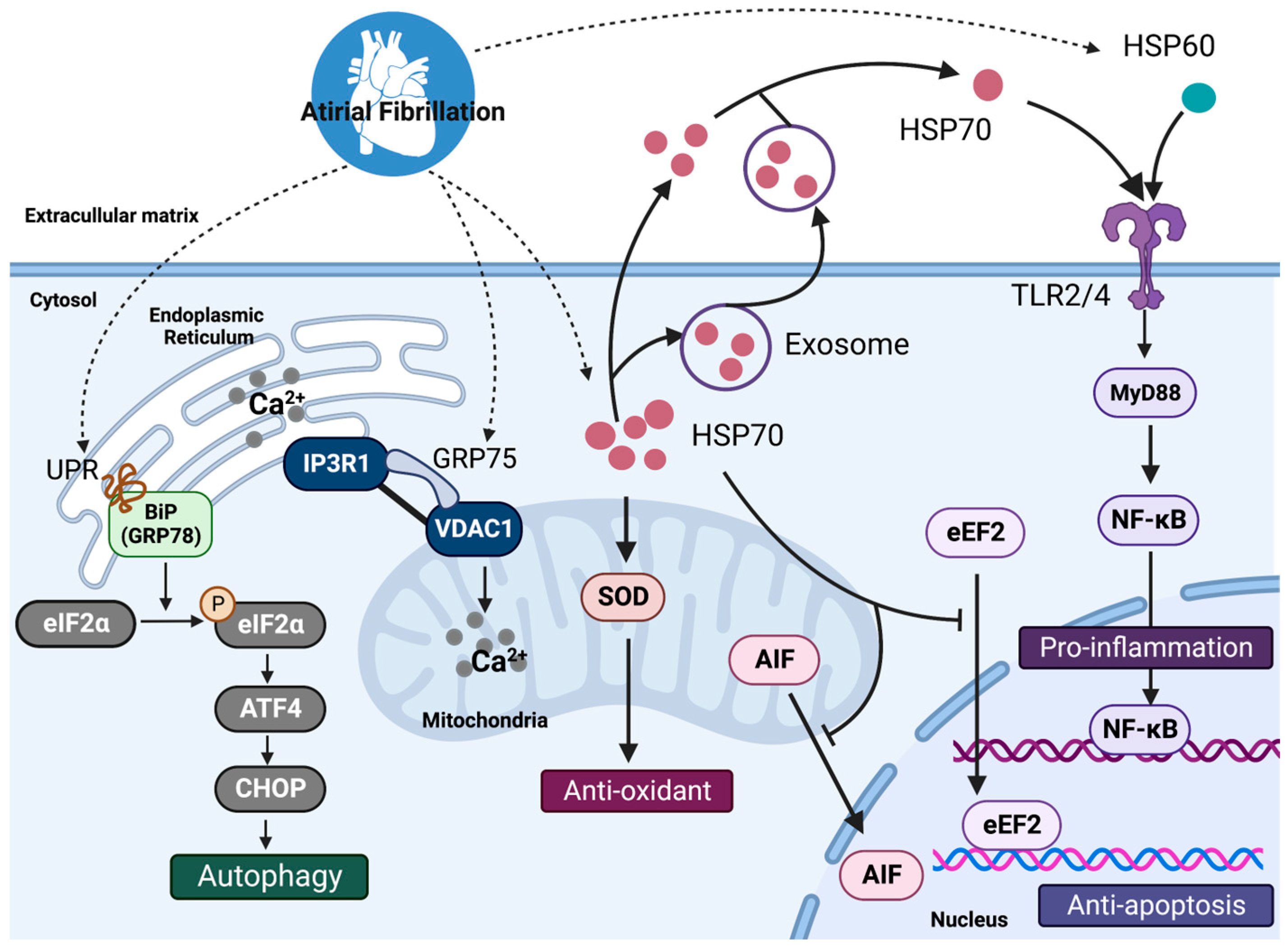
3. Protein–Protein Interactions (PPIs) of HSP Complexes in Atrial Fibrillation
3.1. HSP60 and Atrial Fibrillation
3.2. HSP70 Family and Atrial Fibrillation
3.3. Small HSPs and Atrial Fibrillation
4. Clinical Application of HSPs in Atrial Fibrillation
4.1. HSP60 Predicts Atrial Fibrillation and Indicates the Stability of Sinus Rhythm
4.2. HSP70 Predicts Atrial Fibrillation and Atrial Fibrillation Severity
4.3. HSP70 and the Treatment of Atrial Fibrillation
4.4. HSP90 in Atrial Fibrillation and Other Arrhythmogenic Diseases
4.5. HSPB and the Diagnosis of Atrial Fibrillation

{kind=link}
| Type | Family | Correlation with AF | Therapeutic Target | References |
|---|---|---|---|---|
| HSP70 | HSPA | No significant difference between SR and AF. | Induced by TRC051384 or tubastatin A. | Kornej, J. et al. [59] Allende, M. et al. [64] |
| HSP27 | HSPB | Lower baseline HSPB in patients with AF than in those with SR. | Induced by GGA or L-glutamine. | Brundel, B. J. et al. [9] Brundel, B. J. et al. [41] Zhang, D. et al. [42] Sakabe, M. et al. [76] Hamiel, C. R. et al. [78] |
| HSP60 | HSPC | Higher HSP60 levels in patients with AF than in those with SR. | No treatment strategy for atrial fibrillation targeting HSP60. | Schafler, A. E. et al. [17] Schafler, A. E. et al. [53] Chen, B. X. et al. [57] |
| HSP90 | HSPD | No significant difference between SR and AF. | No treatment strategy for atrial fibrillation targeting HSP90. | No reference. |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kornej, J.; Borschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Reese-Petersen, A.L.; Olesen, M.S.; Karsdal, M.A.; Svendsen, J.H.; Genovese, F. Atrial fibrillation and cardiac fibrosis: A review on the potential of extracellular matrix proteins as biomarkers. Matrix Biol. 2020, 91–92, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, Y.; Barnes, M.E.; Gersh, B.J.; Cha, S.S.; Bailey, K.R.; Abhayaratna, W.P.; Seward, J.B.; Tsang, T.S. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006, 114, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.E.; Wang, K.L.; Lip, G.Y. Stroke prevention in atrial fibrillation: An Asian perspective. Thromb. Haemost. 2014, 111, 789–797. [Google Scholar] [PubMed] [Green Version]
- Krijthe, B.P.; Kunst, A.; Benjamin, E.J.; Lip, G.Y.; Franco, O.H.; Hofman, A.; Witteman, J.C.; Stricker, B.H.; Heeringa, J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013, 34, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Staerk, L.; Wang, B.; Preis, S.R.; Larson, M.G.; Lubitz, S.A.; Ellinor, P.T.; McManus, D.D.; Ko, D.; Weng, L.C.; Lunetta, K.L.; et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: Cohort study based on longitudinal data from the Framingham Heart Study. BMJ 2018, 361, k1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, D.M.; Fynn, S.P.; Walden, A.P.; Hobbs, W.J.; Arya, S.; Garratt, C.J. Repetitive 4-week periods of atrial electrical remodeling promote stability of atrial fibrillation: Time course of a second factor involved in the self-perpetuation of atrial fibrillation. Circulation 2004, 109, 1434–1439. [Google Scholar] [CrossRef] [Green Version]
- van Wijk, S.W.; Ramos, K.S.; Brundel, B. Cardioprotective Role of Heat Shock Proteins in Atrial Fibrillation: From Mechanism of Action to Therapeutic and Diagnostic Target. Int. J. Mol. Sci. 2021, 22, 442. [Google Scholar] [CrossRef]
- Brundel, B.J.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef]
- Currie, R.W. Protein synthesis in perfused rat hearts after in vivo hyperthermia and in vitro cold ischemia. Biochem. Cell Biol. 1988, 66, 13–19. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Azevedo, C.A.B.; da Cunha, R.S.; de Yurre, A.R.; Medei, E.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Heat Shock Proteins: Connectors between Heart and Kidney. Cells 2021, 10, 1939. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Morimoto, R.I. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 2005, 280, 33097–33100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barna, J.; Csermely, P.; Vellai, T. Roles of heat shock factor 1 beyond the heat shock response. Cell. Mol. Life Sci. 2018, 75, 2897–2916. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K.; Torsney, E.; Poloniecki, J.; Camm, A.J.; Xu, Q.; Jahangiri, M. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 2005, 79, 865–871; discussion 871. [Google Scholar] [CrossRef]
- Kirmanoglou, K.; Hannekum, A.; Schafler, A.E. Expression of mortalin in patients with chronic atrial fibrillation. Basic Res. Cardiol. 2004, 99, 404–408. [Google Scholar] [CrossRef]
- Schafler, A.E.; Kirmanoglou, K.; Pecher, P.; Hannekum, A.; Schumacher, B. Overexpression of heat shock protein 60/10 in myocardium of patients with chronic atrial fibrillation. Ann. Thorac. Surg. 2002, 74, 767–770. [Google Scholar] [CrossRef]
- Cai, W.; Rudolph, J.L.; Harrison, S.M.; Jin, L.; Frantz, A.L.; Harrison, D.A.; Andres, D.A. An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol. Biol. Cell 2011, 22, 3231–3241. [Google Scholar] [CrossRef]
- Hohfeld, J.; Hartl, F.U. Role of the chaperonin cofactor Hsp10 in protein folding and sorting in yeast mitochondria. J. Cell Biol. 1994, 126, 305–315. [Google Scholar] [CrossRef]
- Lin, K.M.; Lin, B.; Lian, I.Y.; Mestril, R.; Scheffler, I.E.; Dillmann, W.H. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation 2001, 103, 1787–1792. [Google Scholar] [CrossRef]
- Kirchhoff, S.R.; Gupta, S.; Knowlton, A.A. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation 2002, 105, 2899–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Knowlton, A.A. Cytosolic heat shock protein 60, hypoxia, and apoptosis. Circulation 2002, 106, 2727–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.C.; Stice, J.P.; Chen, L.; Jung, J.S.; Gupta, S.; Wang, Y.; Baumgarten, G.; Trial, J.; Knowlton, A.A. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ. Res. 2009, 105, 1186–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiersma, M.; van Marion, D.M.S.; Wust, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Gong, M.; He, J.; Xie, B.; Zhang, Z.; Meng, L.; Tse, G.; Zhao, Y.; Bao, Q.; Zhang, Y.; et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. 2022, 52, 102289. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, e006458. [Google Scholar] [CrossRef] [Green Version]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, Y.; Wang, Z.; Liu, D.; Xie, B.; Zhang, Y.; Yuan, M.; Tse, G.; Li, G.; Xu, G.; et al. Activation of NADPH oxidase mediates mitochondrial oxidative stress and atrial remodeling in diabetic rabbits. Life Sci. 2021, 272, 119240. [Google Scholar] [CrossRef]
- Choudhury, S.; Bae, S.; Ke, Q.; Lee, J.Y.; Kim, J.; Kang, P.M. Mitochondria to nucleus translocation of AIF in mice lacking Hsp70 during ischemia/reperfusion. Basic Res. Cardiol. 2011, 106, 397–407. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Miao, J.; Wang, S.; Wu, L.; Yan, D.; Li, J.; Guo, W.; Wu, X.; Shen, A. Heat shock protein 70 protects cardiomyocytes through suppressing SUMOylation and nucleus translocation of phosphorylated eukaryotic elongation factor 2 during myocardial ischemia and reperfusion. Apoptosis 2017, 22, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Murakoshi, N.; Igarashi, M.; Hirayama, A.; Ito, Y.; Seo, Y.; Tada, H.; Aonuma, K. PPAR-gamma activator pioglitazone prevents age-related atrial fibrillation susceptibility by improving antioxidant capacity and reducing apoptosis in a rat model. J. Cardiovasc. Electrophysiol. 2012, 23, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xue, L.; Cao, H.; Cui, F.; Dai, T.; Chen, Y. TLR2 was overexpressed independent of IL-6 in patients with valvular atrial fibrillation. J. Biomed. Res. 2011, 25, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, S.; Honda, S.; Watanabe, T.; Suzuki, S.; Ishino, M.; Kitahara, T.; Funayama, A.; Netsu, S.; Sasaki, T.; Shishido, T.; et al. Atrial endothelial impairment through Toll-like receptor 4 signaling causes atrial thrombogenesis. Heart Vessels 2014, 29, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, H.; Orihara, K.; Hamasaki, S.; Ishida, S.; Oketani, N.; Iriki, Y.; Ninomiya, Y.; Okui, H.; Kuwahata, S.; Fujita, S.; et al. The role of infection in the development of non-valvular atrial fibrillation: Up-regulation of Toll-like receptor 2 expression levels on monocytes. J. Cardiol. 2009, 53, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Mathur, S.; Walley, K.R.; Wang, Y.; Indrambarya, T.; Boyd, J.H. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ. J. 2011, 75, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Suarez, J.; Hu, Y.; McDonough, P.M.; Boer, C.; Dix, D.J.; Dillmann, W.H. Deletion of the inducible 70-kDa heat shock protein genes in mice impairs cardiac contractile function and calcium handling associated with hypertrophy. Circulation 2006, 113, 2589–2597. [Google Scholar] [CrossRef] [Green Version]
- Ke, L.; Meijering, R.A.; Hoogstra-Berends, F.; Mackovicova, K.; Vos, M.J.; Van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 2011, 6, e20395. [Google Scholar] [CrossRef]
- Brundel, B.J.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Ke, L.; Mackovicova, K.; Van Der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Hu, X.; Li, J.; Hoogstra-Berends, F.; Zhuang, Q.; Esteban, M.A.; de Groot, N.; Henning, R.H.; Brundel, B. Converse role of class I and class IIa HDACs in the progression of atrial fibrillation. J. Mol. Cell. Cardiol. 2018, 125, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibert, B.; Eckel, B.; Fasquelle, L.; Moulin, M.; Bouhallier, F.; Gonin, V.; Mellier, G.; Simon, S.; Kretz-Remy, C.; Arrigo, A.P.; et al. Knock down of heat shock protein 27 (HspB1) induces degradation of several putative client proteins. PLoS ONE 2012, 7, e29719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundel, B.; Ai, X.; Hills, M.T.; Kuipers, M.F.; Lip, G.Y.H.; de Groot, N.M.S. Atrial fibrillation. Nat. Rev. Dis. Prim. 2022, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- de Vos, C.B.; Pisters, R.; Nieuwlaat, R.; Prins, M.H.; Tieleman, R.G.; Coelen, R.J.; van den Heijkant, A.C.; Allessie, M.A.; Crijns, H.J. Progression from paroxysmal to persistent atrial fibrillation clinical correlates and prognosis. J. Am. Coll. Cardiol. 2010, 55, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Gianfranchi, L.; Brignole, M.; Menozzi, C.; Lolli, G.; Bottoni, N. Determinants of development of permanent atrial fibrillation and its treatment. Europace 1999, 1, 35–39. [Google Scholar] [CrossRef]
- Li, K.H.C.; White, F.A.; Tipoe, T.; Liu, T.; Wong, M.C.; Jesuthasan, A.; Baranchuk, A.; Tse, G.; Yan, B.P. The Current State of Mobile Phone Apps for Monitoring Heart Rate, Heart Rate Variability, and Atrial Fibrillation: Narrative Review. JMIR Mhealth Uhealth 2019, 7, e11606. [Google Scholar] [CrossRef]
- Charitos, E.I.; Purerfellner, H.; Glotzer, T.V.; Ziegler, P.D. Clinical classifications of atrial fibrillation poorly reflect its temporal persistence: Insights from 1,195 patients continuously monitored with implantable devices. J. Am. Coll. Cardiol. 2014, 63 (25 Pt A), 2840–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, N.M.; Houben, R.P.; Smeets, J.L.; Boersma, E.; Schotten, U.; Schalij, M.J.; Crijns, H.; Allessie, M.A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: Epicardial breakthrough. Circulation 2010, 122, 1674–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafler, A.E.; Kirmanoglou, K.; Balbach, J.; Pecher, P.; Hannekum, A.; Schumacher, B. The expression of heat shock protein 60 in myocardium of patients with chronic atrial fibrillation. Basic Res. Cardiol. 2002, 97, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Neufer, P.D.; Ordway, G.A.; Hand, G.A.; Shelton, J.M.; Richardson, J.A.; Benjamin, I.J.; Williams, R.S. Continuous contractile activity induces fiber type specific expression of HSP70 in skeletal muscle. Am. J. Physiol. 1996, 271 (6 Pt 1), C1828–C1837. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xue, L.; Xu, X.; Wu, Y.; Zhu, J.; Chen, L.; Chen, D.; Chen, Y. Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones 2011, 16, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Glaudemans, A.W.; de Vries, E.F.; Galli, F.; Dierckx, R.A.; Slart, R.H.; Signore, A. The use of (18)F-FDG-PET/CT for diagnosis and treatment monitoring of inflammatory and infectious diseases. Clin. Dev. Immunol. 2013, 2013, 623036. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.X.; Xie, B.; Zhou, Y.; Shi, L.; Wang, Y.; Zeng, L.; Liu, X.; Yang, M.F. Association of Serum Biomarkers and Cardiac Inflammation in Patients With Atrial Fibrillation: Identification by Positron Emission Tomography. Front. Cardiovasc. Med. 2021, 8, 735082. [Google Scholar] [CrossRef]
- Rosito, G.A.; Massaro, J.M.; Hoffmann, U.; Ruberg, F.L.; Mahabadi, A.A.; Vasan, R.S.; O’Donnell, C.J.; Fox, C.S. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community-based sample: The Framingham Heart Study. Circulation 2008, 117, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Kornej, J.; Reinhardt, C.; Kosiuk, J.; Arya, A.; Hindricks, G.; Adams, V.; Husser, D.; Bollmann, A. Response of circulating heat shock protein 70 and anti-heat shock protein 70 antibodies to catheter ablation of atrial fibrillation. J. Transl. Med. 2013, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Marion, D.M.V.; Lanters, E.A.; Ramos, K.S.; Li, J.; Wiersma, M.; Baks-te Bulte, L.; JQMMuskens, A.; Boersma, E.; de Groot, N.M.; Brundel, B.J. Evaluating Serum Heat Shock Protein Levels as Novel Biomarkers for Atrial Fibrillation. Cells 2020, 9, 2105. [Google Scholar] [CrossRef]
- Min, T.J.; Jo, W.M.; Shin, S.Y.; Lim, H.E. The protective effect of heat shock protein 70 (Hsp70) in atrial fibrillation in various cardiomyopathy conditions. Heart Vessels 2015, 30, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Abbott, R.; Kannel, W. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke 1991, 22, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, B.A.; Kim, S.; Piccini, J.P.; Fonarow, G.C.; Lopes, R.D.; Thomas, L.; Ezekowitz, M.D.; Ansell, J.; Kowey, P.; Singer, D.E.; et al. Patients, Use and associated risks of concomitant aspirin therapy with oral anticoagulation in patients with atrial fibrillation: Insights from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) Registry. Circulation 2013, 128, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allende, M.; Molina, E.; Lecumberri, R.; Sanchez-Arias, J.A.; Ugarte, A.; Guruceaga, E.; Oyarzabal, J.; Hermida, J. Inducing heat shock protein 70 expression provides a robust antithrombotic effect with minimal bleeding risk. Thromb. Haemost. 2017, 117, 1722–1729. [Google Scholar]
- Dans, A.L.; Connolly, S.J.; Wallentin, L.; Yang, S.; Nakamya, J.; Brueckmann, M.; Ezekowitz, M.; Oldgren, J.; Eikelboom, J.W.; Reilly, P.A.; et al. Concomitant use of antiplatelet therapy with dabigatran or warfarin in the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial. Circulation 2013, 127, 634–640. [Google Scholar] [CrossRef]
- Chen, Z.; Shen, X.; Shen, F.; Zhong, W.; Wu, H.; Liu, S.; Lai, J. TAK1 activates AMPK-dependent cell death pathway in hydrogen peroxide-treated cardiomyocytes, inhibited by heat shock protein-70. Mol. Cell. Biochem. 2013, 377, 35–44. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Zhang, C.; Li, J.; Guo, W.; Yan, D.; Yang, C.; Zhao, J.; Wu, X.; Shi, J. Phosphorylated eEF2 is SUMOylated and induces cardiomyocyte apoptosis during myocardial ischemia reperfusion. J. Cardiol. 2017, 69, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Iwai, C.; Li, P.; Kurata, Y.; Hoshikawa, Y.; Morikawa, K.; Maharani, N.; Higaki, K.; Sasano, T.; Notsu, T.; Ishido, Y.; et al. Hsp90 prevents interaction between CHIP and HERG proteins to facilitate maturation of wild-type and mutant HERG proteins. Cardiovasc. Res. 2013, 100, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef]
- Verschuure, P.; Tatard, C.; Boelens, W.C.; Grongnet, J.F.; David, J.C. Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur. J. Cell Biol. 2003, 82, 523–530. [Google Scholar] [CrossRef]
- Hu, X.; Van Marion, D.M.S.; Wiersma, M.; Zhang, D.; Brundel, B. The protective role of small heat shock proteins in cardiac diseases: Key role in atrial fibrillation. Cell Stress Chaperones 2017, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]
- van Marion, D.M.; Hu, X.; Zhang, D.; Hoogstra-Berends, F.; Seerden, J.G.; Loen, L.; Heeres, A.; Steen, H.; Henning, R.H.; Brundel, B.J. Screening of novel HSP-inducing compounds to conserve cardiomyocyte function in experimental atrial fibrillation. Drug Des. Dev. Ther. 2019, 13, 345–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, M.; Sang, C.; Adachi, H.; Minamiyama, M.; Waza, M.; Tanaka, F.; Doyu, M.; Sobue, G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. USA 2005, 102, 16801–16806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooie, T.; Takahashi, N.; Saikawa, T.; Nawata, T.; Arikawa, M.; Yamanaka, K.; Hara, M.; Shimada, T.; Sakata, T. Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation 2001, 104, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, L.; Qi, X.Y.; Dijkhuis, A.J.; Chartier, D.; Nattel, S.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 2008, 45, 685–693. [Google Scholar] [CrossRef]
- Sakabe, M.; Shiroshita-Takeshita, A.; Maguy, A.; Brundel, B.J.; Fujiki, A.; Inoue, H.; Nattel, S. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc. Res. 2008, 78, 63–70. [Google Scholar] [CrossRef]
- Marunouchi, T.; Inomata, S.; Sanbe, A.; Takagi, N.; Tanonaka, K. Protective effect of geranylgeranylacetone via enhanced induction of HSPB1 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats. Eur. J. Pharmacol. 2014, 730, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Hamiel, C.R.; Pinto, S.; Hau, A.; Wischmeyer, P.E. Glutamine enhances heat shock protein 70 expression via increased hexosamine biosynthetic pathway activity. Am. J. Physiol. Cell Physiol. 2009, 297, C1509–C1519. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Han, X.; Zhang, Z.; Tse, G.; Shao, Q.; Liu, T. Role of Heat Shock Proteins in Atrial Fibrillation: From Molecular Mechanisms to Diagnostic and Therapeutic Opportunities. Cells 2023, 12, 151. https://doi.org/10.3390/cells12010151
Liu D, Han X, Zhang Z, Tse G, Shao Q, Liu T. Role of Heat Shock Proteins in Atrial Fibrillation: From Molecular Mechanisms to Diagnostic and Therapeutic Opportunities. Cells. 2023; 12(1):151. https://doi.org/10.3390/cells12010151
Chicago/Turabian StyleLiu, Daiqi, Xuyao Han, Zhiwei Zhang, Gary Tse, Qingmiao Shao, and Tong Liu. 2023. "Role of Heat Shock Proteins in Atrial Fibrillation: From Molecular Mechanisms to Diagnostic and Therapeutic Opportunities" Cells 12, no. 1: 151. https://doi.org/10.3390/cells12010151