
The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ethical Statement
2.3. Animals
2.4. Bronchoalveolar Lavage Fluid (BALF) White Blood Cell Number and Total Protein Concentration
2.5. Histopathology, Immunohistochemistry and Lung Fibrosis Scoring
2.6. Tissue Collection
2.7. Western Blot Analysis
2.8. RNA Isolation and Quantitative Real-Time PCR (qPCR)
2.9. Lung Mechanics Measurements
2.10. Cell Culture and Protein Extraction
2.11. Endothelial Barrier Function
2.12. Statistical Analysis
3. Results
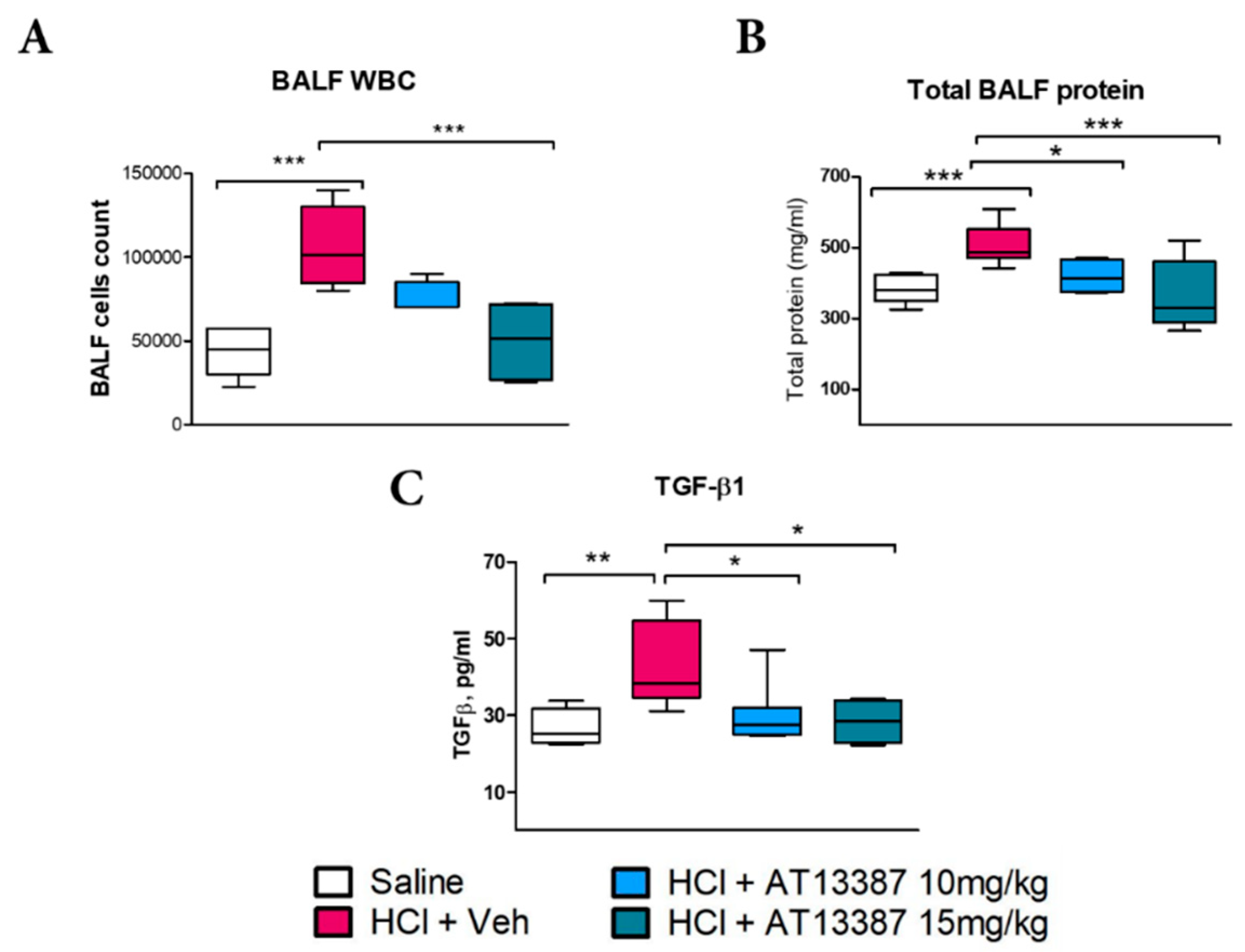
3.1. AT13387 Modulates HCl-Induced Persistent Alveolar Inflammation
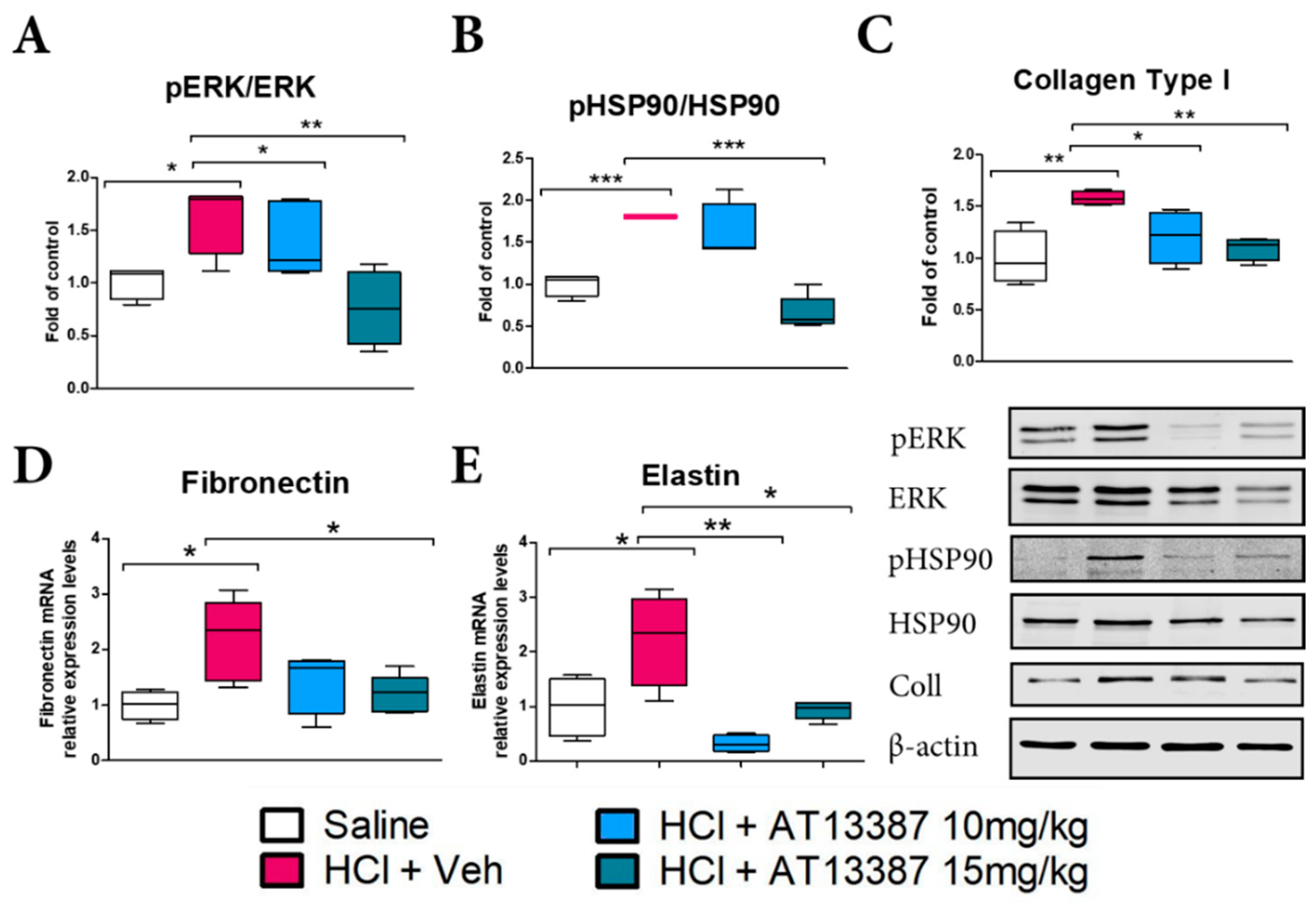
3.2. AT13387 Ameliorates Pulmonary Fibrosis and NLRP3 Staining after HCl Exposure
3.3. AT13387 Blocks Pro-Fibrotic Pathways
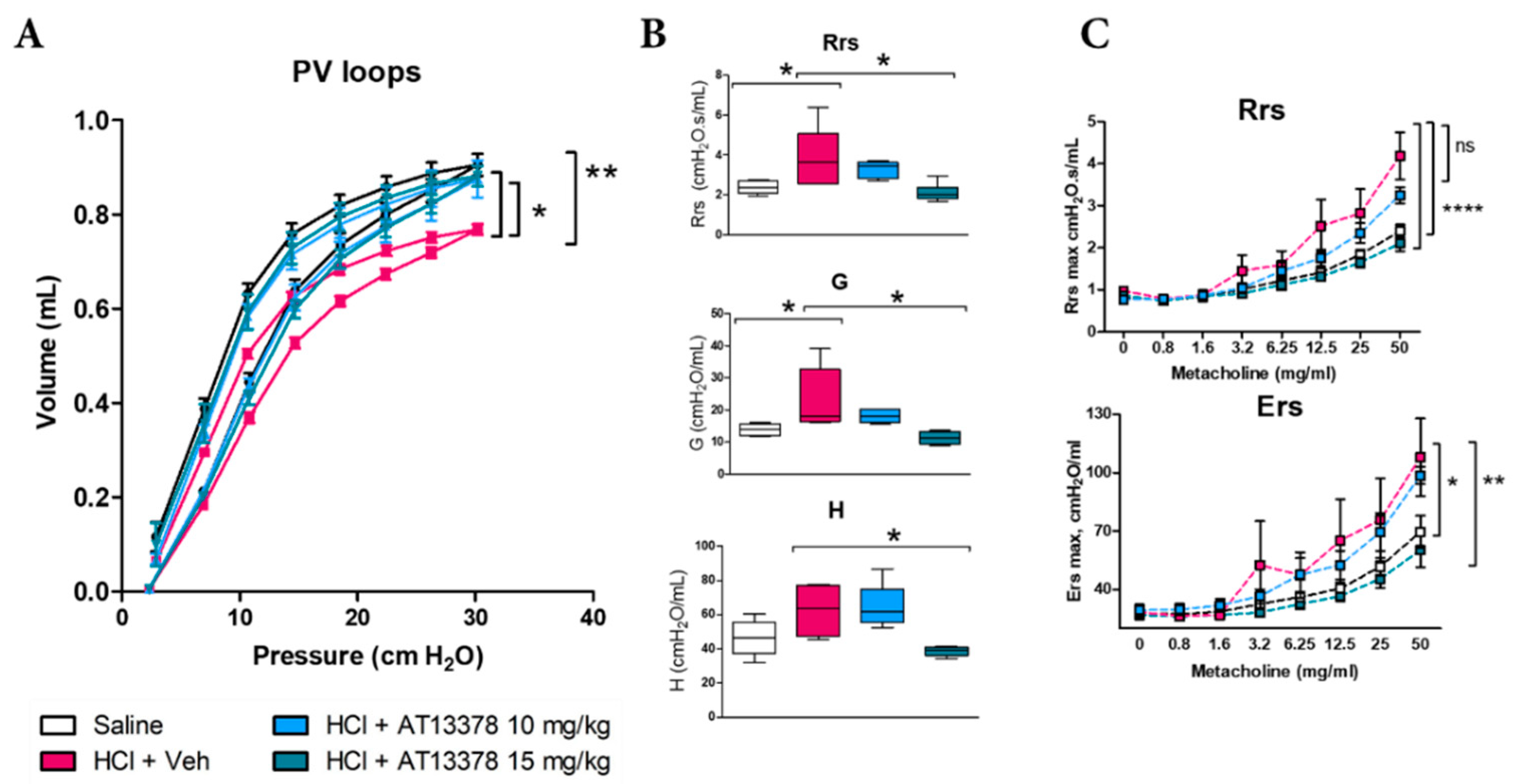
3.4. AT13387 Prevents HCl-Induced Lung Dysfunction and Airway Hyper-Responsiveness to Methacholine
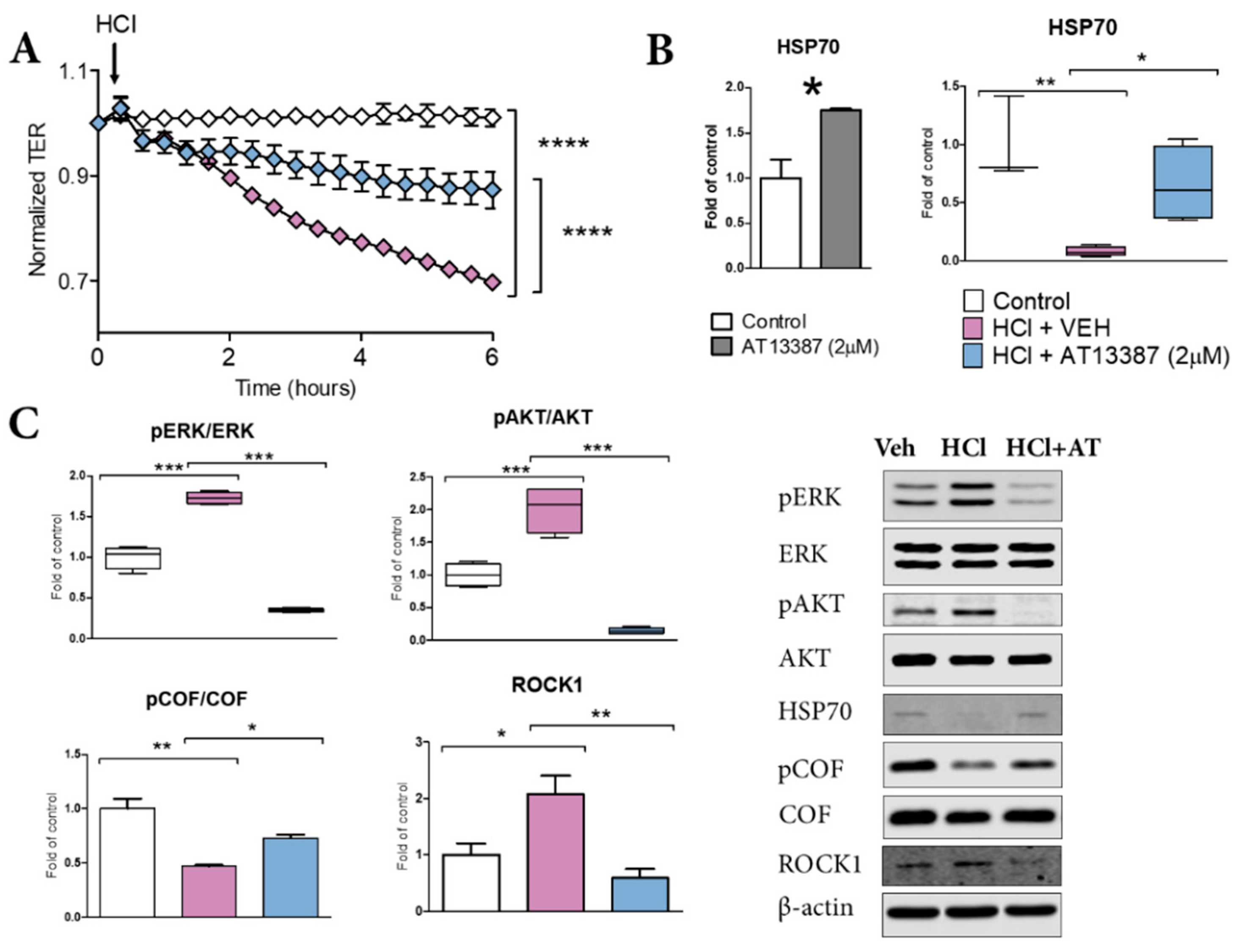
3.5. AT13387 Prevents HCl-Mediated Endothelial Barrier Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem. 1993, 62, 349–384. [Google Scholar] [CrossRef] [PubMed]
- Craig, E.A. The heat shock response. CRC Crit. Rev. Biochem. 1985, 18, 239–280. [Google Scholar] [CrossRef]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. Heat shock protein 90 inhibition in the inflamed lungs. Cell Stress Chaperones 2020, 25, 195–197. [Google Scholar] [CrossRef]
- Handbook, C.E. Hydrochloric acid. SRI Int. 2001, 733.400A–733.3003F. [Google Scholar]
- News, B. Syria Conflict: Government Helicopters ‘Drop Chlorine’ on Aleppo; BBC: London, UK, 7 September 2016. [Google Scholar]
- Kerger, B.D.; Fedoruk, M.J. Pathology, toxicology, and latency of irritant gases known to cause bronchiolitis obliterans disease: Does diacetyl fit the pattern? Toxicol. Rep. 2015, 2, 1463–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilburn, K.H. Effects of a hydrochloric acid spill on neurobehavioral and pulmonary function. J. Occup. Environ. Med. 1996, 38, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.M.; Weiss, M.A.; Bernstein, I.L. Reactive airways dysfunction syndrome (RADS). Persistent asthma syndrome after high level irritant exposures. Chest 1985, 88, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal. Toxicol. 2019, 31, 147–160. [Google Scholar] [CrossRef]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Exp. Lung Res. 2020, 46, 203–216. [Google Scholar] [CrossRef]
- Solopov, P.; Colunga Biancatelli, R.M.L.; Marinova, M.; Drimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Gregory, B.; Catravas, J.D. HSP90 Inhibition and Modulation of the Proteome: Therapeutical Implications for Idiopathic Pulmonary Fibrosis (IPF). Int. J. Mol. Sci. 2020, 21, 5286. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Bellaye, P.-S.; Burgy, O.; Kolb, M. Heat shock protein: A hot topic in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1602152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhead, A.J.; Angove, H.; Carr, M.G.; Chessari, G.; Congreve, M.; Coyle, J.E.; Cosme, J.; Graham, B.; Day, P.J.; Downham, R.; et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem. 2010, 53, 5956–5969. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Dimitropoulou, C.; Catravas, J.D. Age-Dependent Chronic Lung Injury and Pulmonary Fibrosis following Single Exposure to Hydrochloric Acid. Int. J. Mol. Sci. 2021, 22, 8833. [Google Scholar] [CrossRef] [PubMed]
- Solopov, P.; Marinova, M.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Development of chronic lung injury and pulmonary fibrosis in mice following acute exposure to nitrogen mustard. Inhal. Toxicol. 2020, 32, 141–154. [Google Scholar] [CrossRef]
- Solopov, P.; Colunga Biancatelli, R.M.L.; Dimitropoulou, C.; Catravas, J.D. Sex-Related Differences in Murine Models of Chemically Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 5909. [Google Scholar] [CrossRef]
- Catravas, J.D.; Snead, C.; Dimitropoulou, C.; Chang, A.S.Y.; Lucas, R.; Verin, A.D.; Black, S.M. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vasc. Pharm. 2010, 52, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Gregory, B.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction. Cells 2021, 10, 1489. [Google Scholar] [CrossRef] [PubMed]
- Tomcik, M.; Zerr, P.; Pitkowski, J.; Palumbo-Zerr, K.; Avouac, J.; Distler, O.; Becvar, R.; Senolt, L.; Schett, G.; Distler, J.H. Heat shock protein 90 (Hsp90) inhibition targets canonical TGF-β signalling to prevent fibrosis. Ann. Rheum. Dis. 2014, 73, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.; Kim, H.J.; Yu, M.R.; Kim, W.Y.; Kim, J.; Ryu, J.H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Ziyadeh, F. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab. Investig. 2012, 92, 1583–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, S.J.; Yoon, J.H.; Kim, B.H.; Lee, J.H.; Jung, E.U.; Lee, H.S. Heat shock protein 90 inhibitor induces apoptosis and attenuates activation of hepatic stellate cells. J. Pharmacol. Exp. Ther. 2009, 330, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P.; Houghton, P.J.; Alexander, D.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; et al. Initial testing (Stage 1) of AT13387, an HSP90 inhibitor, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2012, 59, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, T.; Paraiso, K.H.T.; Hearn, K.; Rodriguez-Lopez, A.M.; Munck, J.M.; Haarberg, H.E.; Sondak, V.K.; Thompson, N.T.; Azab, M.; Lyons, J.F.; et al. Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol. Cancer Ther. 2014, 13, 2793–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangjam, G.S.; Dimitropoulou, C.; Joshi, A.D.; Barabutis, N.; Shaw, M.C.; Kovalenkov, Y.; Wallace, C.M.; Fulton, D.J.; Patel, V.; Catravas, J.D. Novel mechanism of attenuation of LPS-induced NF-κB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizami, S.; Arunasalam, K.; Green, J.; Cook, J.; Lawrence, C.B.; Zarganes-Tzitzikas, T.; Davis, J.B.; Di Daniel, E.; Brough, D. Inhibition of the NLRP3 inflammasome by HSP90 inhibitors. Immunology 2021, 162, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicher, J.M. The Role of Heat Shock Proteins in Regulating Receptor Signal Transduction. Mol. Pharmacol. 2019, 95, 468. [Google Scholar] [CrossRef] [PubMed]
- Antonov, A.S.; Antonova, G.N.; Fujii, M.; ten Dijke, P.; Handa, V.; Catravas, J.D.; Verin, A.D. Regulation of endothelial barrier function by TGF-β type I receptor ALK5: Potential role of contractile mechanisms and heat shock protein 90. J. Cell. Physiol. 2012, 227, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, H.K.; Gillis, J.L.; Johnson, I.R.D.; Nassar, Z.D.; Moldovan, M.; Levrier, C.; Sadowski, M.C.; Chin, M.Y.; Tomlinson Guns, E.S.; Tarulli, G.; et al. Dysregulated fibronectin trafficking by Hsp90 inhibition restricts prostate cancer cell invasion. Sci. Rep. 2018, 8, 2090. [Google Scholar] [CrossRef]
- Hunter, M.C.; O’Hagan, K.L.; Kenyon, A.; Dhanani, K.C.; Prinsloo, E.; Edkins, A.L. Hsp90 binds directly to fibronectin (FN) and inhibition reduces the extracellular fibronectin matrix in breast cancer cells. PLoS ONE 2014, 9, e86842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.D.; Dimitropoulou, C.; Thangjam, G.; Snead, C.; Feldman, S.; Barabutis, N.; Fulton, D.; Hou, Y.; Kumar, S.; Patel, V.; et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J. Respir. Cell Mol. Biol. 2014, 50, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 2020, 11, 5975. [Google Scholar] [CrossRef] [PubMed]
- Kawana, K.; Miyamoto, Y.; Tanonaka, K.; Han-no, Y.; Yoshida, H.; Takahashi, M.; Takeo, S. Cytoprotective mechanism of heat shock protein 70 against hypoxia/reoxygenation injury. J. Mol. Cell. Cardiol. 2000, 32, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Bidmon-Fliegenschnee, B.; Lederhuber, H.C.; Csaicsich, D.; Pichler, J.; Herzog, R.; Memaran-Dadgar, N.; Huber, W.-D.; Aufricht, C.; Kratochwill, K. Overexpression of Hsp70 confers cytoprotection during gliadin exposure in Caco-2 cells. Pediatr. Res. 2015, 78, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yuan, B.; Dong, W.; Yang, B.; Yang, Y.; Lin, X.; Gong, G. Induction of Heat-Shock Protein 70 Expression by Geranylgeranylacetone Shows Cytoprotective Effects in Cardiomyocytes of Mice under Humid Heat Stress. PLoS ONE 2014, 9, e93536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voellmy, R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones 2004, 9, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colunga Biancatelli, R.M.L.; Solopov, P.; Dimitropoulou, C.; Gregory, B.; Day, T.; Catravas, J.D. The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. Cells 2022, 11, 1046. https://doi.org/10.3390/cells11061046
Colunga Biancatelli RML, Solopov P, Dimitropoulou C, Gregory B, Day T, Catravas JD. The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. Cells. 2022; 11(6):1046. https://doi.org/10.3390/cells11061046
Chicago/Turabian StyleColunga Biancatelli, Ruben M. L., Pavel Solopov, Christiana Dimitropoulou, Betsy Gregory, Tierney Day, and John D. Catravas. 2022. "The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis" Cells 11, no. 6: 1046. https://doi.org/10.3390/cells11061046