
The Role of HSP90 Inhibitors in the Treatment of Cardiovascular Diseases

Abstract
:1. Introduction
2. Subtypes and Molecular Properties of HSP90 and Its Secretory Pathway
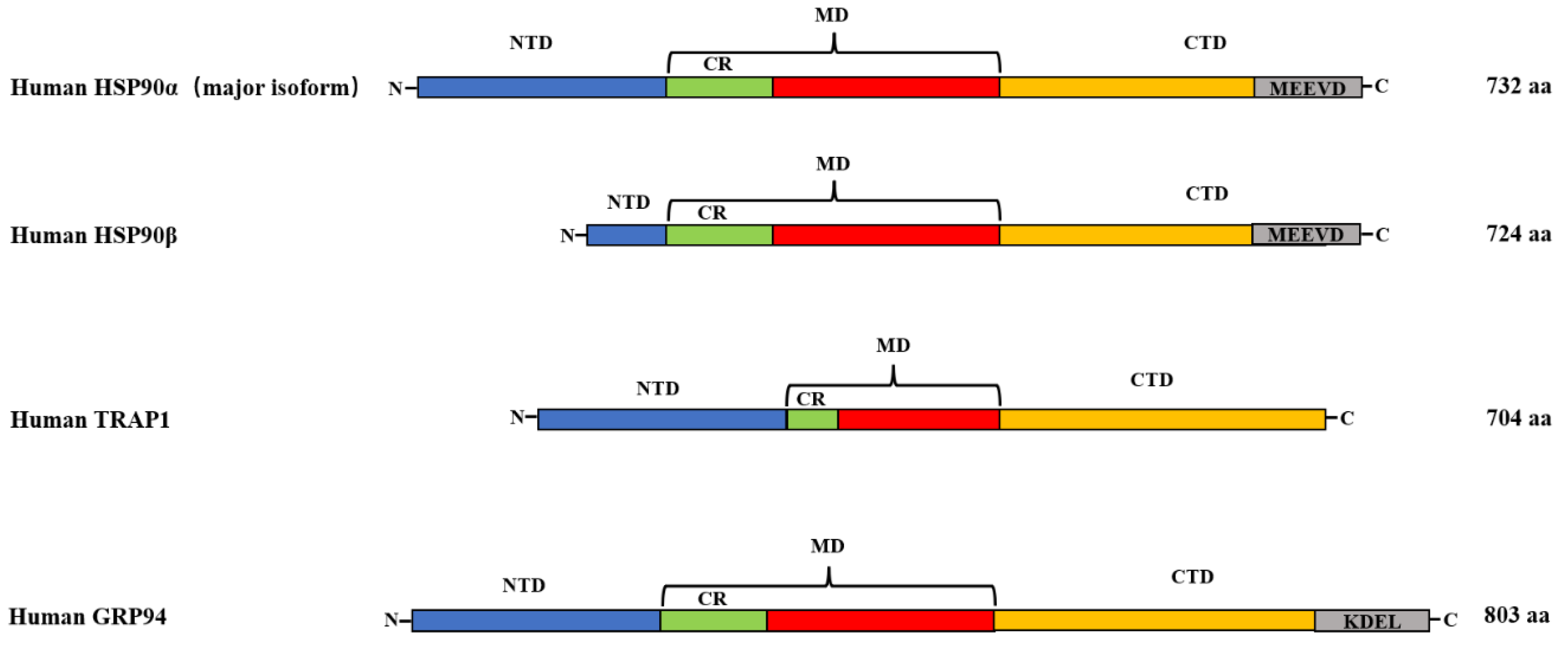
2.1. Structure of HSP90
2.1.1. HSP90α and HSP90β
2.1.2. GRP94
2.1.3. TRAP1
2.2. Potential Pathways for HSP90 to Enter the Bloodstream
2.2.1. Secretion of Viable Cardiomyocytes
2.2.2. HSP90 as a Component of Exosomes Secreted by Cardiac Cells into Blood and HSP90 Being Released by Dying Cardiac Cells
2.2.3. Necrotic Cells Can Passively Release HSPs into the Extracellular Environment
3. HSP90 and Cardiovascular Disease
3.1. HSP90 and Hypertension
3.2. HSP90 and PAH
3.3. HSP90 and Atherosclerosis
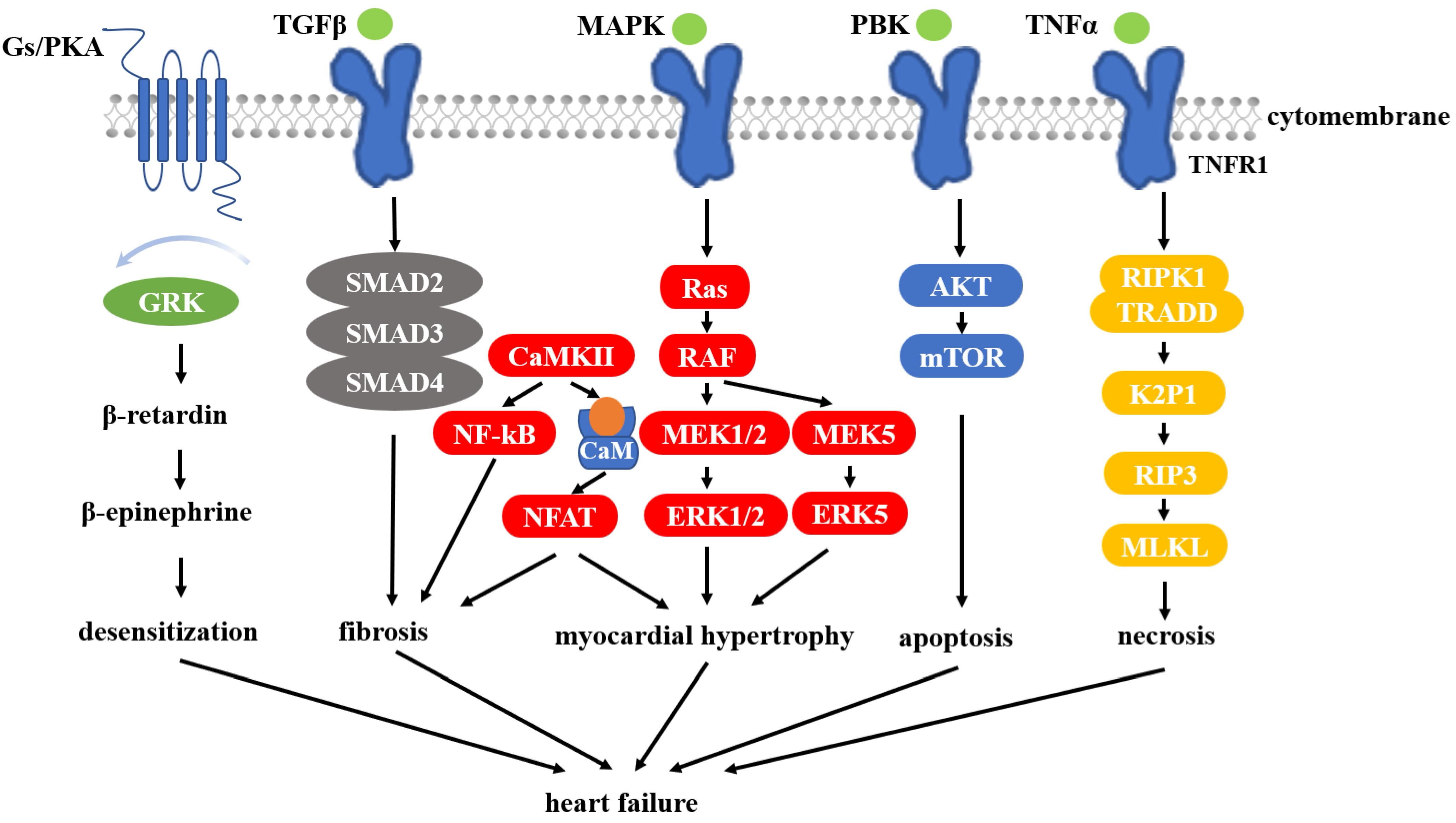
3.4. HSP90 and Heart Failure
4. HSP90 Inhibitors Associated with the Treatment of Cardiovascular Disease
4.1. Geldanamycin
4.2. 17-AGG
4.3. 17-DMAG
4.4. Gamitrinib
4.5. Celastrol
4.6. Inhibitors of HSP90 and Heart Ischemia
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ritossa, F. New Puffing Pattern Induced by Temperature Shock and Dnp in Drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Mitsiades, C.S.; Laubach, J.P.; Lonial, S.; Chanan-Khan, A.A.; Anderson, K.C. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol. 2011, 152, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Mehta, T.A.; Greenman, J.; Ettelaie, C.; Venkatasubramaniam, A.; Chetter, I.C.; McCollum, P.T. Heat shock proteins in vascular disease—A review. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.R.; Deng, S.L.; Lian, Z.X.; Yu, K. Terazosin reduces steroidogenic factor 1 and upregulates heat shock protein 90 expression in LH-induced bovine ovarian theca cells. Free Radic. Biol. Med. 2021, 163, 190–195. [Google Scholar] [CrossRef]
- Deng, S.L.; Sun, T.C.; Yu, K.; Wang, Z.P.; Zhang, B.L.; Zhang, Y.; Wang, X.X.; Lian, Z.X.; Liu, Y.X. Melatonin reduces oxidative damage and upregulates heat shock protein 90 expression in cryopreserved human semen. Free Radic. Biol. Med. 2017, 113, 347–354. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Park, C.J.; Seo, Y.S. Heat Shock Proteins: A Review of the Molecular Chaperones for Plant Immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef]
- Chakafana, G.; Spracklen, T.F.; Kamuli, S.; Zininga, T.; Shonhai, A.; Ntusi, N.A.B.; Sliwa, K. Heat Shock Proteins: Potential Modulators and Candidate Biomarkers of Peripartum Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 633013. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Chebotareva, N.; Vinogradov, A.; Gindis, A.; Tao, E.K.; Moiseev, S. Heat shock protein 90 and NFkB levels in serum and urine in patients with chronic glomerulonephritis. Cell Stress Chaperones 2020, 25, 495–501. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Sarkissian, S.D.; Aceros, H.; Williams, P.M.; Scalabrini, C.; Borie, M.; Noiseux, N. Heat shock protein 90 inhibition and multi-target approach to maximize cardioprotection in ischaemic injury. Br. J. Pharmacol. 2020, 177, 3378–3388. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Lopez-Franco, O.; Blanco-Colio, L.M.; Munoz-Garcia, B.; Ramos-Mozo, P.; Ortega, L.; Egido, J.; Martin-Ventura, J.L. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc. Res. 2010, 86, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.W.; Veal, J.M.; Fadden, R.P.; Barabasz, A.F.; Partridge, J.M.; Barta, T.E.; Dubois, L.G.; Huang, K.H.; Mabbett, S.R.; Silinski, M.A.; et al. Small Molecule Inhibitors of Hsp90 Potently Affect Inflammatory Disease Pathways and Exhibit Activity in Models of Rheumatoid Arthritis. Arthritis Rheum. 2008, 58, 3765–3775. [Google Scholar] [CrossRef]
- Li, D.Y.; Liang, S.; Wen, J.H.; Tang, J.X.; Deng, S.L.; Liu, Y.X. Extracellular HSPs: The Potential Target for Human Disease Therapy. Molecules 2022, 27, 2361. [Google Scholar] [CrossRef]
- Qi, S.; Deng, S.; Lian, Z.; Yu, K. Novel Drugs with High Efficacy against Tumor Angiogenesis. Int. J. Mol. Sci. 2022, 23, 6934. [Google Scholar] [CrossRef]
- Tong, S.Y.; Zhang, L.L.; Joseph, J.; Jiang, X.J. Celastrol pretreatment attenuates rat myocardial ischemia/reperfusion injury by inhibiting high mobility group box 1 protein expression via the PI3K/Akt pathway. Biochem. Biophys. Res. Commun. 2018, 497, 843–849. [Google Scholar] [CrossRef]
- Li, X.Y.; Wu, N.; Zou, L.; Jia, D.L. Protective effect of celastrol on myocardial ischemia-reperfusion injury. Anatol. J. Cardiol. 2017, 18, 384–390. [Google Scholar] [CrossRef]
- Sharma, S.; Mishra, R.; Walker, B.L.; Deshmukh, S.; Zampino, M.; Patel, J.; Anamalai, M.; Simpson, D.; Singh, I.S.; Kaushal, S.; et al. Celastrol, an oral heat shock activator, ameliorates multiple animal disease models of cell death. Cell Stress Chaperones 2015, 20, 185–201. [Google Scholar] [CrossRef] [Green Version]
- Der Sarkissian, S.; Cailhier, J.F.; Borie, M.; Stevens, L.M.; Gaboury, L.; Mansour, S.; Hamet, P.; Noiseux, N. Celastrol protects ischaemic myocardium through a heat shock response with up-regulation of haeme oxygenase-1. Br. J. Pharmacol. 2014, 171, 5265–5279. [Google Scholar] [CrossRef] [Green Version]
- Krishnan-Sivadoss, I.; Mijares-Rojas, I.A.; Villarreal-Leal, R.A.; Torre-Amione, G.; Knowlton, A.A.; Guerrero-Beltran, C.E. Heat shock protein 60 and cardiovascular diseases: An intricate love-hate story. Med. Res. Rev. 2021, 41, 29–71. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, S.; Tokarczyk, G. Lycopene in the Prevention of Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 1957. [Google Scholar] [CrossRef]
- Pham, L.; Kim, E.C.; Ou, W.Q.; Phung, C.D.; Nguyen, T.T.; Pham, T.T.; Poudel, K.; Gautam, M.; Nguyen, H.T.; Jeong, J.H.; et al. Targeting and clearance of senescent foamy macrophages and senescent endothelial cells by antibody-functionalized mesoporous silica nanoparticles for alleviating aorta atherosclerosis. Biomaterials 2021, 269, 120677. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Orekhov, N.A.; Grechko, A.V.; Orekhov, A.N. Heat Shock Protein 90 as Therapeutic Target for CVDs and Heart Ageing. Int. J. Mol. Sci. 2022, 23, 649. [Google Scholar] [CrossRef]
- Carlisle, C.; Prill, K.; Pilgrim, D. Chaperones and the Proteasome System: Regulating the Construction and Demolition of Striated Muscle. Int. J. Mol. Sci. 2018, 19, 32. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, A.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Dunkerly-Eyring, B.L.; Keceli, G.; Ranek, M.J. Phosphorylation Modifications Regulating Cardiac Protein Quality Control Mechanisms. Front. Physiol. 2020, 11, 593585. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Duron, D.I.; Stine, C.; Mishra, S.; Blagg, B.S.J.; Streicher, J.M. The Alpha Isoform of Heat Shock Protein 90 and the Co-chaperones p23 and Cdc37 Promote Opioid Anti-nociception in the Brain. Front. Mol. Neurosci. 2019, 12, 294. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [CrossRef]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160530. [Google Scholar] [CrossRef] [Green Version]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.T.; Panaretou, B.; Yoshida, S.; Mayer, M.P.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.T.; Scroggins, B.T.; Xu, W.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar] [CrossRef]
- Gupta, R.S. Phylogenetic analysis of the 90 kD heat shock family of protein sequences and an examination of the relationship among animals, plants, and fungi species. Mol. Biol. Evol. 1995, 12, 1063–1073. [Google Scholar] [CrossRef]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 2012, 1823, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Koch, G.; Smith, M.; Macer, D.; Webster, P.; Mortara, R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 1986, 86, 217–232. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z. Roles of heat shock protein gp96 in the ER quality control: Redundant or unique function? Mol. Cells 2005, 20, 173–182. [Google Scholar]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Soldano, K.L.; Jivan, A.; Nicchitta, C.V.; Gewirth, D.T. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J. Biol. Chem. 2003, 278, 48330–48338. [Google Scholar] [CrossRef] [Green Version]
- Immormino, R.M.; Dollins, D.E.; Shaffer, P.L.; Soldano, K.L.; Walker, M.A.; Gewirth, D.T. Ligand-induced conformational shift in the N-terminal domain of GRP94, an Hsp90 chaperone. J. Biol. Chem. 2004, 279, 46162–46171. [Google Scholar] [CrossRef] [Green Version]
- Dollins, D.E.; Immormino, R.M.; Gewirth, D.T. Structure of unliganded GRP94, the endoplasmic reticulum Hsp90. Basis for nucleotide-induced conformational change. J. Biol. Chem. 2005, 280, 30438–30447. [Google Scholar] [CrossRef] [Green Version]
- Garg, G.; Khandelwal, A.; Blagg, B.S. Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects. Adv. Cancer Res. 2016, 129, 51–88. [Google Scholar] [CrossRef] [Green Version]
- Hainzl, O.; Lapina, M.C.; Buchner, J.; Richter, K. The Charged Linker Region Is an Important Regulator of Hsp90 Function. J. Biol. Chem. 2009, 284, 22559–22567. [Google Scholar] [CrossRef] [Green Version]
- Biswas, C.; Ostrovsky, O.; Makarewich, C.A.; Wanderling, S.; Gidalevitz, T.; Argon, Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Hsp90 regulation of mitochondrial protein folding: From organelle integrity to cellular homeostasis. Cell. Mol. Life Sci. 2013, 70, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Pant, A.; Chittayil Krishnakumar, K.; Chakkalaparambil Dileep, N.; Yamana, M.; Meenakshisundaran Alamelu, N.; Paithankar, K.; Amash, V.; Amere Subbarao, S. Hsp90 and its mitochondrial homologue TRAP-1 independently regulate hypoxia adaptations in Caenorhabditis elegans. Mitochondrion 2021, 60, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 as a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef]
- Leskovar, A.; Wegele, H.; Werbeck, N.D.; Buchner, J.; Reinstein, J. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J. Biol. Chem. 2008, 283, 11677–11688. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H. TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors. BMB Rep. 2012, 45, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Felts, S.J.; Owen, B.A.L.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef] [Green Version]
- Song, X.M.; Luo, Y.Z. The regulatory mechanism of Hsp90 alpha secretion from endothelial cells and its role in angiogenesis during wound healing. Biochem. Biophys. Res. Commun. 2010, 398, 111–117. [Google Scholar] [CrossRef]
- Hunter-Lavin, C.; Davies, E.L.; Bacelar, M.M.F.V.G.; Marshall, M.J.; Andrew, S.M.; Williams, J.H.H. Hsp70 release from peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun. 2004, 324, 511–517. [Google Scholar] [CrossRef]
- Hightower, L.E.; Guidon, P.T. Selective Release from Cultured Mammalian-Cells of Heat-Shock (Stress) Proteins That Resemble Glia-Axon Transfer Proteins. J. Cell. Physiol. 1989, 138, 257–266. [Google Scholar] [CrossRef]
- Nickel, W.; Seedorf, M. Unconventional Mechanisms of Protein Transport to the Cell Surface of Eukaryotic Cells. Annu. Rev. Cell Dev. Biol. 2008, 24, 287–308. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Tytell, M.; Greenberg, S.G.; Lasek, R.J. Heat Shock-Like Protein Is Transferred from Glia to Axon. Brain Res. 1986, 363, 161–164. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.; Srivastava, P.K. Necrotic but not apoptiotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF kappa B pathway. Cell Stress Chaperones 2000, 5, 373. [Google Scholar]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Skorzynska-Dziduszko, K.E.; Olszewska, A.; Prendecka, M.; Malecka-Massalska, T. Serum Heat Shock Protein 90 Alpha: A New Marker of Hypertension-Induced Endothelial Injury? Adv. Clin. Exp. Med. 2016, 25, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Giles, T.D.; Sander, G.E.; Nossaman, B.D.; Kadowitz, P.J. Impaired Vasodilation in the Pathogenesis of Hypertension: Focus on Nitric Oxide, Endothelial-Derived Hyperpolarizing Factors, and Prostaglandins. J. Clin. Hypertens. 2012, 14, 198–205. [Google Scholar] [CrossRef]
- Chatterjee, A.; Catravas, J.D. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vasc. Pharmacol. 2008, 49, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Buchner, J. Hsp90: Chaperoning signal transduction. J. Cell. Physiol. 2001, 188, 281–290. [Google Scholar] [CrossRef]
- Usman, M.; Ilyas, A.; Syed, B.; Hashim, Z.; Ahmed, A.; Zarina, S. Serum HSP90-Alpha and Oral Squamous Cell Carcinoma: A Prospective Biomarker. Protein Pept. Lett. 2021, 28, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Fawcett, T.W.; Udelsman, R.; Holbrook, N.J. Activation of heat shock transcription factor 1 in rat aorta in response to high blood pressure. Hypertension 1996, 28, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yang, P.; Yi, B.; Huo, Y.; Chen, M.; Zhang, J.; Sun, J. Heat shock protein 90 inhibition by 17-DMAG attenuates abdominal aortic aneurysm formation in mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H841–H852. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, J.; Chen, J.; Lai, J.; Zhu, F.; Xu, X.; Wang, D.W. CYP2J2-Derived EETs Attenuated Angiotensin II-Induced Adventitial Remodeling via Reduced Inflammatory Response. Cell. Physiol. Biochem. 2016, 39, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.Q.; Gao, P.J.; Shen, W.L.; Fan, C.L.; Liu, J.J.; Zhu, D.L. Angiotensin II-stimulated collagen synthesis in aortic adventitial fibroblasts is mediated by connective tissue growth factor. Hypertens. Res. 2008, 31, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Cong, Z.; Wang, X.; Yuan, Y.; Xu, R.; Lu, Z.; Wang, X.; Qi, J. Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovasc. Res. 2020, 116, 1071–1084. [Google Scholar] [CrossRef]
- Oyama, J.; Maeda, T.; Sasaki, M.; Higuchi, Y.; Node, K.; Makino, N. Repetitive hyperthermia attenuates progression of left ventricular hypertrophy and increases telomerase activity in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2092–H2101. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Sakao, S.; Tatsumi, K. Vascular remodeling in pulmonary arterial hypertension: Multiple cancer-like pathways and possible treatment modalities. Int. J. Cardiol. 2011, 147, 4–12. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Paulin, R.; Courboulin, A.; Meloche, J.; Mainguy, V.; Dumas de la Roque, E.; Saksouk, N.; Cote, J.; Provencher, S.; Sussman, M.A.; Bonnet, S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011, 123, 1205–1215. [Google Scholar] [CrossRef]
- Crosswhite, P.; Sun, Z. Molecular mechanisms of pulmonary arterial remodeling. Mol. Med. 2014, 20, 191–201. [Google Scholar] [CrossRef]
- Ma, L.; Chung, W.K. The genetic basis of pulmonary arterial hypertension. Hum. Genet. 2014, 133, 471–479. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Cool, C.; Lee, S.D.; Wright, L.; Geraci, M.W.; Tuder, R.M. Primary pulmonary hypertension between inflammation and cancer. Chest 1998, 114, 225s–230s. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.C.; Caino, M.C.; Lisanti, S.; Ghosh, J.C.; Dohi, T.; Danial, N.N.; Villanueva, J.; Ferrero, S.; Vaira, V.; Santambrogio, L.; et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 2012, 22, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 Accumulation Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wolk, J.H.; Gewitz, M.H.; Mathew, R. Progressive endothelial cell damage in an inflammatory model of pulmonary hypertension. Exp. Lung Res. 2010, 36, 57–66. [Google Scholar] [CrossRef]
- Mathew, R.; Huang, J.; Katta, U.S.; Krishnan, U.; Sandoval, C.; Gewitz, M.H. Immunosuppressant-induced endothelial damage and pulmonary arterial hypertension. J. Pediatr. Hematol. Oncol. 2011, 33, 55–58. [Google Scholar] [CrossRef]
- Murata, T.; Sato, K.; Hori, M.; Ozaki, H.; Karaki, H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J. Biol. Chem. 2002, 277, 44085–44092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R. Cell-specific dual role of caveolin-1 in pulmonary hypertension. Pulm. Med. 2011, 2011, 573432. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Pfister, S.L.; Slaughter, J.C.; Kaplowitz, M.R.; Zhang, Y.; Zeng, H.; Frye, N.R.; Aschner, J.L. Protein complex formation with heat shock protein 90 in chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1190–H1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Frid, M.; Gewitz, M.H.; Fallon, J.T.; Brown, D.; Krafsur, G.; Stenmark, K.; Mathew, R. Hypoxia-induced pulmonary hypertension and chronic lung disease: Caveolin-1 dysfunction an important underlying feature. Pulm. Circ. 2019, 9, 2045894019837876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Iturbe, B.; Johnson, R.J. Heat shock proteins and cardiovascular disease. Physiol. Int. 2018, 105, 19–37. [Google Scholar] [CrossRef]
- Mayerl, C.; Lukasser, M.; Sedivy, R.; Niederegger, H.; Seiler, R.; Wick, G. Atherosclerosis research from past to present—On the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch. 2006, 449, 96–103. [Google Scholar] [CrossRef]
- Williams, J.W.; Huang, L.H.; Randolph, G.J. Cytokine Circuits in Cardiovascular Disease. Immunity 2019, 50, 941–954. [Google Scholar] [CrossRef]
- Businaro, R.; Profumo, E.; Tagliani, A.; Buttari, B.; Leone, S.; D’Amati, G.; Ippoliti, F.; Leopizzi, M.; D’Arcangelo, D.; Capoano, R.; et al. Heat-shock protein 90: A novel autoantigen in human carotid atherosclerosis. Atherosclerosis 2009, 207, 74–83. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Martin-Ventura, J.L.; Blanco-Colio, L.M.; Egido, J.; Michel, J.B.; Meilhac, O. Heat-shock proteins in cardiovascular disease. Adv. Clin. Chem. 2011, 54, 1–43. [Google Scholar] [CrossRef]
- Binder, R.J.; Srivastava, P.K. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 6128–6133. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Hinagata, J.; Tanaka, T.; Imanishi, T.; Wada, Y.; Kodama, T.; Doi, T. HSP90, HSP70, and GAPDH directly interact with the cytoplasmic domain of macrophage scavenger receptors. Biochem. Biophys. Res. Commun. 2002, 290, 858–864. [Google Scholar] [CrossRef]
- Xu, Q.; Metzler, B.; Jahangiri, M.; Mandal, K. Molecular chaperones and heat shock proteins in atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H506–H514. [Google Scholar] [CrossRef]
- Mu, H.; Wang, L.; Zhao, L. HSP90 inhibition suppresses inflammatory response and reduces carotid atherosclerotic plaque formation in ApoE mice. Cardiovasc. Ther. 2017, 35, e12243. [Google Scholar] [CrossRef]
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Meagher, P.B.; Lee, X.A.; Lee, J.; Visram, A.; Friedberg, M.K.; Connelly, K.A. Cardiac Fibrosis: Key Role of Integrins in Cardiac Homeostasis and Remodeling. Cells 2021, 10, 770. [Google Scholar] [CrossRef]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Tamura, S.; Marunouchi, T.; Tanonaka, K. Heat-shock protein 90 modulates cardiac ventricular hypertrophy via activation of MAPK pathway. J. Mol. Cell. Cardiol. 2019, 127, 134–142. [Google Scholar] [CrossRef]
- Datta, R.; Bansal, T.; Rana, S.; Datta, K.; Datta Chaudhuri, R.; Chawla-Sarkar, M.; Sarkar, S. Myocyte-Derived Hsp90 Modulates Collagen Upregulation via Biphasic Activation of STAT-3 in Fibroblasts during Cardiac Hypertrophy. Mol. Cell. Biol. 2017, 37, e00611–e00616. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Merino, D.; Gomez, J.M.; Nistal, J.F.; Hurle, M.A.; Cortajarena, A.L.; Villar, A.V. Extracellular heat shock protein 90 binding to TGFbeta receptor I participates in TGFbeta-mediated collagen production in myocardial fibroblasts. Cell. Signal. 2016, 28, 1563–1579. [Google Scholar] [CrossRef]
- Roberts, R.J.; Hallee, L.; Lam, C.K. The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases. J. Pers. Med. 2021, 11, 1373. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kim, M.; Min, H.K.; Lee, Y.U.; Kwon, D.H.; Lee, M.; Lee, S.; Kook, T.; Joung, H.; Nam, K.I.; et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc. Res. 2019, 115, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodriguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gomez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohji, G.; Hidayat, S.; Nakashima, A.; Tokunaga, C.; Oshiro, N.; Yoshino, K.; Yokono, K.; Kikkawa, U.; Yonezawa, K. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J. Biochem. 2006, 139, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Giulino-Roth, L.; van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of Hsp90 Suppresses PI3K/AKT/mTOR Signaling and Has Antitumor Activity in Burkitt Lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [Green Version]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Wu, J.X.; Sha, J.Z.; Yang, B.; Sun, J.R.; Bao, E.D. Heat shock protein 90 relieves heat stress damage of myocardial cells by regulating Akt and PKM2 signaling in vivo. Int. J. Mol. Med. 2020, 45, 1888–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, V.; Rothermel, B.A. Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell. Cardiol. 2017, 103, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef]
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]
- Minami, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The calmodulin-binding domain of the mouse 90-kDa heat shock protein. J. Biol. Chem. 1993, 268, 9604–9610. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; He, L.; Xiang, Y.; Tian, C.; Li, C.; Tan, P.; Jing, J.; Tian, Y.; Du, L.; et al. Discovery of Small-Molecule Inhibitors of the HSP90-Calcineurin-NFAT Pathway against Glioblastoma. Cell Chem. Biol. 2019, 26, 352–365. [Google Scholar] [CrossRef]
- Salazar, N.C.; Chen, J.; Rockman, H.A. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 2007, 1768, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Benovic, J.L. G protein-coupled receptor kinase interaction with Hsp90 mediates kinase maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef] [Green Version]
- Dzimiri, N.; Al-Bahnasi, K.; Al-Halees, Z. Myocardial hypertrophy is not a prerequisite for changes in early gene expression in left ventricular volume overload. Fundam. Clin. Pharmacol. 2004, 18, 39–44. [Google Scholar] [CrossRef]
- Salim, S.; Eikenburg, D.C. Role of 90-kDa heat shock protein (Hsp 90) and protein degradation in regulating neuronal levels of G protein-coupled receptor kinase 3. J. Pharmacol. Exp. Ther. 2007, 320, 1106–1112. [Google Scholar] [CrossRef] [Green Version]
- Mielczarek-Palacz, A.; Sikora, J.; Kondera-Anasz, Z.; Smycz, M. Changes in concentrations of tumor necrosis factor TNF and its soluble receptors type 1 (sTNF-r1) and type 2 (sTNF-R2) in serum of patients with ST-segment elevation myocardial infarction. Wiad. Lek. 2011, 64, 71–74. [Google Scholar]
- Monden, Y.; Kubota, T.; Inoue, T.; Tsutsumi, T.; Kawano, S.; Ide, T.; Tsutsui, H.; Sunagawa, K. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H743–H753. [Google Scholar] [CrossRef]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; Di Loreto, C.; Beltrami, C.A.; Krajewski, S.; et al. Apoptosis in the failing human heart. N. Engl. J. Med. 1997, 336, 1131–1141. [Google Scholar] [CrossRef]
- Zhe-Wei, S.; Li-Sha, G.; Yue-Chun, L. The Role of Necroptosis in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 721. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Cook, W.D.; Moujalled, D.M.; Ralph, T.J.; Lock, P.; Young, S.N.; Murphy, J.M.; Vaux, D.L. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014, 21, 1600–1612. [Google Scholar] [CrossRef]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.C.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; van Delft, M.F.; Liu, Z.; Conos, S.A.; Zhang, J.G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; Liu, Z.G. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 2000, 275, 10519–10526. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, T.; Kalai, M.; van Loo, G.; Declercq, W.; Vandenabeele, P. Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J. Biol. Chem. 2003, 278, 5622–5629. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xu, T.; Cao, Y.; Wang, H.; Li, L.; Chen, S.; Wang, X.; Shen, Z. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5017–5022. [Google Scholar] [CrossRef] [Green Version]
- Marunouchi, T.; Nishiumi, C.; Iinuma, S.; Yano, E.; Tanonaka, K. Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur. J. Pharmacol. 2021, 898, 173987. [Google Scholar] [CrossRef]
- Marunouchi, T.; Ito, T.; Onda, S.; Kyo, L.; Takahashi, K.; Uchida, M.; Yano, E.; Tanonaka, K. Effects of 17-AAG on the RIP1/RIP3/MLKL pathway during the development of heart failure following myocardial infarction in rats. J. Pharmacol. Sci. 2021, 147, 192–199. [Google Scholar] [CrossRef]
- Caceres, R.A.; Chavez, T.; Maestro, D.; Palanca, A.R.; Bolado, P.; Madrazo, F.; Aires, A.; Cortajarena, A.L.; Villar, A.V. Reduction of cardiac TGFbeta-mediated profibrotic events by inhibition of Hsp90 with engineered protein. J. Mol. Cell. Cardiol. 2018, 123, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Fernandez-Garcia, C.E.; Gomez-Guerrero, C.; Lopez-Franco, O.; Muñoz-Garcia, B.; Egido, J.; Blanco-Colio, L.M.; Martin-Ventura, J.L. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc. Res. 2012, 95, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.H.; Skinner, J.P.; Bird, M.J.; Formosa, L.E.; Zhang, J.G.; Kluck, R.M.; Belz, G.T.; Chong, M.M. A Role for the Mitochondrial Protein Mrpl44 in Maintaining OXPHOS Capacity. PLoS ONE 2015, 10, e0134326. [Google Scholar] [CrossRef]
- Kawano, S.; Kubota, T.; Monden, Y.; Tsutsumi, T.; Inoue, T.; Kawamura, N.; Tsutsui, H.; Sunagawa, K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1337–H1344. [Google Scholar] [CrossRef] [Green Version]
- DeBoer, C.; Meulman, P.A.; Wnuk, R.J.; Peterson, D.H. Geldanamycin, a new antibiotic. J. Antibiot. 1970, 23, 442–447. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Huang, Z.; Li, Q.J.; Zhong, G.Q.; Meng, J.J.; Wang, D.X.; Tu, R.H. Role of HSP90 in suppressing TLR4-mediated inflammation in ischemic postconditioning. Clin. Hemorheol. Microcirc. 2020, 76, 51–62. [Google Scholar] [CrossRef]
- Michela, P.; Velia, V.; Aldo, P.; Ada, P. Role of connexin 43 in cardiovascular diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Uesugi, M.; Takase, K.; Miyamoto, N.; Sawada, K. Hsp90 inhibitor geldanamycin attenuates the cytotoxicity of sunitinib in cardiomyocytes via inhibition of the autophagy pathway. Toxicol. Appl. Pharmacol. 2017, 329, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Assadi, H. Heat acclimation-mediated cross-tolerance in cardioprotection Do HSP70 and HIF-1 alpha play a role? Anal. Card. Dev. Embryo Old Age 2010, 1188, 199–206. [Google Scholar] [CrossRef]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef]
- Fukuyo, Y.; Hunt, C.R.; Horikoshi, N. Geldanamycin and its anti-cancer activities. Cancer Lett. 2010, 290, 24–35. [Google Scholar] [CrossRef]
- Le Brazidec, J.Y.; Kamal, A.; Busch, D.; Thao, L.; Zhang, L.; Timony, G.; Grecko, R.; Trent, K.; Lough, R.; Salazar, T.; et al. Synthesis and biological evaluation of a new class of geldanamycin derivatives as potent inhibitors of Hsp90. J. Med. Chem. 2004, 47, 3865–3873. [Google Scholar] [CrossRef]
- Kim, J.; Jang, S.W.; Park, E.; Oh, M.; Park, S.; Ko, J. The role of heat shock protein 90 in migration and proliferation of vascular smooth muscle cells in the development of atherosclerosis. J. Mol. Cell. Cardiol. 2014, 72, 157–167. [Google Scholar] [CrossRef]
- Marunouchi, T.; Nakashima, M.; Ebitani, S.; Umezu, S.; Karasawa, K.; Yano, E.; Tanonaka, K. Hsp90 Inhibitor Attenuates the Development of Pathophysiological Cardiac Fibrosis in Mouse Hypertrophy via Suppression of the Calcineurin-NFAT and c-Raf-Erk Pathways. J. Cardiovasc. Pharmacol. 2021, 77, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update from 2014. Curr. Drug Targets 2020, 21, 302–317. [Google Scholar] [CrossRef]
- Ambade, A.; Catalano, D.; Lim, A.; Mandrekar, P. Inhibition of heat shock protein (molecular weight 90 kDa) attenuates proinflammatory cytokines and prevents lipopolysaccharide-induced liver injury in mice. Hepatology 2012, 55, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.G.; Lee, S.C.; Kim, O.H.; Kim, K.H.; Song, K.Y.; Lee, S.K.; Choi, B.J.; Jeong, W.; Kim, S.J. HSP90 inhibitor 17-DMAG exerts anticancer effects against gastric cancer cells principally by altering oxidant-antioxidant balance. Oncotarget 2017, 8, 56473–56489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.L.; Shen, H.H.; Cheng, P.Y.; Chu, Y.J.; Hwang, H.R.; Lam, K.K.; Lee, Y.M. 17-DMAG, an HSP90 Inhibitor, Ameliorates Multiple Organ Dysfunction Syndrome via Induction of HSP70 in Endotoxemic Rats. PLoS ONE 2016, 11, e0155583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, C.; Mori, A.; Moser, C.; Lang, S.A.; Dayoub, R.; Weiss, T.S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; Stoeltzing, O. Effect of heat-shock protein-90 (HSP90) inhibition on human hepatocytes and on liver regeneration in experimental models. Surgery 2010, 147, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Plescia, J.; Song, H.Y.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.Y.; Tan, H.W.; Jia, Y.F.; Li, D. Therapeutic effect of tripterine on adjuvant arthritis in rats. J. Ethnopharmacol. 2008, 118, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Pinna, G.F.; Fiorucci, M.; Reimund, J.M.; Taquet, N.; Arondel, Y.; Muller, C.D. Celastrol inhibits pro-inflammatory cytokine secretion in Crohn’s disease biopsies. Biochem. Biophys. Res. Commun. 2004, 322, 778–786. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [Green Version]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.Y.; Stegmaier, K.; Raj, S.M.; et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.A.; Kawahara, T.L.A.; Matsumoto, G.; Kim, S.J.; Gu, W.X.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [Green Version]
- Gray, P.J., Jr.; Prince, T.; Cheng, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 2008, 8, 491–495. [Google Scholar] [CrossRef]
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol Attenuates Angiotensin II-Induced Cardiac Remodeling by Targeting STAT3. Circ. Res. 2020, 126, 1007–1023. [Google Scholar] [CrossRef]
- Aceros, H.; Sarkissian, S.D.; Borie, M.; Stevens, L.M.; Mansour, S.; Noiseux, N. Celastrol-type HSP90 modulators allow for potent cardioprotective effects. Life Sci. 2019, 227, 8–19. [Google Scholar] [CrossRef]
- Wang, D.X.; Huang, Z.; Li, Q.J.; Zhong, G.Q.; He, Y.; Huang, W.Q.; Cao, X.L.; Tu, R.H.; Meng, J.J. Involvement of HSP90 in ischemic postconditioning-induced cardioprotection by inhibition of the complement system, JNK and inflammation. Acta Cir. Bras. 2020, 35, e202000105. [Google Scholar] [CrossRef] [Green Version]
- Tu, R.H.; Wang, D.X.; Zhong, G.Q.; Meng, J.J.; Wen, H.; Jie, F.; Bi, Q.; He, Y. New targets of morphine postconditioning protection of the myocardium in ischemia/reperfusion injury: Involvement of HSP90/Akt and C5a/NF-kappaB. Open Med. 2021, 16, 1552–1563. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Disease/Model | Detection Indicator | References |
|---|---|---|---|
| Geldanamycin | Ventricular hypertrophy; myocardial fibrosis; heart failure | TGF-β; NF-κB; Cx43 | [150,151,152] |
| 17-AGG | Atherosclerosis; pulmonary hypertension; heart failure | RIP1; RIP3; MLKL; c-Raf; ERK | [142,149] |
| 17-DMAG | Atherosclerosis; myocardial fibrosis | ERK; NADPH oxidase; NF-κB; Drp1 | [108,117,153,154] |
| Gamitrinib | Pulmonary hypertension | MRPL; ERAL1 | [87,155] |
| Celastrol | Myocardial infarction; ischemic cardiomyopathy | NF-κB; HO-1 | [156] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, S.; Yi, G.; Yu, K.; Feng, C.; Deng, S. The Role of HSP90 Inhibitors in the Treatment of Cardiovascular Diseases. Cells 2022, 11, 3444. https://doi.org/10.3390/cells11213444
Qi S, Yi G, Yu K, Feng C, Deng S. The Role of HSP90 Inhibitors in the Treatment of Cardiovascular Diseases. Cells. 2022; 11(21):3444. https://doi.org/10.3390/cells11213444
Chicago/Turabian StyleQi, Shiyu, Guang Yi, Kun Yu, Chong Feng, and Shoulong Deng. 2022. "The Role of HSP90 Inhibitors in the Treatment of Cardiovascular Diseases" Cells 11, no. 21: 3444. https://doi.org/10.3390/cells11213444