
Renoprotective Effect of KLF2 on Glomerular Endothelial Dysfunction in Hypertensive Nephropathy

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. hPGEC Identification
2.2. Animal Model of Hypertensive CKD: 5/6 Nephrectomy Mouse Model
2.3. Human Samples
2.4. Hypertensive Injury in hPGECs Induced via a Pressurizing Device
2.5. Histology and Immunohistochemistry
2.6. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
2.7. Confocal Microscopy Examination
2.8. Regulation of KLF2
2.8.1. Upregulation of KLF2 Expression via Simvastatin Treatment
2.8.2. Downregulation of KLF2 Expression Using Pressurizing Devices that Employ Rotational Force
2.8.3. Transfection of siRNA in hPGECs
2.9. FACS
2.10. ELISA
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
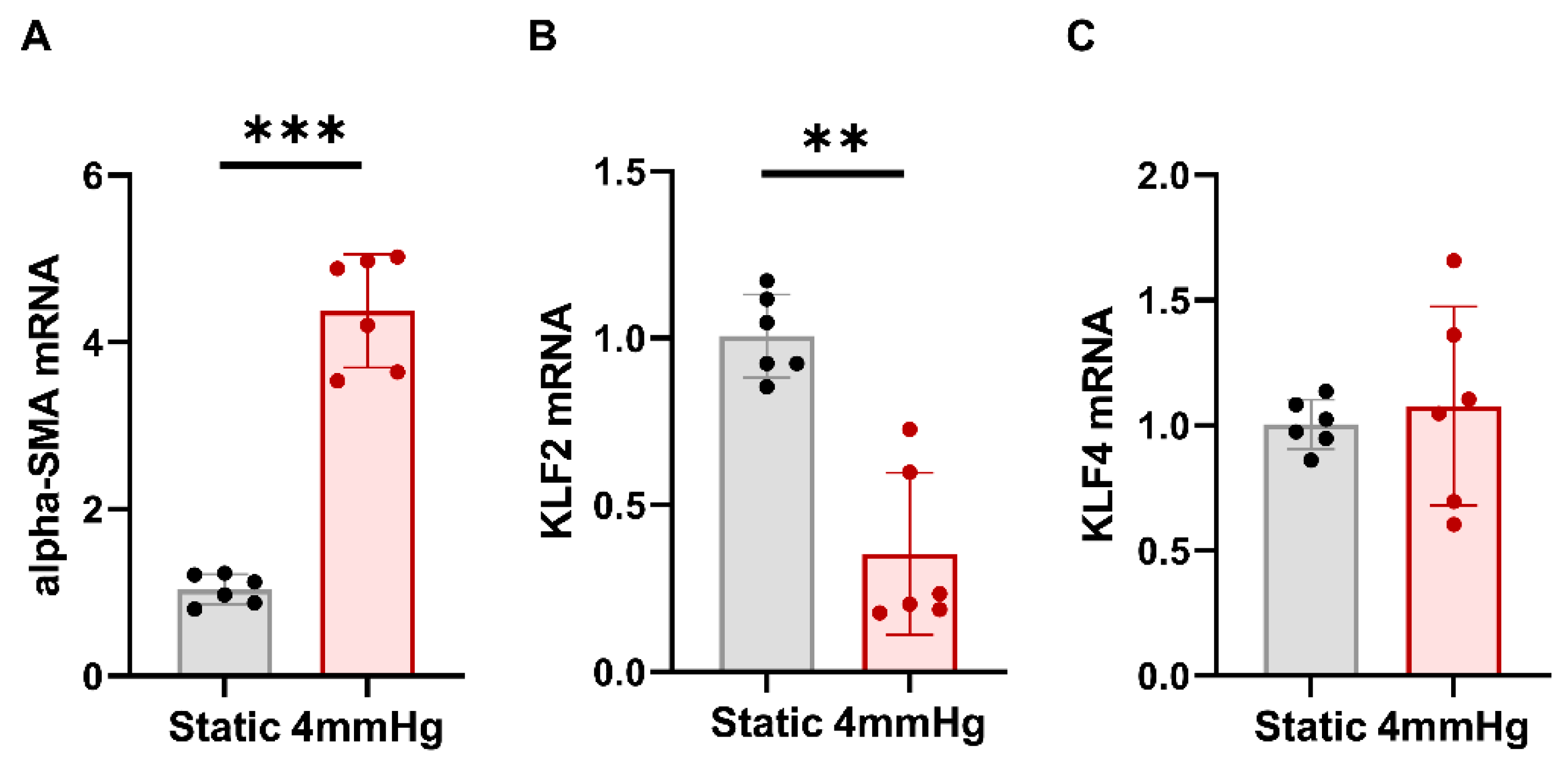
3.1. Effects of Mechanical Pressurization by Rotational Force on Glomerular Endothelial Cells and Reduction in KLF2 Expression
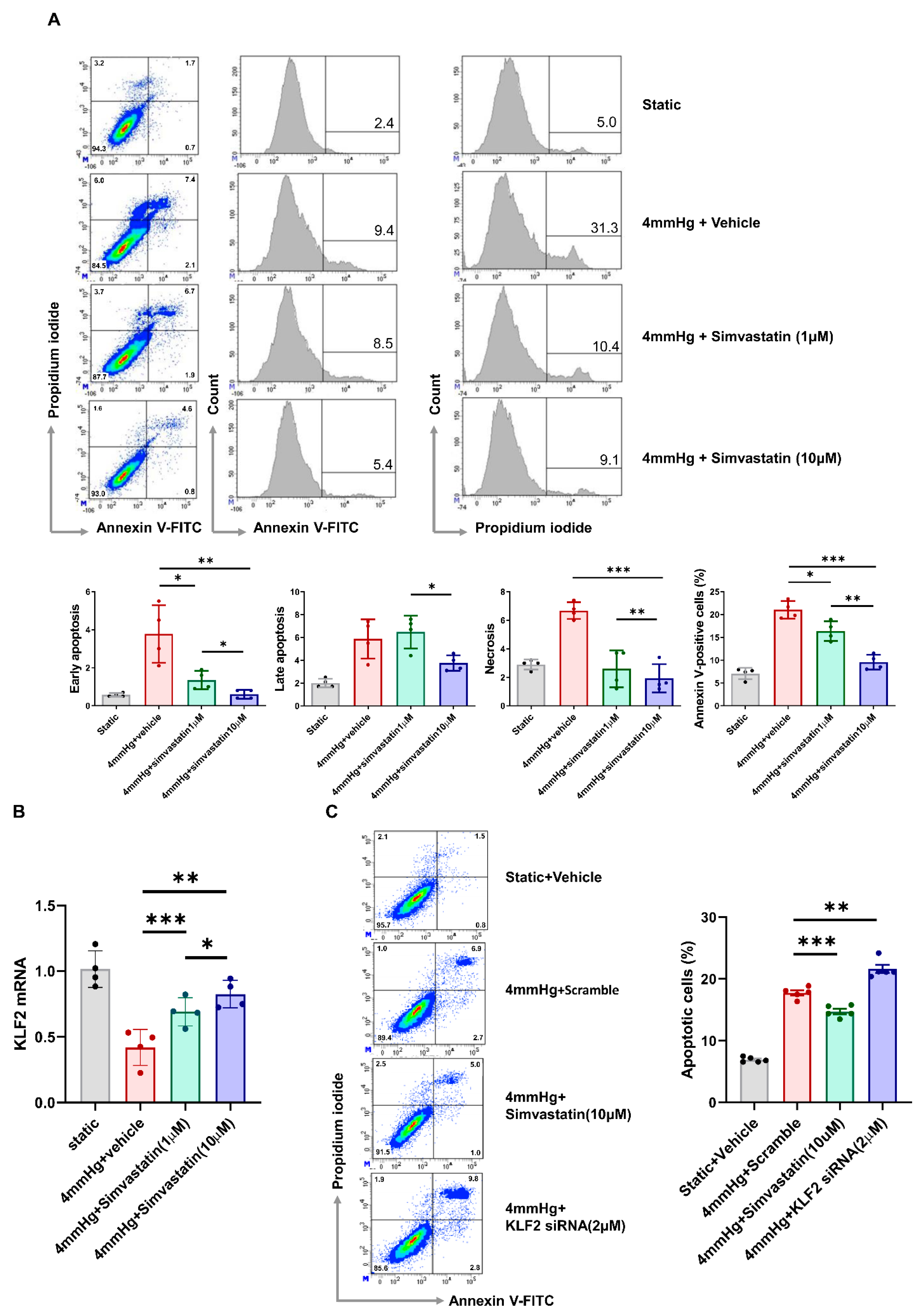
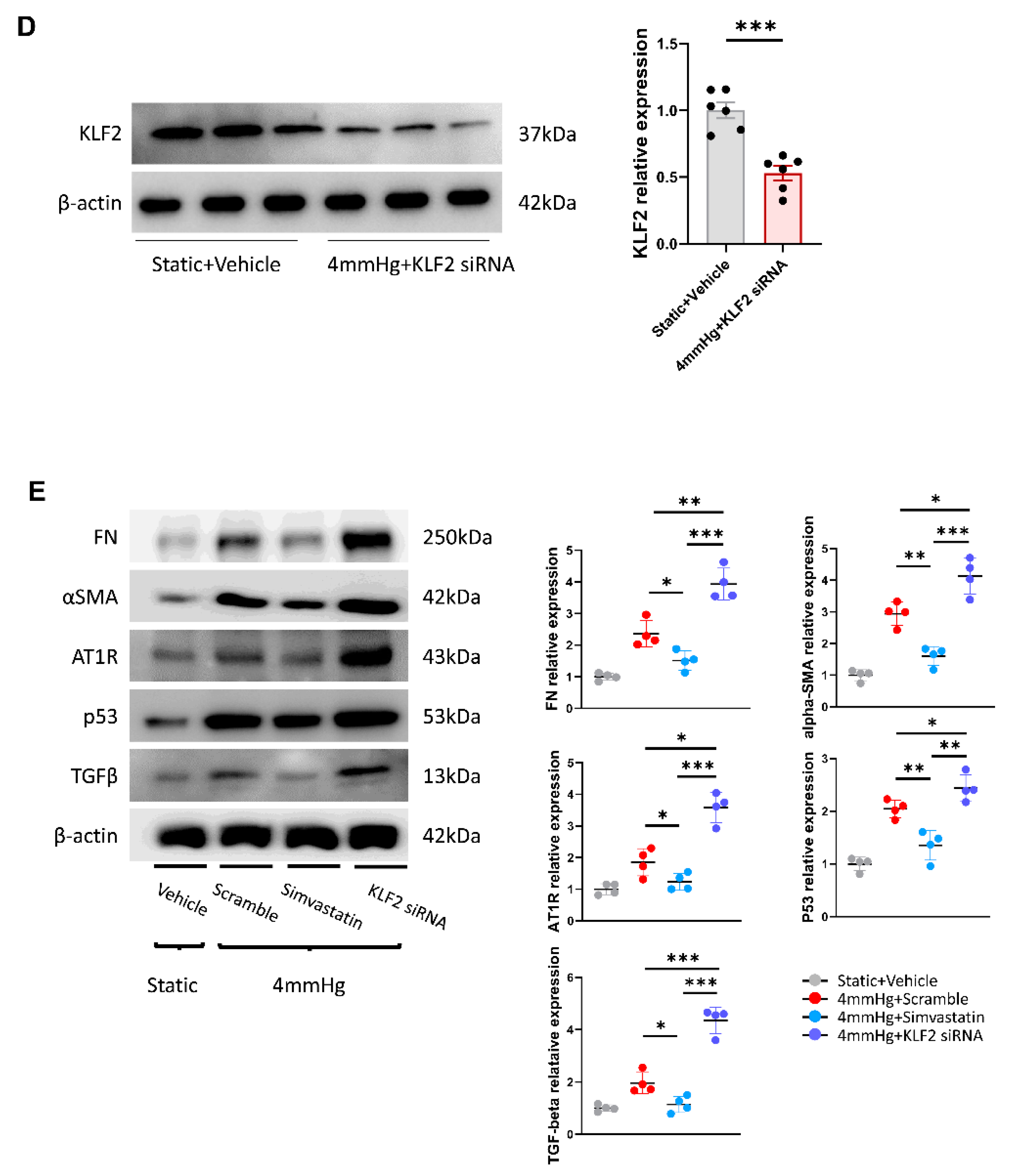
3.2. Pressure-Induced Apoptosis and Its Change through KLF2 Upregulation or Knockdown
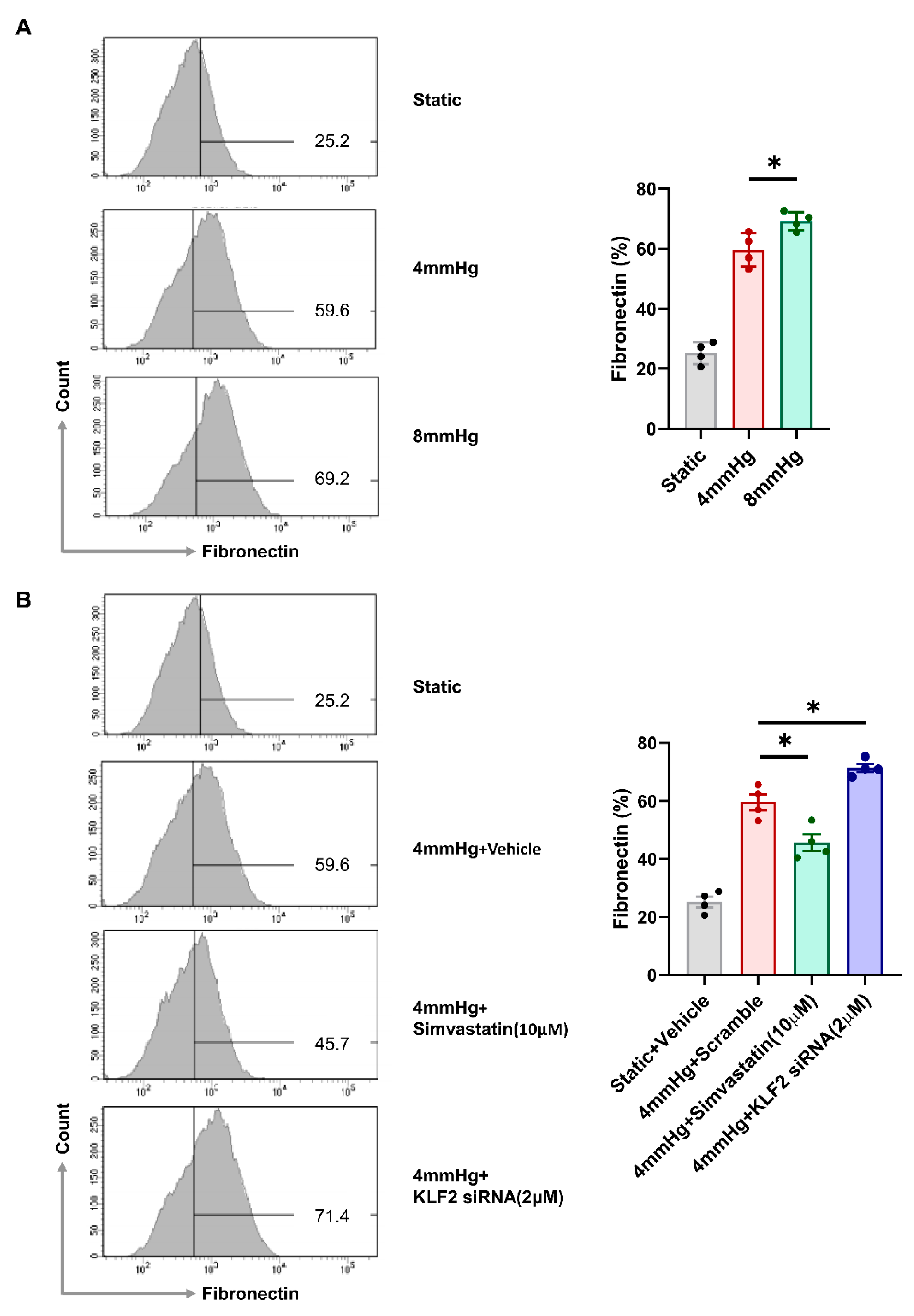
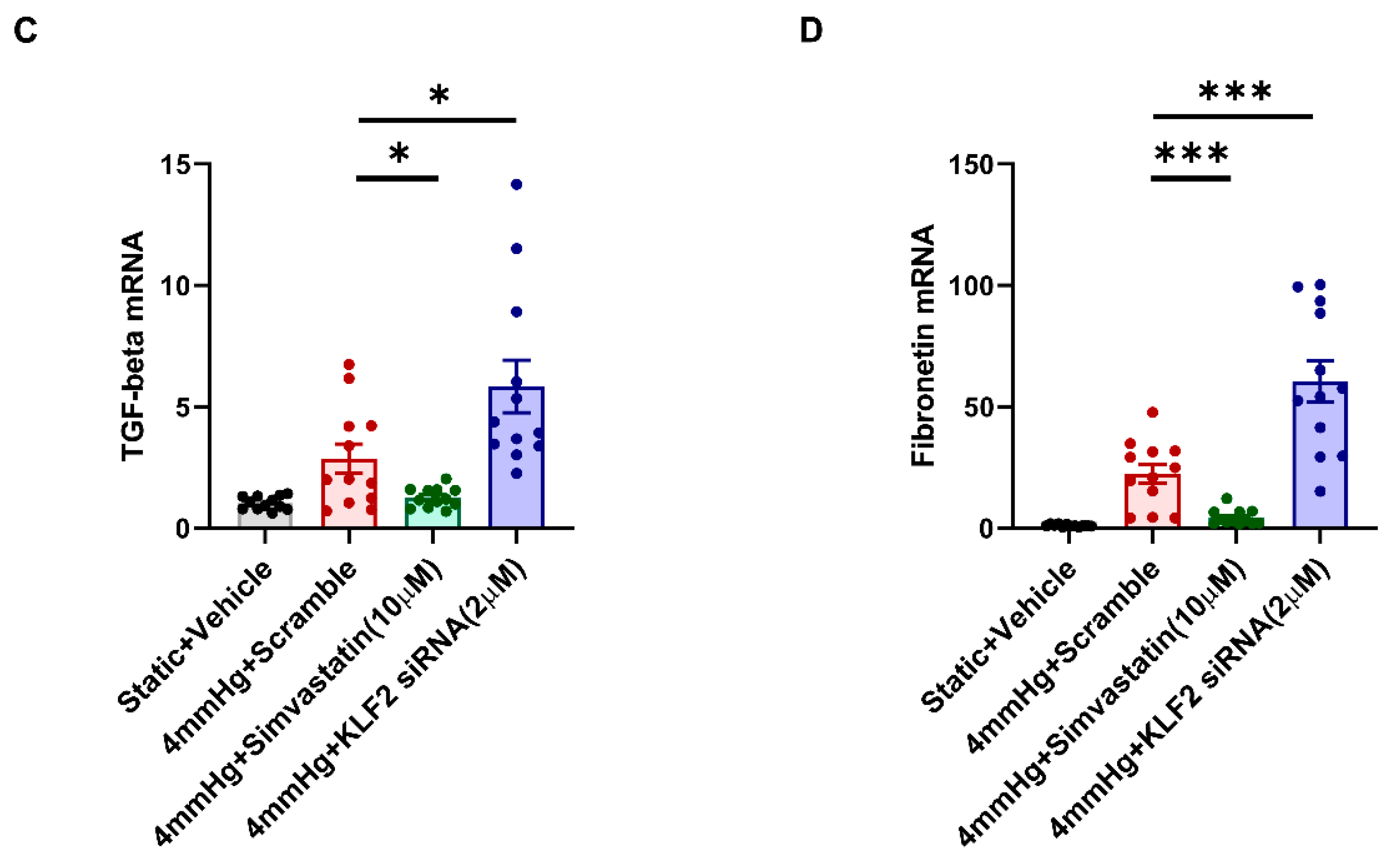
3.3. Induction of Fibrotic Markers by Pressure
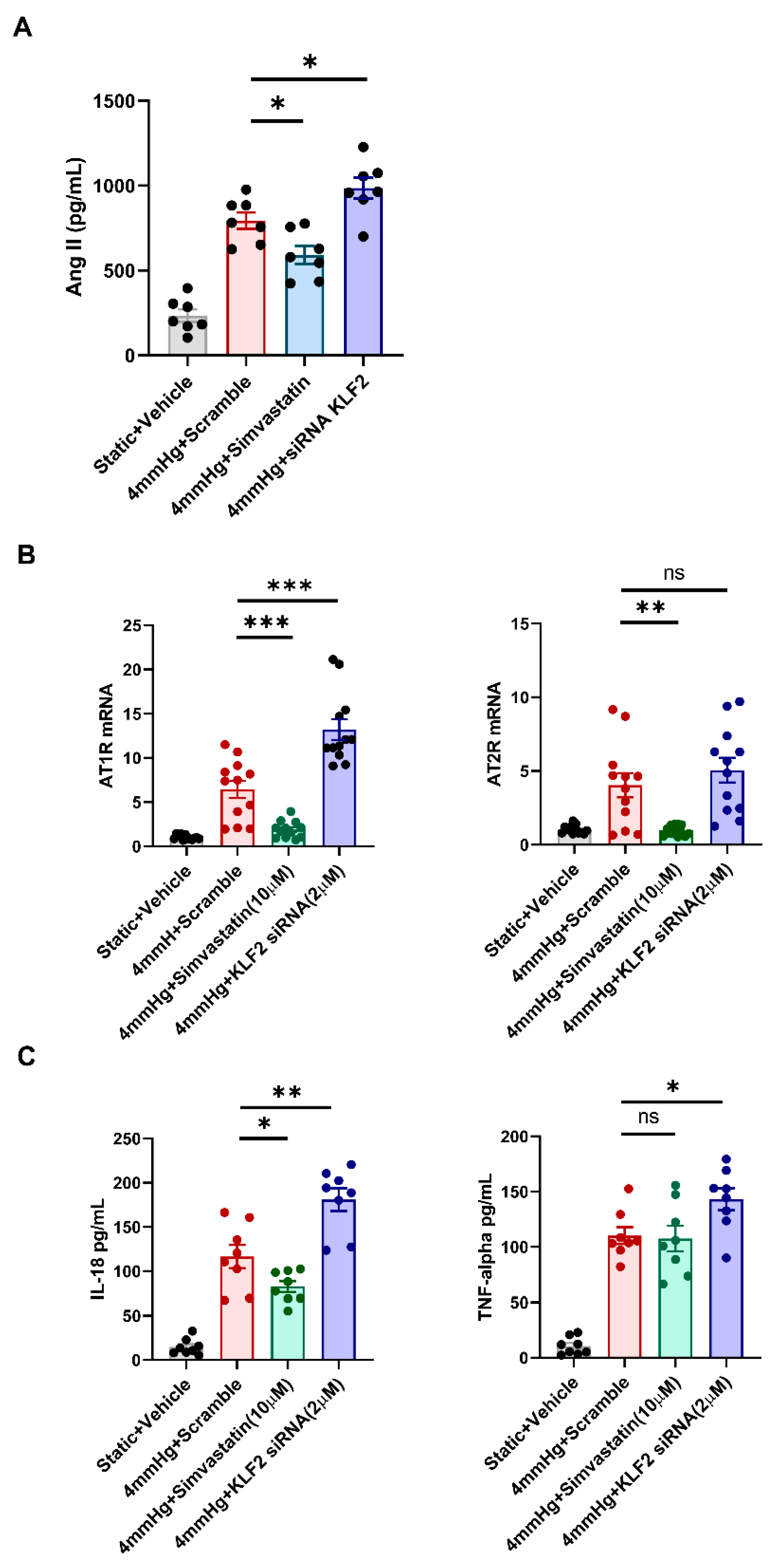
3.4. Changes in the Levels of Angiotensin II, AT1R, AT2R, and Proinflammatory Markers in Response to KLF2 Upregulation and Knockdown under Pressurized Conditions
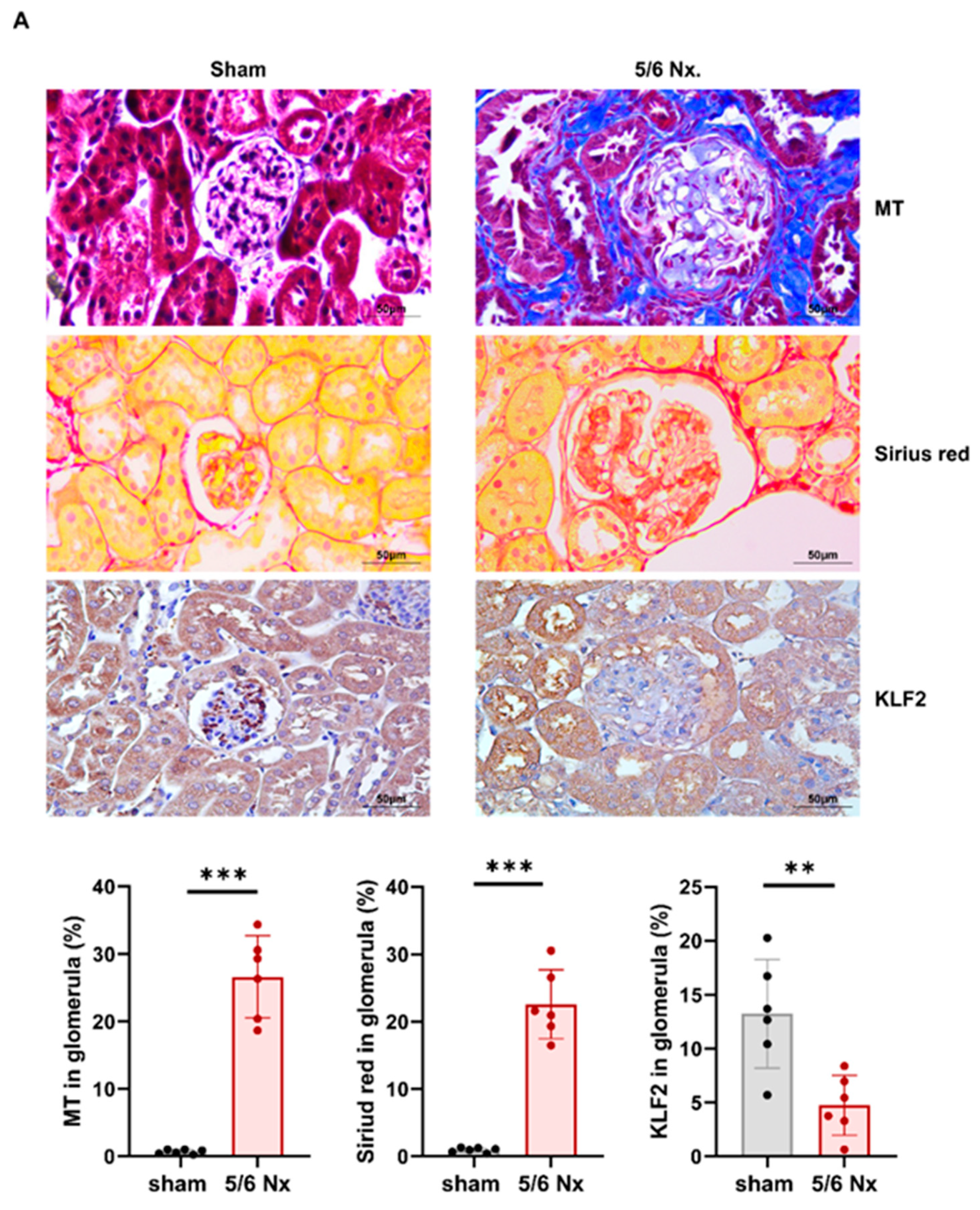
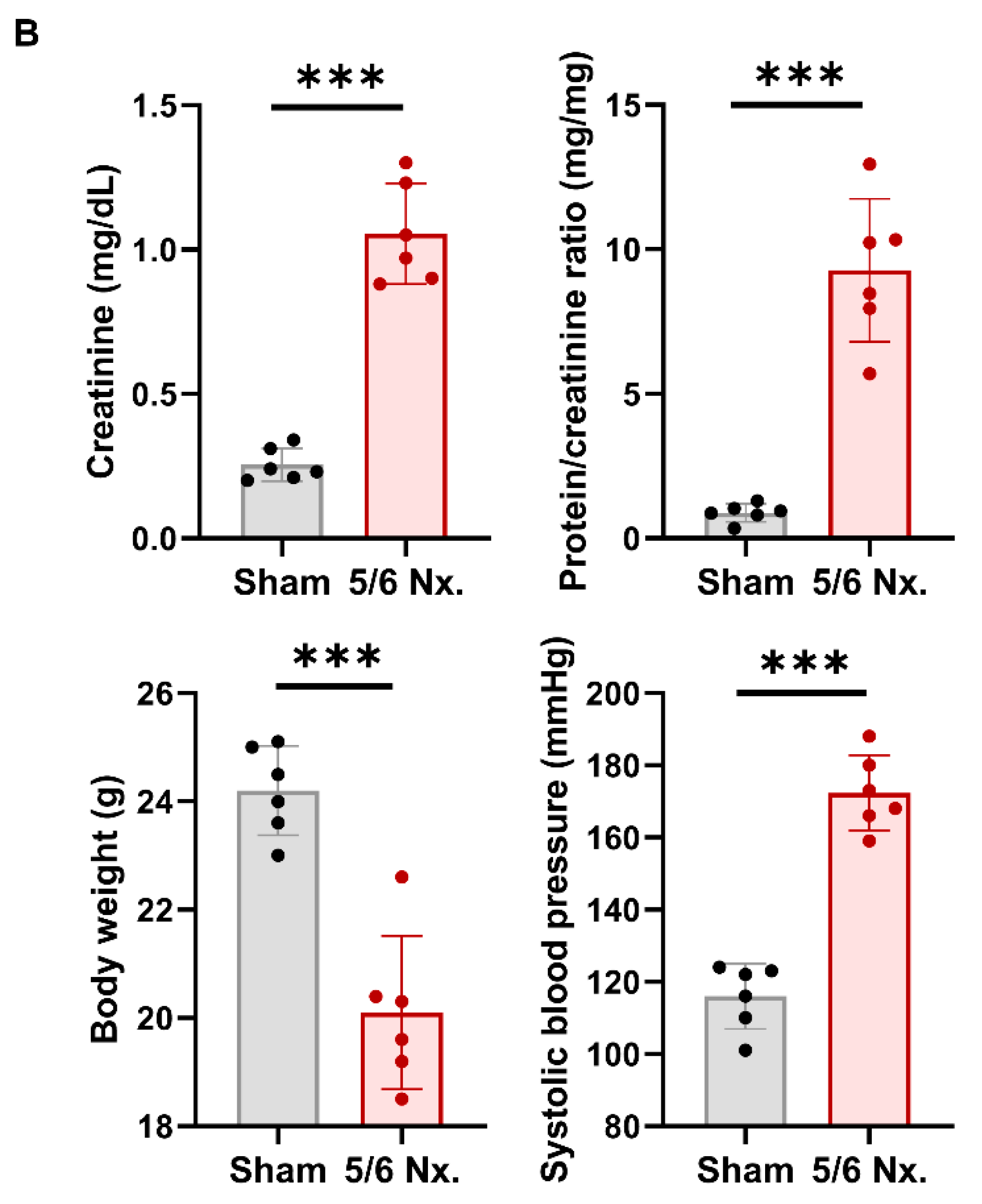
3.5. Hypertensive Kidney Injury Reduces KLF2 Expression in 5/6 Nephrectomy Mouse Model
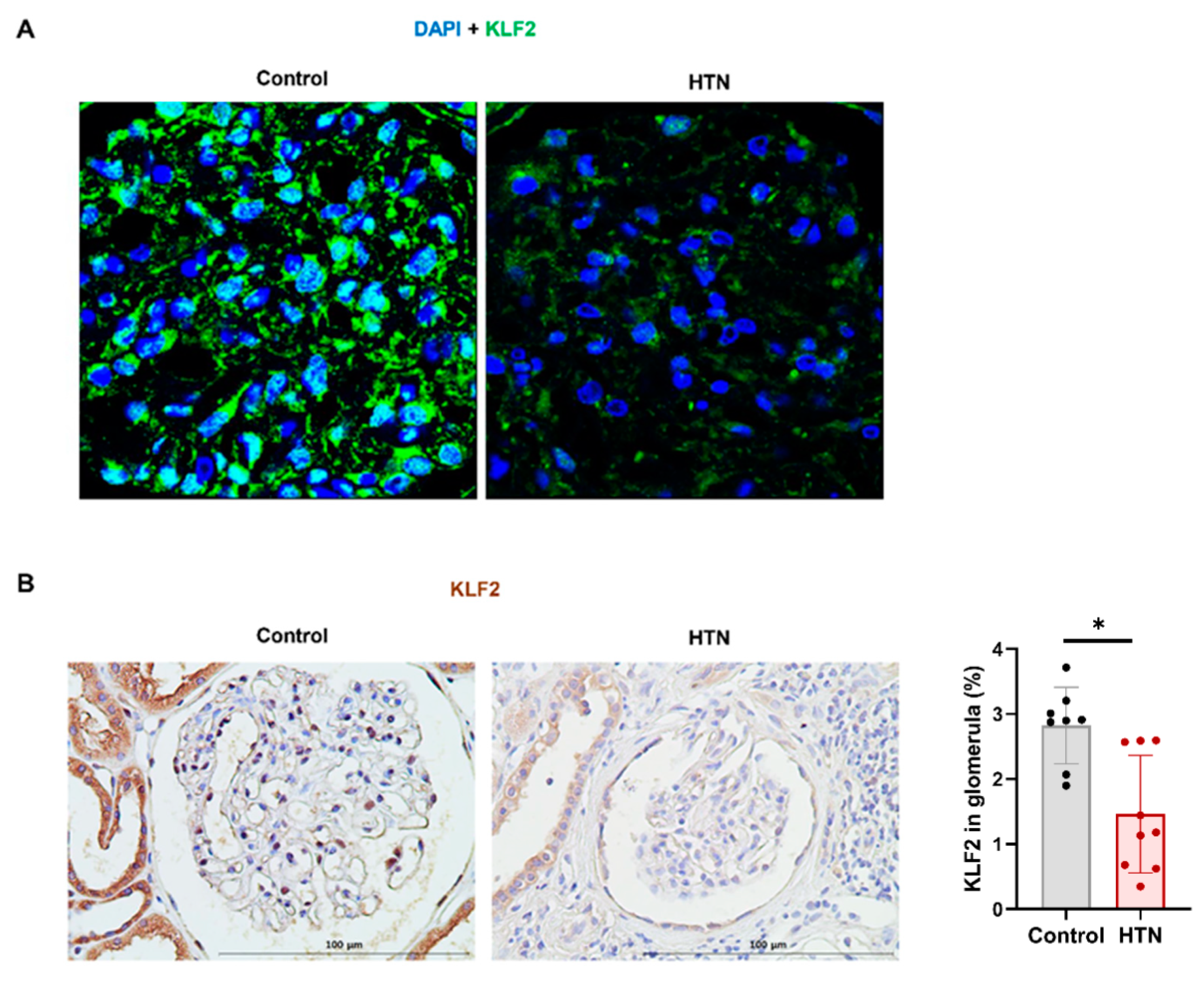
3.6. Decrease in KLF2 Expression in Glomerular Endothelial Cells in Patients with Hypertensive Nephropathy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wieschaus, E.; Nusslein-Volhard, C.; Kluding, H. Krüppel, a gene whose activity is required early in the zygotic genome for normal embryonic segmentation. Dev. Biol. 1984, 104, 172–186. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamashita, M.; Iwai, M.; Hayashi, M. Endothelial Krüppel-Like Factor 4 Mediates the Protective Effect of Statins against Ischemic AKI. J. Am. Soc. Nephrol. 2015, 27, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Sakamaki, Y.; Azegami, T.; Oguchi, H.; Tokuyama, H.; Wakino, S.; Hayashi, K.; Itoh, H. Renin-angiotensin blockade resets podocyte epigenome through Kruppel-like Factor 4 and attenuates proteinuria. Kidney Int. 2015, 88, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallipattu, S.K.; Horne, S.J.; D’Agati, V.; Narla, G.; Liu, R.; Frohman, M.A.; Dickman, K.; Chen, E.Y.; Ma’Ayan, A.; Bialkowska, A.B.; et al. Krüppel-like factor 6 regulates mitochondrial function in the kidney. J. Clin. Investig. 2015, 125, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sui, X.; Hu, X.; Hu, Z. Overexpression of KLF5 inhibits puromycin-induced apoptosis of podocytes. Mol. Med. Rep. 2018, 18, 3843–3849. [Google Scholar] [CrossRef] [Green Version]
- Mallipattu, S.K.; Liu, R.; Zheng, F.; Narla, G.; Ma’Ayan, A.; Dikman, S.; Jain, M.K.; Saleem, M.; D’Agati, V.; Klotman, P.; et al. Krüppel-like Factor 15 (KLF15) Is a Key Regulator of Podocyte Differentiation. J. Biol. Chem. 2012, 287, 19122–19135. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.-Y.; Kim, J.E.; Lee, S.; Choi, J.W.; Kim, Y.C.; Han, S.S.; Lee, H.; Cha, R.H.; Lee, J.P.; Lee, J.W.; et al. Krüppel-like factor 15 is a key suppressor of podocyte fibrosis under rotational force-driven pressure. Exp. Cell Res. 2019, 386, 111706. [Google Scholar] [CrossRef]
- Han, S.S.; Yu, M.; Yoo, K.D.; Lee, J.P.; Kim, D.K.; Kim, Y.S.; Yang, S.H. Loss of KLF15 accelerates chronic podocyte injury. Int. J. Mol. Med. 2018, 42, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Jha, P.; Das, H. KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation. Int. J. Mol. Sci. 2017, 18, 2383. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go With the Flow”: How Krüppel-Like Factor 2 Regulates the Vasoprotective Effects of Shear Stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, R.; SenBanerjee, S.; Lin, Z.; Mir, S.; Hamik, A.; Wang, P.; Mukherjee, P.; Mukhopadhyay, D.; Jain, M.K. Inhibition of Vascular Permeability Factor/Vascular Endothelial Growth Factor-mediated Angiogenesis by the Kruppel-like Factor KLF2. J. Biol. Chem. 2005, 280, 28848–28851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, F.; Hamdulay, S.S.; Kinderlerer, A.R.; Boyle, J.J.; Lidington, E.A.; Yamaguchi, T.; Soares, M.P.; Haskard, D.O.; Randi, A.M.; Mason, J.C. Statin-mediated cytoprotection of human vascular endothelial cells: A role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J. Thromb. Haemost. 2007, 5, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A., Jr.; Balasubramanian, V.; García-Cardeña, G.; Jain, M.K. Kruppel-Like Factor 2 (KLF2) Regulates Endothelial Thrombotic Function. Circ. Res. 2005, 96, e48–e57. [Google Scholar] [CrossRef] [Green Version]
- Pathak, R.; Shao, L.; Chafekar, S.M.; Feng, W.; Ponnappan, U.; Fink, L.M.; Zhou, D.; Hauer-Jensen, M. IKKβ regulates endothelial thrombomodulin in a Klf2-dependent manner. J. Thromb. Haemost. 2014, 12, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Hamik, A.; Jain, R.; Kumar, A.; Jain, M.K. Kruppel-Like Factor 2 Inhibits Protease Activated Receptor-1 Expression and Thrombin-Mediated Endothelial Activation. Arter. Thromb. Vasc. Biol. 2006, 26, 1185. [Google Scholar] [CrossRef] [Green Version]
- Dekker, R.J.; Van Soest, S.; Fontijn, R.D.; Salamanca, S.; De Groot, P.G.; van Bavel, E.; Pannekoek, H.; Horrevoets, A.J.G. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef]
- Wang, N.; Miao, H.; Li, Y.-S.; Zhang, P.; Haga, J.H.; Hu, Y.; Young, A.; Yuan, S.; Nguyen, P.; Wu, C.-C.; et al. Shear stress regulation of Krüppel-like factor 2 expression is flow pattern-specific. Biochem. Biophys. Res. Commun. 2006, 341, 1244–1251. [Google Scholar] [CrossRef]
- Lin, Z.; Natesan, V.; Shi, H.; Dong, F.; Kawanami, D.; Mahabeleshwar, G.H.; Atkins, G.B.; Nayak, L.; Cui, Y.; Finigan, J.H.; et al. Kruppel-Like Factor 2 Regulates Endothelial Barrier Function. Arter. Thromb. Vasc. Biol. 2010, 30, 1952–1959. [Google Scholar] [CrossRef]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.-W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Dharmashankar, K.; Widlansky, M.E. Vascular Endothelial Function and Hypertension: Insights and Directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelial Dysfunction as a Target for Prevention of Cardiovascular Disease. Diabetes Care 2009, 32, S314–S321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnuchamy, B.; Khalil, R.A. Cellular mediators of renal vascular dysfunction in hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1001–R1018. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Fliser, D.; Siebels, M. Pathophysiology of Hypertensive Renal Damage. Am. J. Hypertens. 1993, 6, 241S–244S. [Google Scholar] [CrossRef]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.-S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [Green Version]
- Crowley, S.D.; Tharaux, P.-L.; Audoly, L.P.; Coffman, T.M. Exploring type I angiotensin (AT1) receptor functions through gene targeting. Acta Physiol. Scand. 2004, 181, 561–570. [Google Scholar] [CrossRef]
- Devereux, R.B.; Dahlöf, B.; Gerdts, E.; Boman, K.; Nieminen, M.S.; Papademetriou, V.; Rokkedal, J.; Harris, K.E.; Edelman, J.M.; Wachtell, K. Regression of Hypertensive Left Ventricular Hypertrophy by Losartan Compared With Atenolol. Circulation 2004, 110, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Mazzolai, L.; Pedrazzini, T.; Nicoud, F.; Gabbiani, G.; Brunner, H.-R.; Nussberger, J. Increased Cardiac Angiotensin II Levels Induce Right and Left Ventricular Hypertrophy in Normotensive Mice. Hypertension 2000, 35, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Oliverio, M.I.; Mannon, P.J.; Best, C.F.; Maeda, N.; Smithies, O.; Coffman, T.M. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc. Natl. Acad. Sci. USA 1995, 92, 3521–3525. [Google Scholar] [CrossRef] [Green Version]
- Oliverio, M.I.; Best, C.F.; Kim, H.S.; Arendshorst, W.J.; Smithies, O.; Coffman, T.M. Angiotensin II responses in AT1A receptor-deficient mice: A role for AT1B receptors in blood pressure regulation. Am. J. Physiol. Content 1997, 272, F515–F520. [Google Scholar] [CrossRef]
- Masilamani, S.; Kim, G.-H.; Mitchell, C.; Wade, J.B.; Knepper, M.A. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J. Clin. Investig. 1999, 104, R19–R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangemi, S.; Mallamace, A.; Minciullo, P.; Santoro, D.; Merendino, R.; Savica, V.; Bellinghieri, G. Involvement of Interleukin-18 in Patients on Maintenance Haemodialysis. Am. J. Nephrol. 2002, 22, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Kanmatsuse, K. Elevated interleukin-18 levels in the urine of nephrotic patients. Nephron 2001, 88, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Trøseid, M.; Seljeflot, I.; Hjerkinn, E.M.; Arnesen, H. Interleukin-18 Is a Strong Predictor of Cardiovascular Events in Elderly Men With the Metabolic Syndrome. Diabetes Care 2009, 32, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL -1β and IL -18: Inflammatory markers or mediators of hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602. [Google Scholar] [CrossRef]
- Dalekos, G.; Elisaf, M.; Bairaktari, E.; Tsolas, O.; Siamopoulos, K. Increased serum levels of interleukin-1β in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J. Lab. Clin. Med. 1997, 129, 300–308. [Google Scholar] [CrossRef]
- Rabkin, S.W. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat. Rev. Cardiol. 2009, 6, 192–199. [Google Scholar] [CrossRef]
- Thomas, J.M.; Ling, Y.H.; Huuskes, B.; Jelinic, M.; Sharma, P.; Saini, N.; Ferens, D.M.; Diep, H.; Krishnan, S.M.; Kemp-Harper, B.K.; et al. IL-18 (Interleukin-18) Produced by Renal Tubular Epithelial Cells Promotes Renal Inflammation and Injury During Deoxycorticosterone/Salt-Induced Hypertension in Mice. Hypertension 2021, 78, 1296–1309. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, M.B.; Griffiths, R.; Mao, A.; Song, Y.-S.; Karlovich, N.S.; Sparks, M.; Jin, H.; Wu, M.; Lin, E.E.; et al. Tumor Necrosis Factor-α Produced in the Kidney Contributes to Angiotensin II–dependent Hypertension. Hypertension 2014, 64, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Mehaffey, E.; Majid, D.S.A. Tumor necrosis factor-α, kidney function, and hypertension. Am. J. Physiol. Renal Physiol. 2017, 313, F1005–F1008. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, B.; Colston, J.T.; de la Rosa, S.D.; Rao, P.P.; Freeman, G.L. TNF-α and H2O2 induce IL-18 and IL-18Rβ expression in cardiomyocytes via NF-κB activation. Biochem. Biophys. Res. Commun. 2003, 303, 1152–1158. [Google Scholar] [CrossRef]
- Puren, A.J.; Fantuzzi, G.; Gu, Y.; Su, M.S.; A Dinarello, C. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J. Clin. Investig. 1998, 101, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Chen, H.; Wei, C.; Zhang, W.; Li, Z.; Jain, M.K.; Chuang, P.Y.; Chen, H.; Wang, Y.; Mallipattu, S.K.; et al. Reduced Krüppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. 2015, 87, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Zhong, X.; Yu, G.; Shao, S.; Yang, Q. Vascular protective effects of KLF2 on Aβ-induced toxicity: Implications for Alzheimer’s disease. Brain Res. 2017, 1663, 174–183. [Google Scholar] [CrossRef]
- Zhong, F.; Mallipattu, S.K.; Estrada, C.; Menon, M.; Salem, F.; Jain, M.K.; Chen, H.; Wang, Y.; Lee, K.; He, J.C. Reduced Krüppel-Like Factor 2 Aggravates Glomerular Endothelial Cell Injury and Kidney Disease in Mice with Unilateral Nephrectomy. Am. J. Pathol. 2016, 186, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Agustian, P.A.; Bockmeyer, C.L.; Modde, F.; Wittig, J.; Heinemann, F.M.; Brundiers, S.; Dämmrich, M.E.; Schwarz, A.; Birschmann, I.; Suwelack, B.; et al. Glomerular mRNA Expression of Prothrombotic and Antithrombotic Factors in Renal Transplants With Thrombotic Microangiopathy. Transplantation 2013, 95, 1242–1248. [Google Scholar] [CrossRef]
- Bidani, A.K.; Griffin, K.A. Pathophysiology of Hypertensive Renal Damage. Hypertension 2004, 44, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Brantsma, A.H.; Bakker, S.J.; de Zeeuw, D.; de Jong, P.E.; Gansevoort, R.T. Urinary Albumin Excretion as a Predictor of the Development of Hypertension in the General Population. J. Am. Soc. Nephrol. 2006, 17, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Kestenbaum, B.; Rudser, K.D.; De Boer, I.H.; Peralta, C.A.; Fried, L.F.; Shlipak, M.G.; Palmas, W.; Stehman-Breen, C.; Siscovick, D.S. Differences in kidney function and incident hypertension: The multi-ethnic study of atherosclerosis. Ann. Intern. Med. 2008, 148, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Lerman, L.O.; Kurtz, T.W.; Touyz, R.M.; Ellison, D.H.; Chade, A.R.; Crowley, S.D.; Mattson, D.L.; Mullins, J.J.; Osborn, J.; Eirin, A.; et al. Animal Models of Hypertension: A Scientific Statement From the American Heart Association. Hypertension 2019, 73, e87–e120. [Google Scholar] [CrossRef] [Green Version]
- Healey, C.; Forgione, P.; Lounsbury, K.M.; Corrow, K.; Osler, T.; Ricci, M.A.; Stanley, A. A new in vitro model of venous hypertension: The effect of pressure on dermal fibroblasts. J. Vasc. Surg. 2003, 38, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Zhang, X.; Wen, X.; Wu, T.; Wang, W.; Yang, M.; Wang, J.; Fang, M.; Lin, B.; Lin, H. Development of a Functional Glomerulus at the Organ Level on a Chip to Mimic Hypertensive Nephropathy. Sci. Rep. 2016, 6, 31771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; An, J.N.; Kim, J.H.; Choi, Y.-W.; Oh, S.; Kwon, S.H.; Lee, M.-Y.; Lee, J.; Jeong, J.-G.; Lim, C.S.; et al. Soluble cMet levels in urine are a significant prognostic biomarker for diabetic nephropathy. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- An, J.N.; Yang, S.H.; Kim, Y.C.; Hwang, J.H.; Park, J.Y.; Kim, D.K.; Kim, J.H.; Kim, D.W.; Hur, D.G.; Oh, Y.K.; et al. Periostin induces kidney fibrosis after acute kidney injury via the p38 MAPK pathway. Am. J. Physiol. Renal Physiol. 2019, 316, F426–F437. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Iskandar, S.S.; Appel, R.G. The link between hypertension and nephrosclerosis. Am. J. Kidney Dis. 1995, 25, 207–221. [Google Scholar] [CrossRef]
- Jones, D.W.; Hall, J.E. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure and Evidence from New Hypertension Trials. Hypertension 2004, 43, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Rong, S.; Feng, Y.; Zhao, L.; Hong, J.; Wang, R.; Yuan, W. Simvastatin attenuates renal ischemia/reperfusion injury from oxidative stress via targeting Nrf2/HO-1 pathway. Exp. Ther. Med. 2017, 14, 4460–4466. [Google Scholar] [CrossRef] [Green Version]
- Tuuminen, R.; Nykänen, A.I.; Saharinen, P.; Gautam, P.; Keränen, M.A.I.; Arnaudova, R.; Rouvinen, E.; Helin, H.; Tammi, R.; Rilla, K.; et al. Donor Simvastatin Treatment Prevents Ischemia-Reperfusion and Acute Kidney Injury by Preserving Microvascular Barrier Function. Am. J. Transplant. 2013, 13, 2019–2034. [Google Scholar] [CrossRef]
- Oberleithner, H. Aldosterone makes human endothelium stiff and vulnerable. Kidney Int. 2005, 67, 1680–1682. [Google Scholar] [CrossRef] [Green Version]
- Becher, U.M.; Endtmann, C.; Tiyerili, V.; Nickenig, G.; Werner, N. Endothelial Damage and Regeneration: The Role of the Renin-Angiotensin-Aldosterone System. Curr. Hypertens. Rep. 2011, 13, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Brodsky, S.V.; Noiri, E. NO bioavailability, endothelial dysfunction, and acute renal failure: New insights into pathophysiology. Semin. Nephrol. 2004, 24, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, J.; Yao, L.; Li, Z.; Ohno, S. Research progress in acute hypertensive renal injury by “in vivo cryotechnique”. J. Transl. Intern. Med. 2019, 7, 132–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.-H.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Renal Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Meng, Y.; Liu, Q.; Xuan, M.; Zhang, L.; Deng, B.; Zhang, K.; Liu, Z.; Lei, T. Injury to the Endothelial Surface Layer Induces Glomerular Hyperfiltration Rats with Early-Stage Diabetes. J. Diabetes Res. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fledderus, J.; Boon, R.; Volger, O.L.; Hurttila, H.; Ylä-Herttuala, S.; Pannekoek, H.; Levonen, A.-L.; Horrevoets, A.J.G. KLF2 Primes the Antioxidant Transcription Factor Nrf2 for Activation in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2008, 28, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; Garcia-Pagan, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Qin, S.; Wu, R.; Zhou, X.; Tang, X.; Zhang, S.; Zhao, Q.; Wang, H.; Liu, Y.; Han, X.; et al. Role of MiR-126a-3p in Endothelial Injury in Endotoxic Mice. Crit. Care Med. 2016, 44, e639–e650. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Y.; Liu, J.; Chen, X.; Duan, Y.; Wang, X.; Shen, Y.; Kuang, Y.; Zhuang, T.; Tomlinson, B.; et al. Endothelial Klf2-Foxp1-TGFβ signal mediates the inhibitory effects of simvastatin on maladaptive cardiac remodeling. Theranostics 2021, 11, 1609–1625. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Pascolo, L.; Zito, G.; Zupin, L.; Luppi, S.; Giolo, E.; Martinelli, M.; De Rocco, D.; Crovella, S.; Ricci, G. Renin Angiotensin System, COVID-19 and Male Fertility: Any Risk for Conceiving? Microorganisms 2020, 8, 1492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-S.; Schuman, I.H.; Jaimes, E.A.; Raij, L. Renoprotection by statins is linked to a decrease in renal oxidative stress, TGF-β, and fibronectin with concomitant increase in nitric oxide bioavailability. Am. J. Physiol. Renal Physiol. 2008, 295, F53–F59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II Enhances Interleukin-18 Mediated Inflammatory Gene Expression in Vascular Smooth Muscle Cells. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, C.; Gerdes, N.; Liu, C.; Liao, M.; Liu, J.; Shi, M.; He, A.; Zhou, Y.; Sukhova, G.K.; et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 2015, 21, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Sense (5→3) | Antisense (5→3) |
|---|---|---|
| GAPDH | TCGACAGTCAGCCGCATCT | CCGTTGACTCCGACCTTCA |
| αSMA | GATGGGCATCTATCAGATAC | AAGCATTTCTGATGGTGATG |
| KLF2 | GCAAGACCTACACCAAGAGTTCG | CATGTGCCGTTTCATGTGC |
| KLF4 | AGGGGGTGACTGGAAGTTGT | TTGCACATCTGAAACCACAG |
| TGFβ | CCCAGCATCTGCAAAGCTC | GTCAATGTACAGCTGCCGCA |
| Fibronectin | CCACCCCCATAAGGCATAGG | GTAGGGGTCAAAGCACGAGTCATC |
| AT1R | CCGCATTTAACTGCTCACACA | ATCATGTAGTAGAGAACAGGAATTGCTT |
| AT2R | CGGAATTCATGAGCTGCGTTAATCC | AACTGCAGTTAAGACACAAAGGTCTCCA |
| Variables | Total (n = 17) | Normal (n = 9) | Hypertensive Nephropathy (n = 8) | p-Value |
|---|---|---|---|---|
| Age | 43.6 ± 12.3 | 40.4 ± 10.8 | 47.3 ± 13.4 | 0.115 |
| Men (%) | 12 (63.2) | 4 (40.0) | 8 (88.9) | 0.027 |
| BMI (kg/m2) | 25.1 ± 3.3 | 25.0 ± 3.4 | 25.2 ± 3.4 | 0.894 |
| Systolic blood pressure (mmHg) | 151.0 ± 47.0 | 116.9 ± 6.2 | 188.9 ± 43.1 | <0.001 |
| Diastolic blood pressure (mmHg) | 95.6 ± 31.1 | 76.4 ± 7.2 | 116.9 ± 33.9 | 0.002 |
| eGFR (mL/min/1.73 m2) | 72.7 ± 45.0 | 107.4 ± 26.0 | 29.3 ± 13.6 | <0.001 |
| Urine protein–creatinine ratio (g/gCr) | 0.5 (0.1, 1.8) | 0.3 (0.1, 1.9) | 1.1 (0.2, 2.6) | 0.247 |
| KLF2 expression in glomerular endothelial cells (%) | 2.5 ± 2.1 | 3.5 ± 2.5 | 1.5 ± 0.9 | 0.037 |
| Antihypertensive drug (%) | 9 (52.9) | 1 (11.1) | 8 (88.9) | <0.001 |
| ACEI or ARB (%) | 7 (41.2) | 1 (11.1) | 6 (75.0) | 0.008 |
| Beta blocker | 4 (23.5) | 0 (0.0) | 4 (50.0) | 0.015 |
| Calcium channel blocker | 7 (41.2) | 0 (0.0) | 7 (87.5) | <0.001 |
| Diuretics | 1 (5.9) | 0 (0.0) | 1 (12.5) | 0.274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, E.; Yu, M.-Y.; Moon, J.-J.; Kim, J.-E.; Lee, S.; Han, S.-W.; Park, D.-J.; Kim, Y.-S.; Yang, S.-H. Renoprotective Effect of KLF2 on Glomerular Endothelial Dysfunction in Hypertensive Nephropathy. Cells 2022, 11, 762. https://doi.org/10.3390/cells11050762
Bae E, Yu M-Y, Moon J-J, Kim J-E, Lee S, Han S-W, Park D-J, Kim Y-S, Yang S-H. Renoprotective Effect of KLF2 on Glomerular Endothelial Dysfunction in Hypertensive Nephropathy. Cells. 2022; 11(5):762. https://doi.org/10.3390/cells11050762
Chicago/Turabian StyleBae, Eunjin, Mi-Yeon Yu, Jong-Joo Moon, Ji-Eun Kim, Saram Lee, Sang-Woong Han, Dong-Jun Park, Yon-Su Kim, and Seung-Hee Yang. 2022. "Renoprotective Effect of KLF2 on Glomerular Endothelial Dysfunction in Hypertensive Nephropathy" Cells 11, no. 5: 762. https://doi.org/10.3390/cells11050762