
Long-Term Dynamic Changes of NMDA Receptors Following an Excitotoxic Challenge

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Neuronal Striatal Cultures
2.3. NADPH-Diaphorase Staining
2.4. Sample Collection for mRNA Analysis
2.5. qRT-PCR Analysis
2.6. Bioinformatic Analysis
2.7. Live-Cell Imaging
2.8. Ca2+i Imaging Experiments
2.9. Analysis of Spontaneous Ca2+i Transients
2.10. Neuronal Striatal Culture Immunofluorescence
2.11. Assessment of Neuronal Injury
2.12. Spectroscopic Analysis
2.13. Statistical Analysis
3. Results
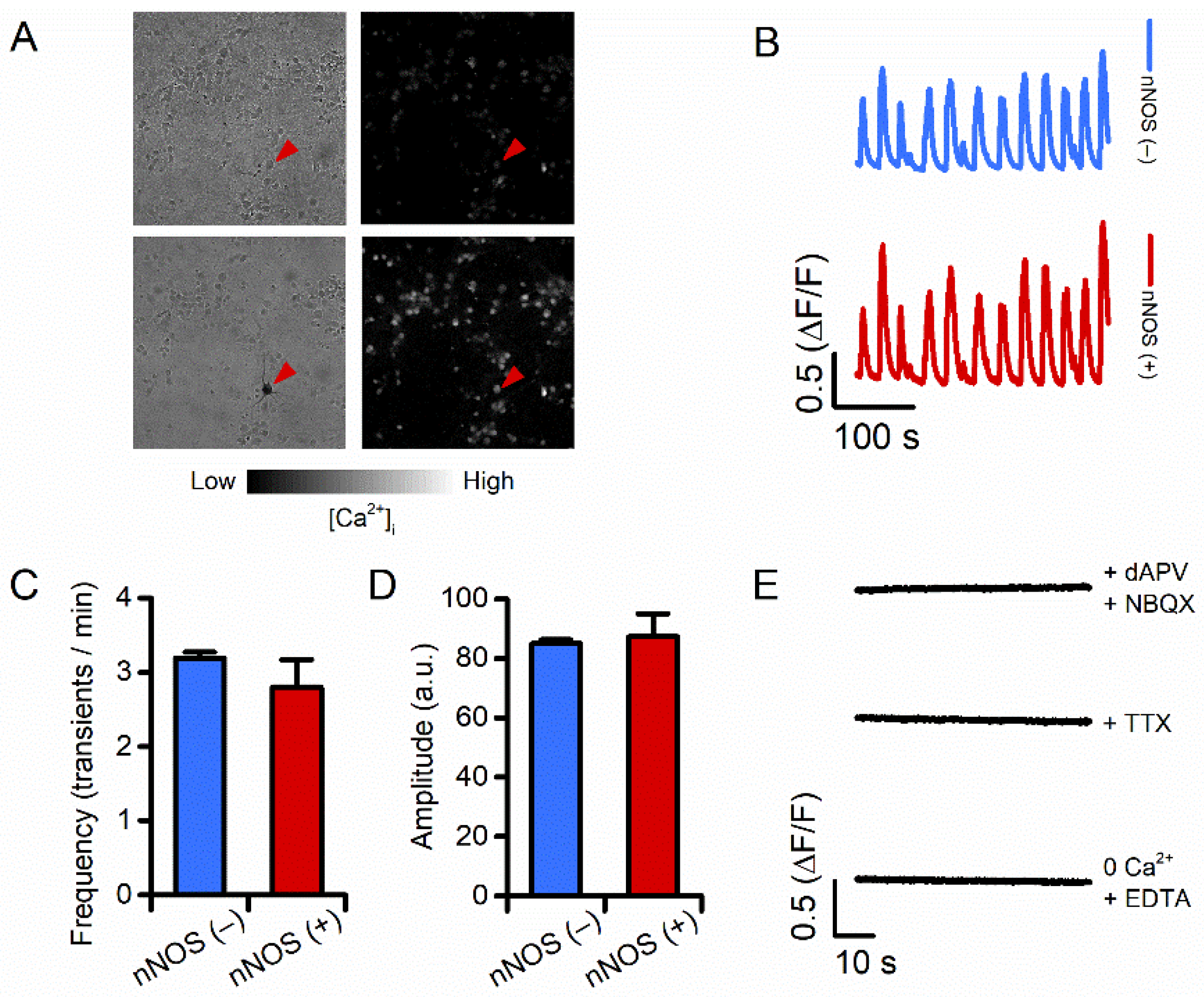
3.1. Spontaneous Ca2+ Transients of nNOS (+) Neurons Are Identical to the Ones of nNOS (−) Neurons
3.2. Compared to nNOS (−) Neurons, nNOS (+) Neurons show Identical Ca2+ Rises following Synaptic and Extrasynaptic NMDAR Activation
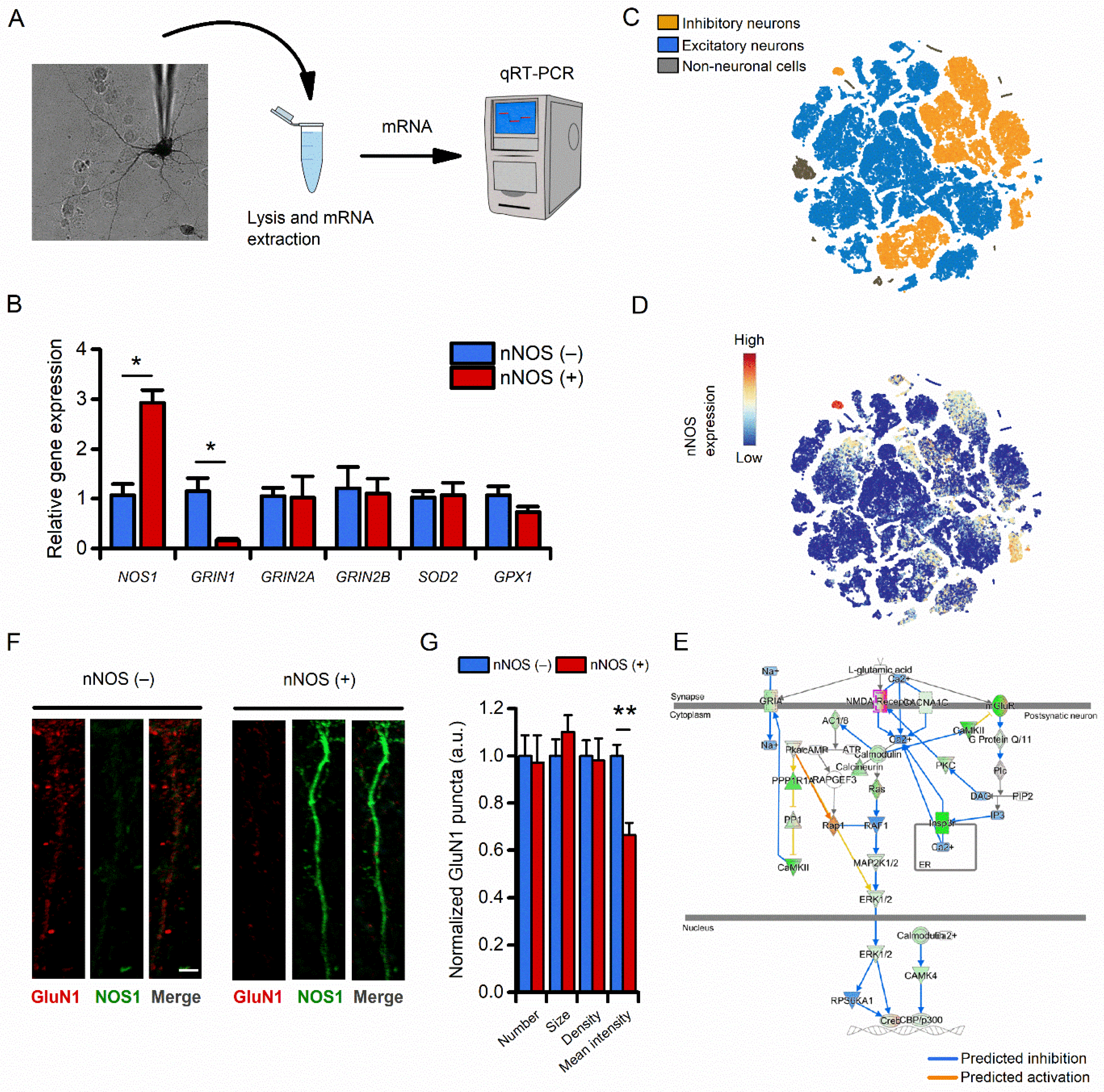
3.3. Compared to nNOS (−) Neurons, nNOS (+) Neurons Show Reduced Transcriptomic and Protein Expression of the NMDAR Subunit 1
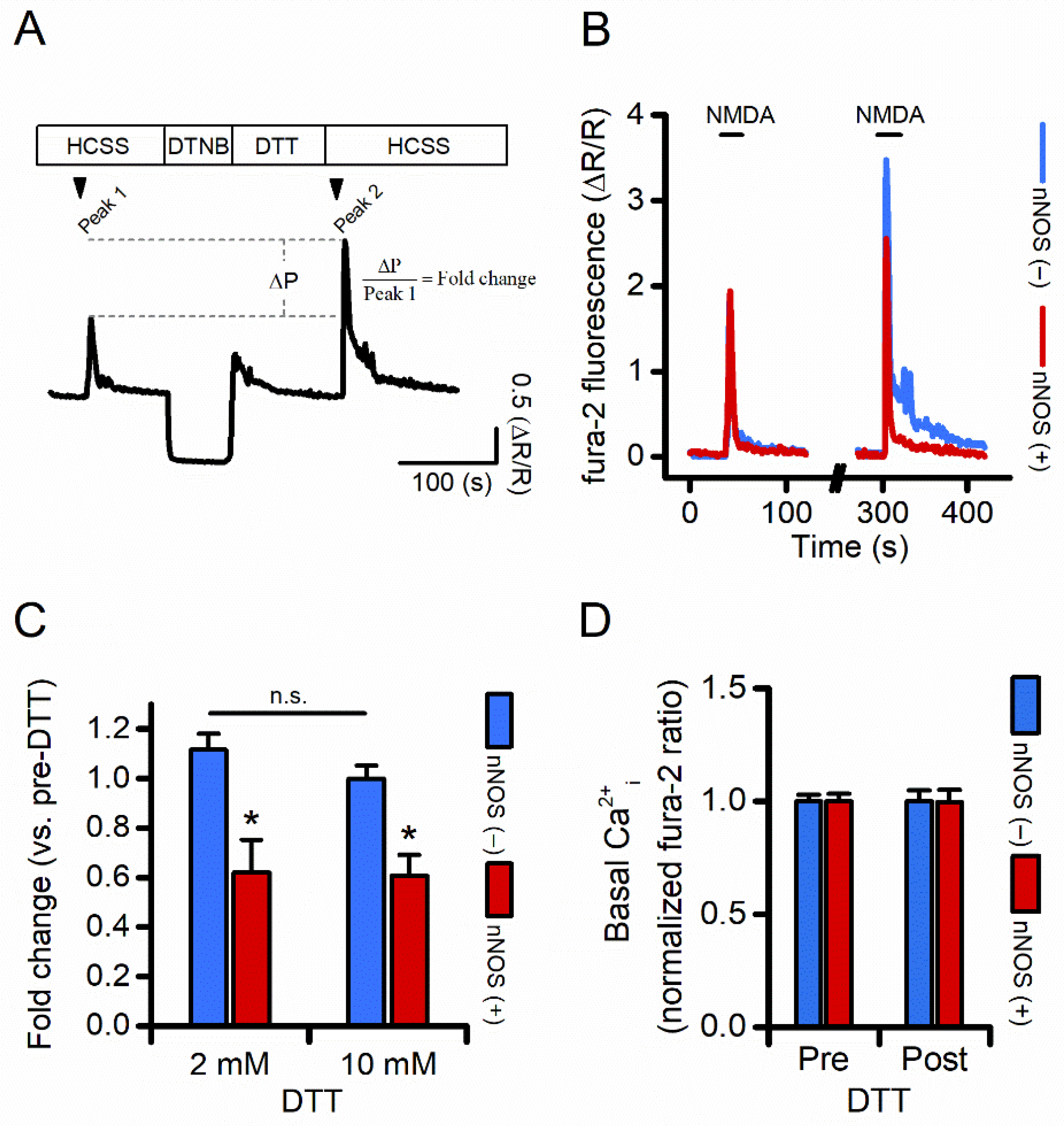
3.4. Compared to nNOS (−), nNOS (+) Neurons Show Reduced NMDAR-Driven [Ca2+]i Rises Following Receptor Reduction
3.5. Long-Term Dynamic Changes in NMDAR Levels Could Account for nNOS (+) Neurons Survival Following an Excitotoxic Challenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 1104. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Swanson, R.A.; Wang, J. Superoxide and non-ionotropic signaling in neuronal excitotoxicity. Front. Neurosci. 2020, 14, 861. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef]
- Beal, M.F. Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Ann. Neurol. 1998, 44, S110–S114. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Granzotto, A.; Sensi, S.L. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015, 81, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Sensi, S.L. Intracellular zinc mobilization is required for nNOS (+) neuron loss. Role of zinc in the excitotoxic cascade. J. Cell. Neurosci. Oxidative Stress 2019, 11, 6. [Google Scholar] [CrossRef]
- Koh, J.Y.; Peters, S.; Choi, D.W. Neurons containing NADPH-diaphorase are selectively resistant to quinolinate toxicity. Science 1986, 234, 73–76. [Google Scholar] [CrossRef]
- Koh, J.Y.; Choi, D.W. Vulnerability of cultured cortical neurons to damage by excitotoxins: Differential susceptibility of neurons containing NADPH-diaphorase. J. Neurosci. 1988, 8, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- Uemura, Y.; Kowall, N.W.; Beal, M.F. Selective sparing of NADPH-diaphorase-somatostatin-neuropeptide Y neurons in ischemic gerbil striatum. Ann. Neurol. 1990, 27, 620–625. [Google Scholar] [CrossRef]
- Weiss, J.H.; Turetsky, D.; Wilke, G.; Choi, D.W. AMPA/kainate receptor-mediated damage to NADPH-diaphorase-containing neurons is Ca2+ dependent. Neurosci. Lett. 1994, 167, 93–96. [Google Scholar] [CrossRef]
- Dawson, T.M.; Bredt, D.S.; Fotuhi, M.; Hwang, P.M.; Snyder, S.H. Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc. Natl. Acad. Sci. USA 1991, 88, 7797–7801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, B.T.; Michael, G.J.; Knigge, K.M.; Vincent, S.R. Neuronal NADPH diaphorase is a nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 2811–2814. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Kowall, N.W.; Beal, M.F.; Richardson, E.P., Jr.; Bird, E.D.; Martin, J.B. Selective sparing of a class of striatal neurons in Huntington’s disease. Science 1985, 230, 561–563. [Google Scholar] [CrossRef]
- Graveland, G.A.; Williams, R.S.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef]
- Mufson, E.J.; Brandabur, M.M. Sparing of NADPH-diaphorase striatal neurons in Parkinson’s and Alzheimer’s diseases. Neuroreport 1994, 5, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Canzoniero, L.M.T.; Granzotto, A.; Turetsky, D.M.; Choi, D.W.; Dugan, L.L.; Sensi, S.L. nNOS(+) striatal neurons, a subpopulation spared in Huntington’s Disease, possess functional NMDA receptors but fail to generate mitochondrial ROS in response to an excitotoxic challenge. Front. Physiol. 2013, 4, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzotto, A.; Canzoniero, L.M.T.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 2020, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivin, J.A.; Choi, D.W. Stroke Therapy. Sci. Am. 1991, 265, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kim, S.H.; Choi, H.B.; Min, C.; Gwag, B.J. High Abundance of GluR1 mRNA and Reduced Q/R Editing of GluR2 mRNA in Individual NADPH-Diaphorase Neurons. Mol. Cell. Neurosci. 2001, 17, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Allen Brain Institute. Available online: https://portal.brain-map.org/ (accessed on 3 January 2022).
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Tasic, B.; Yao, Z.; Graybuck, L.T.; Smith, K.A.; Nguyen, T.N.; Bertagnolli, D.; Goldy, J.; Garren, E.; Economo, M.N.; Viswanathan, S.; et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 2018, 563, 72–78. [Google Scholar] [CrossRef]
- Csernansky, C.A.; Canzoniero, L.M.T.; Sensi, S.L.; Yu, S.P.; Choi, D.W. Delayed application of aurintricarboxylic acid reduces glutamate-induced cortical neuronal injury. J. Neurosci. Res. 1994, 38, 101–108. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Frazzini, V.; Guarnieri, S.; Bomba, M.; Navarra, R.; Morabito, C.; Mariggiò, M.A.A.; Sensi, S.L.L. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 2016, 7, e2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzotto, A.; Bomba, M.; Castelli, V.; Navarra, R.; Massetti, N.; D’Aurora, M.; Onofrj, M.; Cicalini, I.; del Boccio, P.; Gatta, V.; et al. Inhibition of de novo ceramide biosynthesis affects aging phenotype in an in vitro model of neuronal senescence. Aging 2019, 11, 6336–6357. [Google Scholar] [CrossRef] [PubMed]
- Isopi, E.; Granzotto, A.; Corona, C.; Bomba, M.; Ciavardelli, D.; Curcio, M.; Canzoniero, L.M.T.L.M.T.; Navarra, R.; Lattanzio, R.; Piantelli, M.; et al. Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer’s disease without reducing amyloid and tau pathology. Neurobiol. Dis. 2015, 81, 214–224. [Google Scholar] [CrossRef]
- Augood, S.J.; McGowan, E.M.; Emson, P.C. Expression of N-methyl-d-aspartate receptor subunit NR1 messenger RNA by identified striatal somatostatin cells. Neuroscience 1994, 59, 7–12. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Standaert, D.G.; Testa, C.M.; Penney, J.B., Jr.; Young, A.B. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 1995, 15, 5297–5307. [Google Scholar] [CrossRef] [Green Version]
- Price, R.H., Jr.; Mayer, B.; Beitz, A.J. Nitric oxide synthase neurons in rat brain express more NMDA receptor mRNA than non-NOS neurons. Neuroreport 1993, 4, 807–810. [Google Scholar] [CrossRef]
- Weiss, S.W.; Albers, D.S.; Iadarola, M.J.; Dawson, T.M.; Dawson, V.L.; Standaert, D.G. NMDAR1 glutamate receptor subunit isoforms in neostriatal, neocortical, and hippocampal nitric oxide synthase neurons. J. Neurosci. 1998, 18, 1725–1734. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Aizenman, E.; Lipton, S.A.; Loring, R.H. Selective modulation of NMDA responses by reduction and oxidation. Neuron 1989, 2, 1257–1263. [Google Scholar] [CrossRef]
- Aizenman, E.; Hartnett, K.A.; Reynolds, I.J. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron 1990, 5, 841–846. [Google Scholar] [CrossRef]
- Aizenman, E.; Loring, R.H.; Reynolds, I.J.; Rosenberg, P.A. The Redox Biology of Excitotoxic Processes: The NMDA Receptor, TOPA Quinone, and the Oxidative Liberation of Intracellular Zinc. Front. Neurosci. 2020, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Hogins, J.; Crawford, D.C.; Zorumski, C.F.; Mennerick, S. Excitotoxicity triggered by Neurobasal culture medium. PLoS ONE 2011, 6, e25633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggioni, D.; Monfrini, M.; Ravasi, M.; Tredici, G.; Scuteri, A. Neurobasal medium toxicity on mature cortical neurons. Neuroreport 2015, 26, 320–324. [Google Scholar] [CrossRef]
- Olney, J.W.; Zorumski, C.; Price, M.T.; Labruyere, J. L-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science 1990, 248, 596–599. [Google Scholar] [CrossRef]
- Turetsky, D.M.; Canzoniero, L.M.T.; Sensi, S.L.; Weiss, J.H.; Goldberg, M.P.; Choi, D.W. Cortical neurones exhibiting kainate-activated co2+ uptake are selectively vulnerable to ampa/kainate receptor-mediated toxicity. Neurobiol. Dis. 1994, 1, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Paoletti, P. Allosteric modulators of NMDA receptors: Multiple sites and mechanisms. Curr. Opin. Pharmacol. 2015, 20, 14–23. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Gonzalez-Zulueta, M.; Ensz, L.M.; Mukhina, G.; Lebovitz, R.M.; Zwacka, R.M.; Engelhardt, J.F.; Oberley, L.W.; Dawson, V.L.; Dawson, T.M. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J. Neurosci. 1998, 18, 2040–2055. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Angélica Carrasco, M.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Betzen, C.; White, R.; Zehendner, C.M.; Pietrowski, E.; Bender, B.; Luhmann, H.J.; Kuhlmann, C.R.W. Oxidative stress upregulates the NMDA receptor on cerebrovascular endothelium. Free Radic. Biol. Med. 2009, 47, 1212–1220. [Google Scholar] [CrossRef]
- Hota, S.K.; Hota, K.B.; Prasad, D.; Ilavazhagan, G.; Singh, S.B. Oxidative-stress-induced alterations in Sp factors mediate transcriptional regulation of the NR1 subunit in hippocampus during hypoxia. Free Radic. Biol. Med. 2010, 49, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadia, S.; Soriano, F.X.; Léveillé, F.; Martel, M.-A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Biegon, A.; Fry, P.A.; Paden, C.M.; Alexandrovich, A.; Tsenter, J.; Shohami, E. Dynamic changes in N-methyl-d-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. USA 2004, 101, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Roche, K.W.; Standley, S.; McCallum, J.; Dune Ly, C.; Ehlers, M.D.; Wenthold, R.J. Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 2001, 4, 794–802. [Google Scholar] [CrossRef]
- Scott, D.B.; Michailidis, I.; Mu, Y.; Logothetis, D.; Ehlers, M.D. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J. Neurosci. 2004, 24, 7096–7109. [Google Scholar] [CrossRef] [Green Version]
- Nong, Y.; Huang, Y.-Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, C.; Yang, Q.; Jiao, M.; Qiu, S. Endocytosis of GluN2B-containing NMDA receptors mediates NMDA-induced excitotoxicity. Mol. Pain 2017, 13, 1744806917701921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Aizenman, E.; Sinor, J.D.; Brimecombe, J.C.; Herin, G.A. Alterations of N-methyl-D-aspartate receptor properties after chemical ischemia. J. Pharmacol. Exp. Ther. 2000, 295, 572–577. [Google Scholar] [PubMed]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granzotto, A.; d’Aurora, M.; Bomba, M.; Gatta, V.; Onofrj, M.; Sensi, S.L. Long-Term Dynamic Changes of NMDA Receptors Following an Excitotoxic Challenge. Cells 2022, 11, 911. https://doi.org/10.3390/cells11050911
Granzotto A, d’Aurora M, Bomba M, Gatta V, Onofrj M, Sensi SL. Long-Term Dynamic Changes of NMDA Receptors Following an Excitotoxic Challenge. Cells. 2022; 11(5):911. https://doi.org/10.3390/cells11050911
Chicago/Turabian StyleGranzotto, Alberto, Marco d’Aurora, Manuela Bomba, Valentina Gatta, Marco Onofrj, and Stefano L. Sensi. 2022. "Long-Term Dynamic Changes of NMDA Receptors Following an Excitotoxic Challenge" Cells 11, no. 5: 911. https://doi.org/10.3390/cells11050911