
Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Ethical Statements
2.2. Experimental Groups
2.3. Sample Collection
2.4. Antibodies and Reagents
2.5. Coagulation, Endothelium and Liver
2.6. Peripheral Blood Analysis (ROTEM)
2.7. Histological Analysis
2.8. Immunohistochemistry and Image Analysis
2.9. Immunofluorescence
2.10. Transmission Electron Microscopy
2.11. Real-Time PCR
2.12. Western Blot
2.13. Statistical Analysis
3. Results
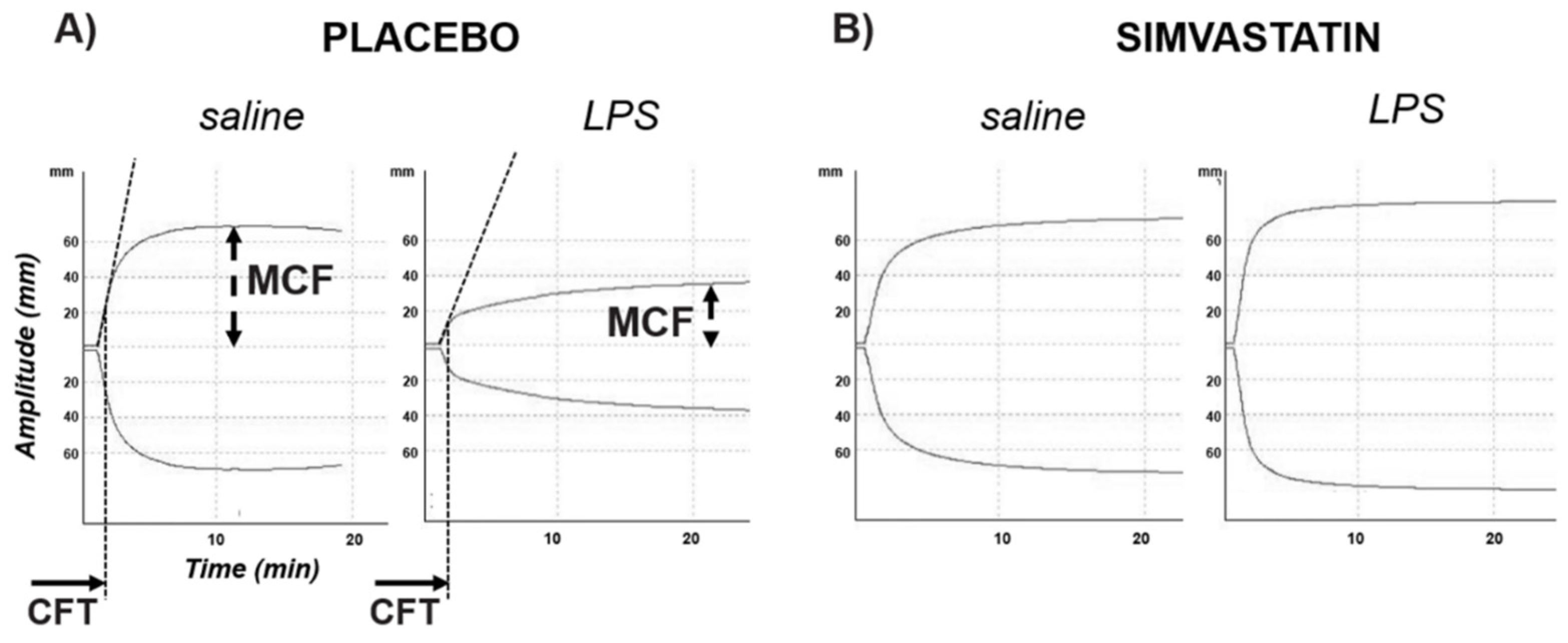
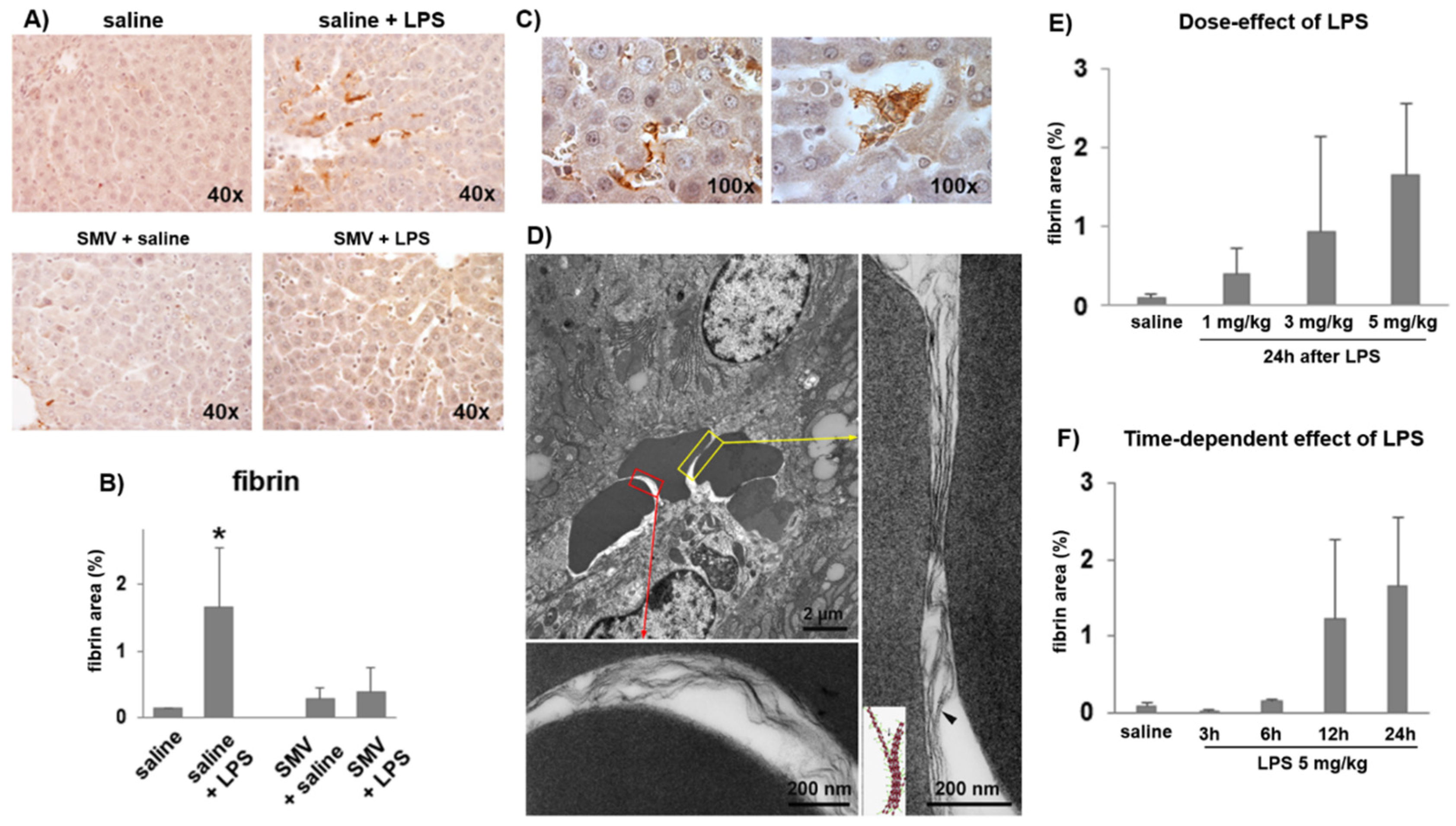
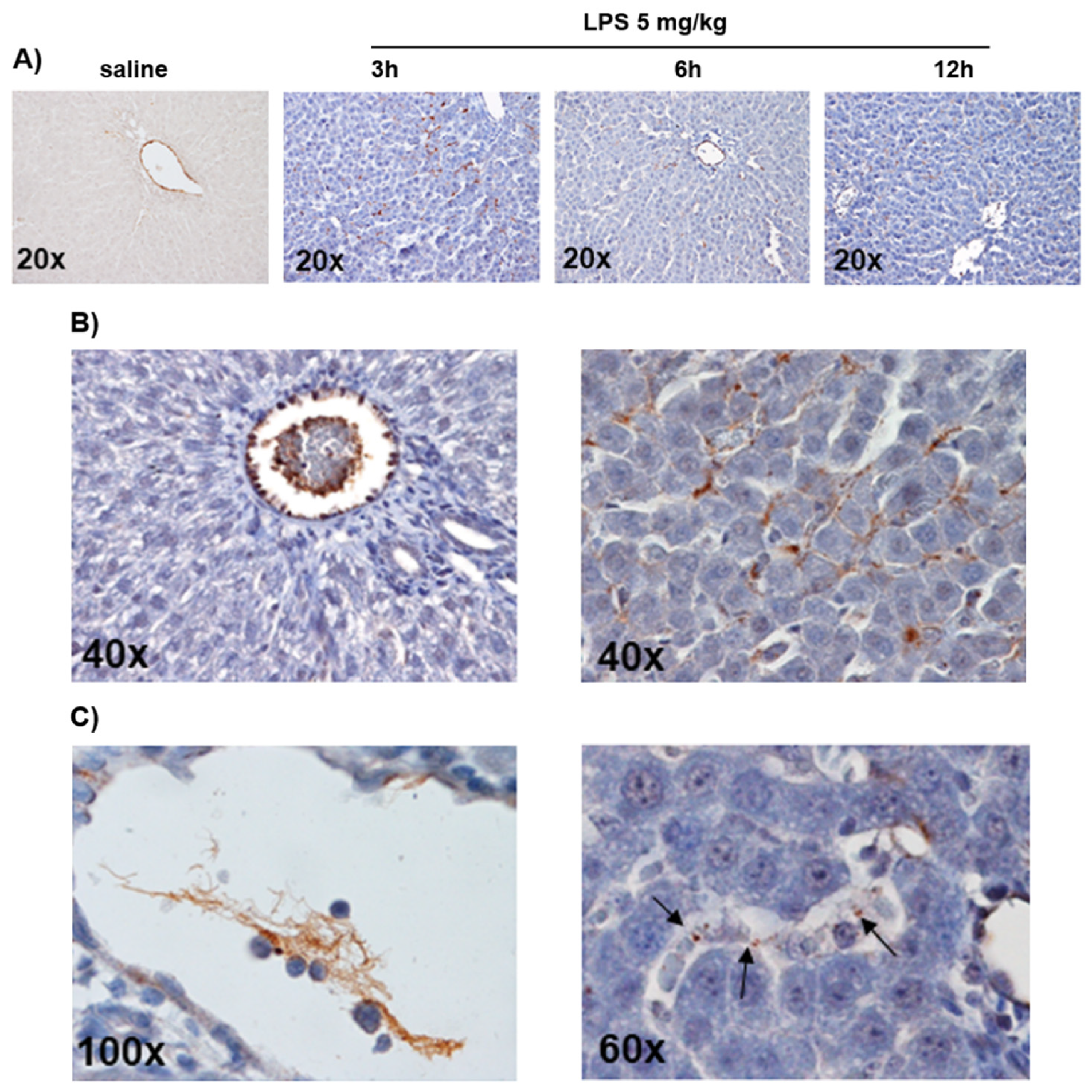
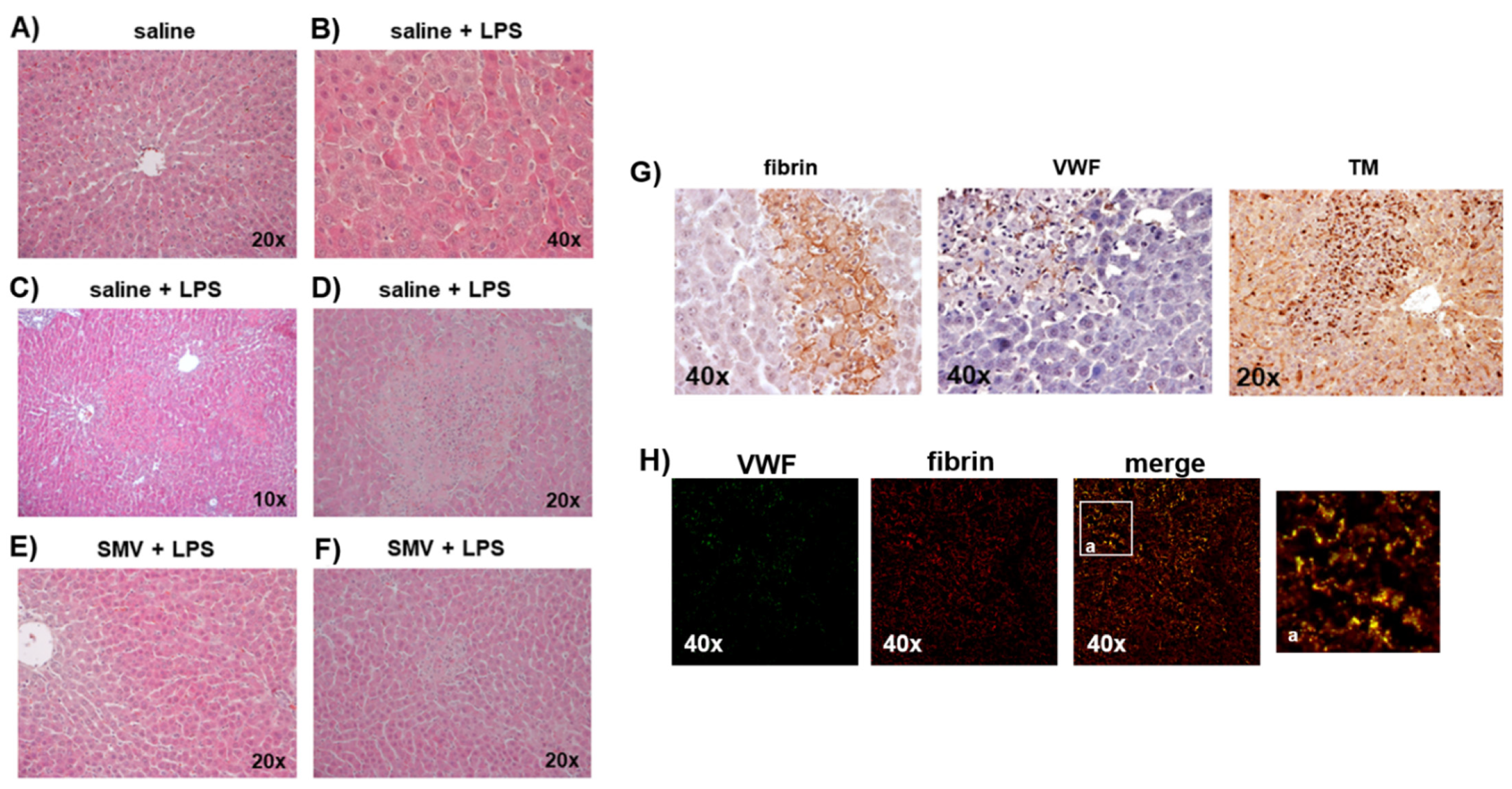
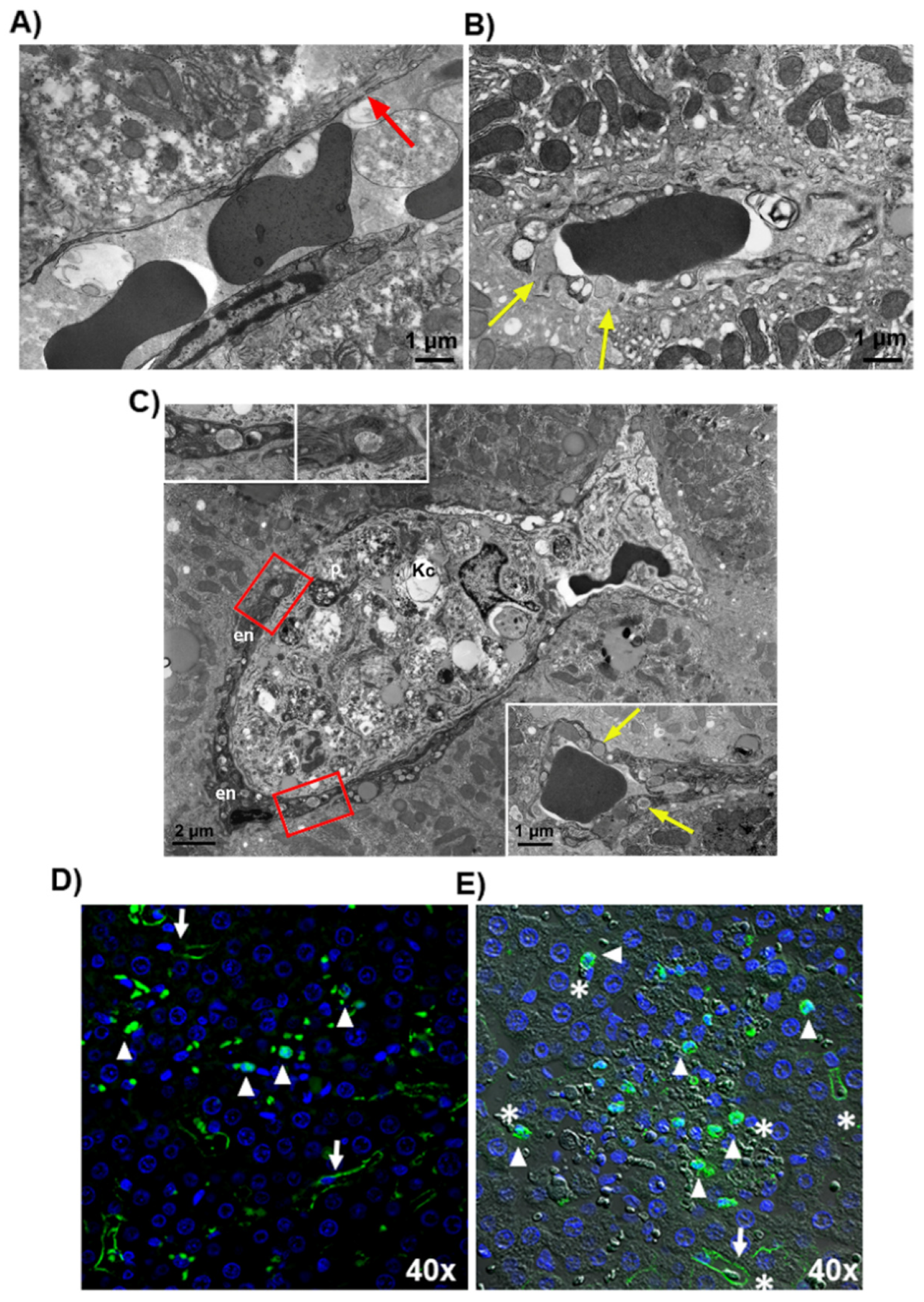
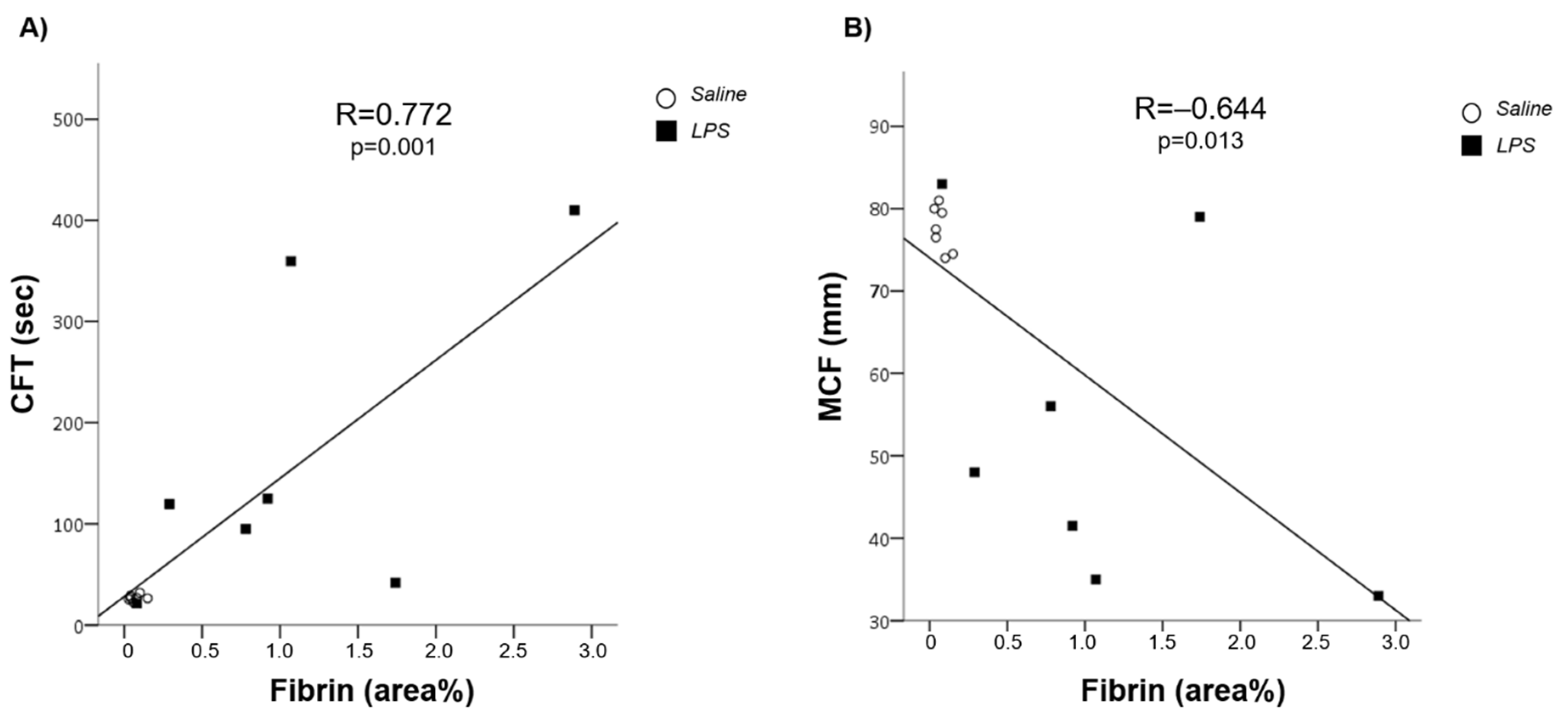
3.1. LPS-Induced Coagulopathy and Liver Microthrombosis
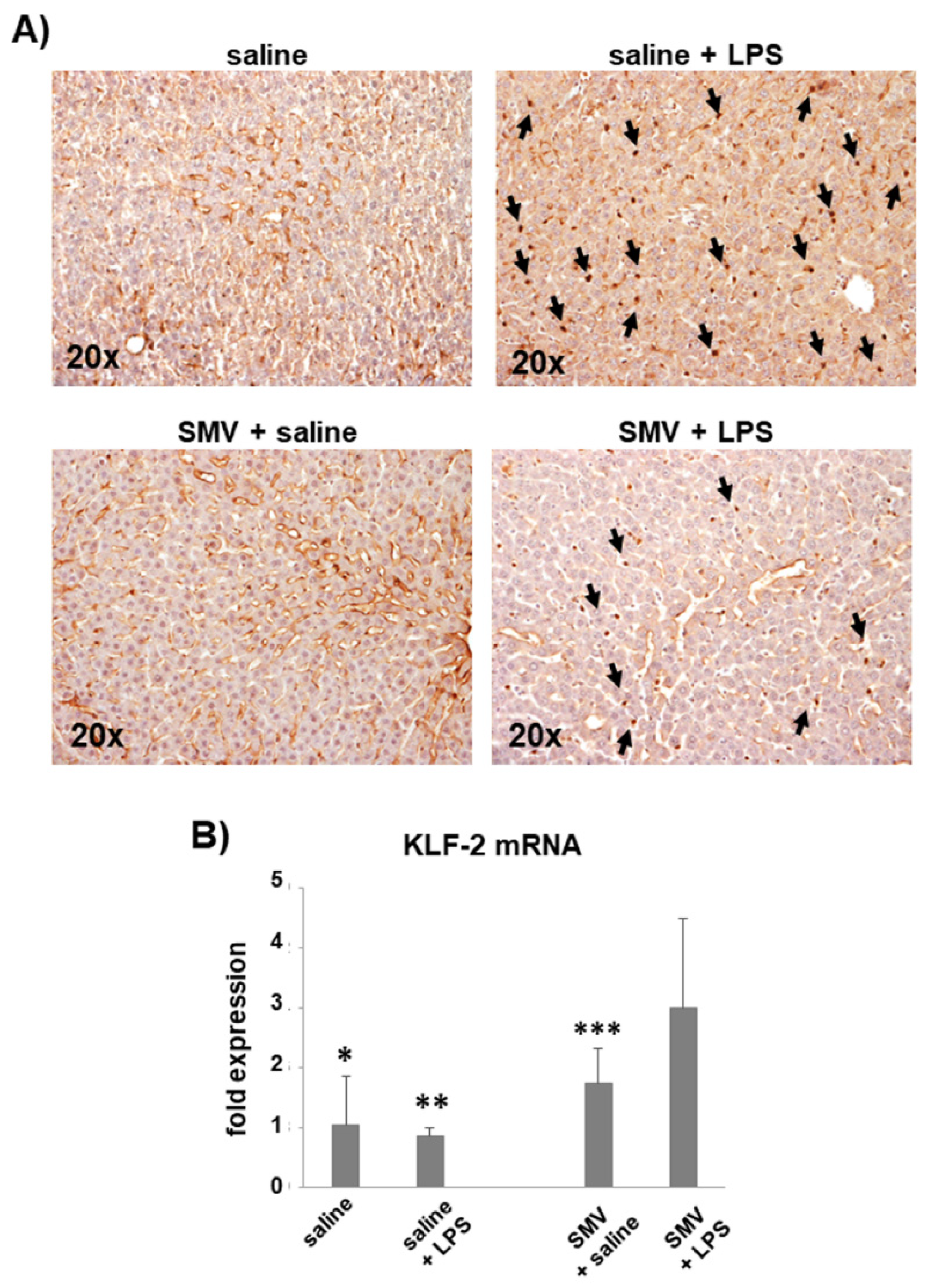
3.2. Microthrombosis-Associated Liver Damage
3.3. Simvastatin Effects on LPS-Induced Coagulopathy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing Sepsis as a Global Health Priority—A WHO Resolution. N. Engl. J. Med. 2017, 377, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, S.; Li, S. The Role of the Liver in Sepsis. Int. Rev. Immunol. 2014, 33, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, L.; Jordan, B.; Druml, W.; Bauer, P.; Metnitz, P.G.H.; Austrian Epidemiologic Study on Intensive Care, ASDI Study Group. Incidence and Prognosis of Early Hepatic Dysfunction in Critically Ill Patients—A Prospective Multicenter Study. Crit. Care Med. 2007, 35, 1099–1104. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Coagulation and Sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef]
- Lyons, P.G.; Micek, S.T.; Hampton, N.; Kollef, M.H. Sepsis-Associated Coagulopathy Severity Predicts Hospital Mortality. Crit. Care Med. 2018, 46, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Sivapalaratnam, S. Disseminated Intravascular Coagulation: An Update on Pathogenesis and Diagnosis. Expert Rev. Hematol. 2018, 11, 663–672. [Google Scholar] [CrossRef]
- Umemura, Y.; Yamakawa, K.; Ogura, H.; Yuhara, H.; Fujimi, S. Efficacy and Safety of Anticoagulant Therapy in Three Specific Populations with Sepsis: A Meta-Analysis of Randomized Controlled Trials. J. Thromb. Haemost. 2016, 14, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraldes, J.G.; Rodríguez-Vilarrupla, A.; Graupera, M.; Zafra, C.; García-Calderó, H.; García-Pagán, J.C.; Bosch, J. Simvastatin Treatment Improves Liver Sinusoidal Endothelial Dysfunction in CCl4 Cirrhotic Rats. J. Hepatol. 2007, 46, 1040–1046. [Google Scholar] [CrossRef]
- La Mura, V.; Pasarín, M.; Meireles, C.Z.; Miquel, R.; Rodríguez-Vilarrupla, A.; Hide, D.; Gracia-Sancho, J.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Effects of Simvastatin Administration on Rodents with Lipopolysaccharide-Induced Liver Microvascular Dysfunction. Hepatology 2013, 57, 1172–1181. [Google Scholar] [CrossRef]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. The Transcription Factor KLF2 Mediates Hepatic Endothelial Protection and Paracrine Endothelial-Stellate Cell Deactivation Induced by Statins. J. Hepatol. 2013, 58, 98–103. [Google Scholar] [CrossRef]
- Tripathi, D.M.; Vilaseca, M.; Lafoz, E.; Garcia-Calderó, H.; Haute, G.V.; Fernández-Iglesias, A.; de Oliveira, J.R.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Simvastatin Prevents Progression of Acute on Chronic Liver Failure in Rats With Cirrhosis and Portal Hypertension. Gastroenterology 2018, 155, 1564–1577. [Google Scholar] [CrossRef] [Green Version]
- Zafra, C.; Abraldes, J.G.; Turnes, J.; Berzigotti, A.; Fernández, M.; Garca-Pagán, J.C.; Rodés, J.; Bosch, J. Simvastatin Enhances Hepatic Nitric Oxide Production and Decreases the Hepatic Vascular Tone in Patients with Cirrhosis. Gastroenterology 2004, 126, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García-Pagán, J.C.; Bosch, J. Simvastatin Lowers Portal Pressure in Patients with Cirrhosis and Portal Hypertension: A Randomized Controlled Trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Villanueva, C.; Aracil, C.; Turnes, J.; Hernandez-Guerra, M.; Genesca, J.; Rodriguez, M.; Castellote, J.; García-Pagán, J.C.; Torres, F.; et al. Addition of Simvastatin to Standard Therapy for the Prevention of Variceal Rebleeding Does Not Reduce Rebleeding but Increases Survival in Patients With Cirrhosis. Gastroenterology 2016, 150, 1160–1170.e3. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Gracia-Sancho, J.; Abraldes, J.G. Cirrhosis as New Indication for Statins. Gut 2020, 69, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Kume, M.; Hayashi, T.; Yuasa, H.; Tanaka, H.; Nishioka, J.; Ido, M.; Gabazza, E.C.; Kawarada, Y.; Suzuki, K. Bacterial Lipopolysaccharide Decreases Thrombomodulin Expression in the Sinusoidal Endothelial Cells of Rats -- a Possible Mechanism of Intrasinusoidal Microthrombus Formation and Liver Dysfunction. J. Hepatol. 2003, 38, 9–17. [Google Scholar] [CrossRef]
- Martin, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the Vascular Endothelium: Insights into Functional, Regulatory, and Therapeutic Aspects. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1585–H1597. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, Z.M. Structure of von Willebrand Factor and Its Function in Platelet Adhesion and Thrombus Formation. Best Pract. Res. Clin. Haematol. 2001, 14, 257–279. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular Mechanisms of NET Formation and Degradation Revealed by Intravital Imaging in the Liver Vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [Green Version]
- Weiler-Guettler, H.; Christie, P.D.; Beeler, D.L.; Healy, A.M.; Hancock, W.W.; Rayburn, H.; Edelberg, J.M.; Rosenberg, R.D. A Targeted Point Mutation in Thrombomodulin Generates Viable Mice with a Prethrombotic State. J. Clin. Investig. 1998, 101, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmar, B.; Menger, M.D. The Hepatic Microcirculation: Mechanistic Contributions and Therapeutic Targets in Liver Injury and Repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef]
- McCarron, R.M.; Doron, D.A.; Sire´n, A.-L.; Feuerstein, G.; Heldman, E.; Pollard, H.B.; Spatz, M.; Hallenbeck, J.M. Agonist-Stimulated Release of von Willebrand Factor and Procoagulant Factor VIII in Rats with and without Risk Factors for Stroke. Brain Res. 1994, 647, 265–272. [Google Scholar] [CrossRef]
- Beier, J.I.; Luyendyk, J.P.; Guo, L.; von Montfort, C.; Staunton, D.E.; Arteel, G.E. Fibrin Accumulation Plays a Critical Role in the Sensitization to Lipopolysaccharide-Induced Liver Injury Caused by Ethanol in Mice. Hepatology 2009, 49, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauckmann, S.; Effenberger-Neidnicht, K.; Nagel, M.; Mayer, C.; Peters, J.; Hartmann, M. Lipopolysaccharide-Induced Hemolysis Is Abolished by Inhibition of Thrombin Generation but Not Inhibition of Platelet Aggregation. Inflammation 2019, 42, 1767–1776. [Google Scholar] [CrossRef]
- Blasi, A.; Calvo, A.; Prado, V.; Reverter, E.; Reverter, J.C.; Hernández-Tejero, M.; Aziz, F.; Amoros, A.; Cardenas, A.; Fernández, J. Coagulation Failure in Patients With Acute-on-Chronic Liver Failure and Decompensated Cirrhosis: Beyond the International Normalized Ratio. Hepatology 2018, 68, 2325–2337. [Google Scholar] [CrossRef] [Green Version]
- Bombeli, T.; Mueller, M.; Haeberli, A. Anticoagulant Properties of the Vascular Endothelium. Thromb. Haemost. 1997, 77, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Van Der Poll, T. Thrombomodulin in Sepsis. Minerva Anestesiol. 2013, 79, 294–298. [Google Scholar]
- Loghmani, H.; Conway, E.M. Exploring Traditional and Nontraditional Roles for Thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murao, S.; Yamakawa, K. A Systematic Summary of Systematic Reviews on Anticoagulant Therapy in Sepsis. J. Clin. Med. 2019, 8, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.-L.; Francois, B.; Zabolotskikh, I.; Daga, M.K.; Lascarrou, J.-B.; Kirov, M.Y.; Pettilä, V.; Wittebole, X.; Meziani, F.; Mercier, E.; et al. Effect of a Recombinant Human Soluble Thrombomodulin on Mortality in Patients With Sepsis-Associated Coagulopathy: The SCARLET Randomized Clinical Trial. JAMA 2019, 321, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-C.; Lee, M.-T.G.; Hsu, T.-C.; Porta, L.; Chang, S.-S.; Yo, C.-H.; Tsai, K.-C.; Lee, M. A Population-Based Cohort Study on the Drug-Specific Effect of Statins on Sepsis Outcome. Chest 2018, 153, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Janda, S.; Young, A.; Fitzgerald, J.M.; Etminan, M.; Swiston, J. The Effect of Statins on Mortality from Severe Infections and Sepsis: A Systematic Review and Meta-Analysis. J. Crit. Care 2010, 25, 656.e7–656.e22. [Google Scholar] [CrossRef] [PubMed]
- Skarlovnik, A.; Janić, M.; Lunder, M.; Turk, M.; Šabovič, M. Coenzyme Q10 Supplementation Decreases Statin-Related Mild-to-Moderate Muscle Symptoms: A Randomized Clinical Study. Med. Sci. Monit. 2014, 20, 2183–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.; Miller, J.A.L. Statins for Sepsis: A Cautionary Note. Intensive Care Med. 2006, 32, 795. [Google Scholar] [CrossRef]
- Cohen, D.E.; Anania, F.A.; Chalasani, N. National Lipid Association Statin Safety Task Force Liver Expert Panel An Assessment of Statin Safety by Hepatologists. Am. J. Cardiol. 2006, 97, 77C–81C. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Uschner, F.E.; Magdaleno, F.; Klein, S.; Trebicka, J. Rationale for the Use of Statins in Liver Disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2017, 312, G407–G412. [Google Scholar] [CrossRef] [Green Version]
- Cione, E.; Piegari, E.; Gallelli, G.; Caroleo, M.C.; Lamirata, E.; Curcio, F.; Colosimo, F.; Cannataro, R.; Ielapi, N.; Colosimo, M.; et al. Expression of MMP-2, MMP-9, and NGAL in Tissue and Serum of Patients with Vascular Aneurysms and Their Modulation by Statin Treatment: A Pilot Study. Biomolecules 2020, 10, 359. [Google Scholar] [CrossRef] [Green Version]
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in Patients with Cirrhosis Increase Mortality Four-Fold and Should Be Used in Determining Prognosis. Gastroenterology 2010, 139, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.M.; Schultze, A.E.; Jean, P.A.; Roth, R.A. Platelet Participation in Liver Injury from Gram-Negative Bacterial Lipopolysaccharide in the Rat. Shock 1995, 4, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Bakhtiari, K.; Adelmeijer, J.; Meijers, J.C.M.; Porte, R.J.; Stravitz, R.T. Intact Thrombin Generation and Decreased Fibrinolytic Capacity in Patients with Acute Liver Injury or Acute Liver Failure. J. Thromb. Haemost. 2012, 10, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Mannucci, P.M. The Coagulopathy of Chronic Liver Disease. N. Engl. J. Med. 2011, 365, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gando, S.; Levi, M.; Toh, C.-H. Disseminated Intravascular Coagulation. Nat. Rev. Dis. Primers 2016, 2, 16037. [Google Scholar] [CrossRef]
- Violi, F.; Calvieri, C.; Ferro, D.; Pignatelli, P. Statins as Antithrombotic Drugs. Circulation 2013, 127, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Anticoagulant Effects of Statins and Their Clinical Implications. Thromb. Haemost. 2014, 111, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Frenette, P.S. The Vessel Wall and Its Interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLACEBO | SIMVASTATIN | |||||
|---|---|---|---|---|---|---|
| Saline | LPS 5 mg/kg | p | Saline | LPS 5 mg/kg | p | |
| CT (sec) | 53 (41–62) | 67 (51–94) | 0.126 | 53 (42–65) | 50 (46–66) | 1.000 |
| CFT (min) | 27 (23–33) | 146 (42–410) | 0.004 | 27 (26–32) | 95 (22–120) | 0.700 |
| MCF (mm) | 77 (70–81) | 40 (33–79) | 0.030 | 78 (74–80) | 56 (48–83) | 0.700 |
| MCF-t (sec) | 1742 (578–1891) | 3336 (2095–3460 | 0.004 | 1581 (1533–2008) | 2720 (1267–3096) | 0.700 |
| Alfa-angle (°) | 85 (84–86) | 77 (70–82) | 0.004 | 85 (84–85) | 80 (79–86) | 0.700 |
| Max-V (velocity) | 46 (45–59) | 19 (12–34) | 0.004 | 50 (40–53) | 23 (22–58) | 0.700 |
| AUC | 7574 (6935–8048) | 4023 (3305–7906) | 0.052 | 7659 (7292–7905) | 6415 (4691–8139) | 0.700 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Mura, V.; Gagliano, N.; Arnaboldi, F.; Sartori, P.; Procacci, P.; Denti, L.; Liguori, E.; Bitto, N.; Ristagno, G.; Latini, R.; et al. Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia. Cells 2022, 11, 1148. https://doi.org/10.3390/cells11071148
La Mura V, Gagliano N, Arnaboldi F, Sartori P, Procacci P, Denti L, Liguori E, Bitto N, Ristagno G, Latini R, et al. Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia. Cells. 2022; 11(7):1148. https://doi.org/10.3390/cells11071148
Chicago/Turabian StyleLa Mura, Vincenzo, Nicoletta Gagliano, Francesca Arnaboldi, Patrizia Sartori, Patrizia Procacci, Luca Denti, Eleonora Liguori, Niccolò Bitto, Giuseppe Ristagno, Roberto Latini, and et al. 2022. "Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia" Cells 11, no. 7: 1148. https://doi.org/10.3390/cells11071148