
Molecular Mechanism of Ferroptosis in Orthopedic Diseases

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
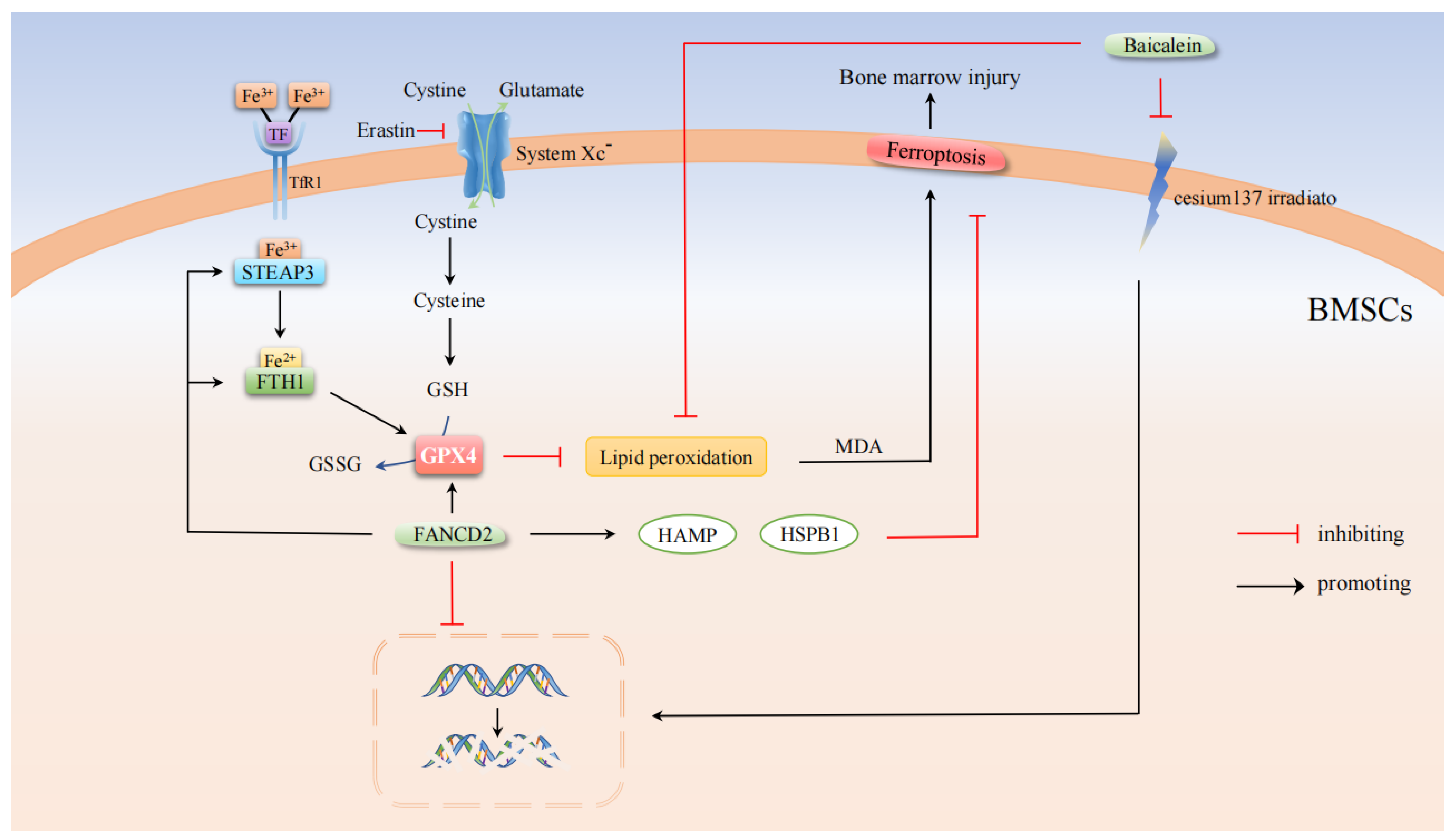
2. Bone Marrow Injury
3. Osteoporosis
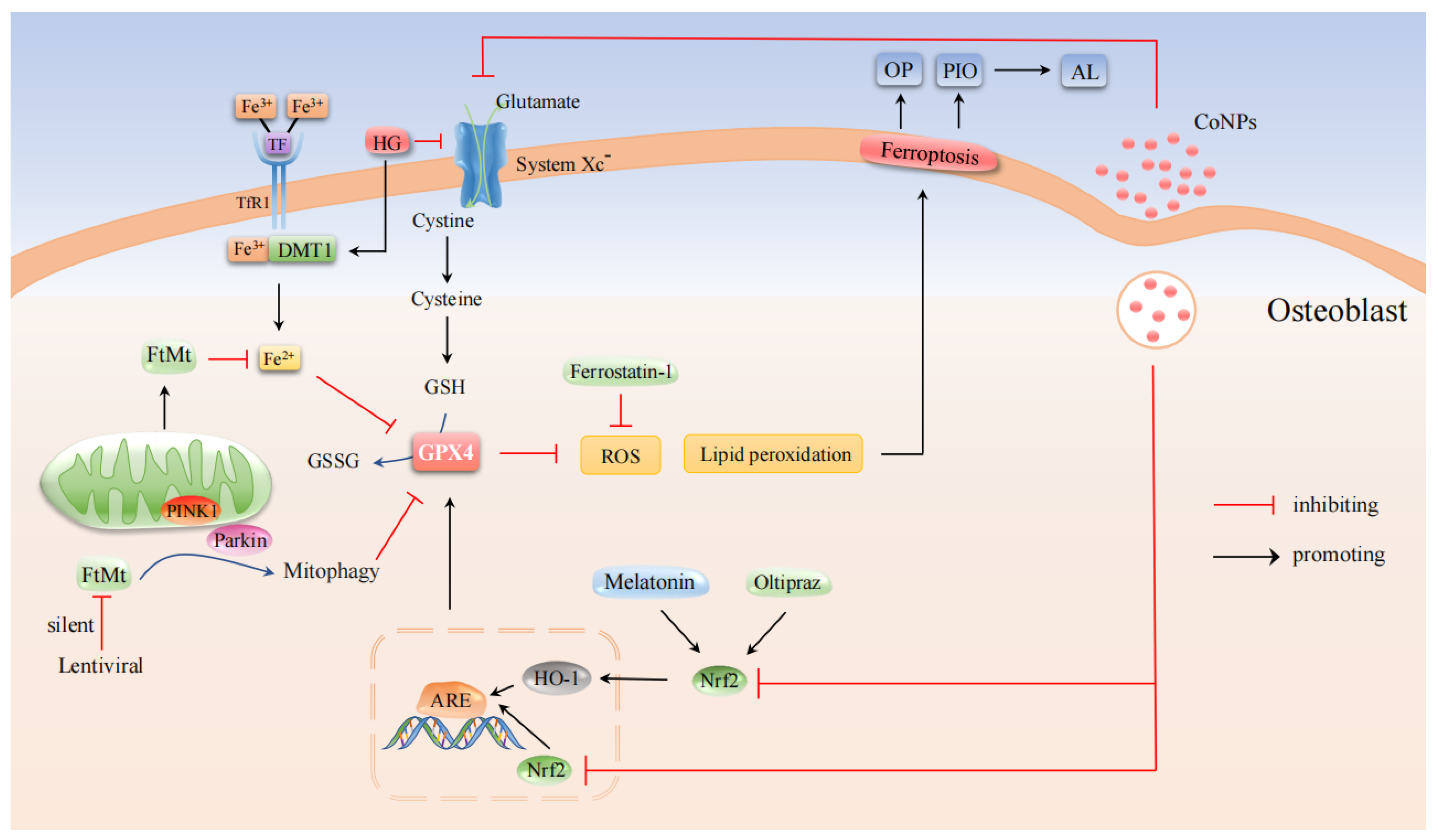
3.1. OP-Associated Osteoblasts with Ferroptosis
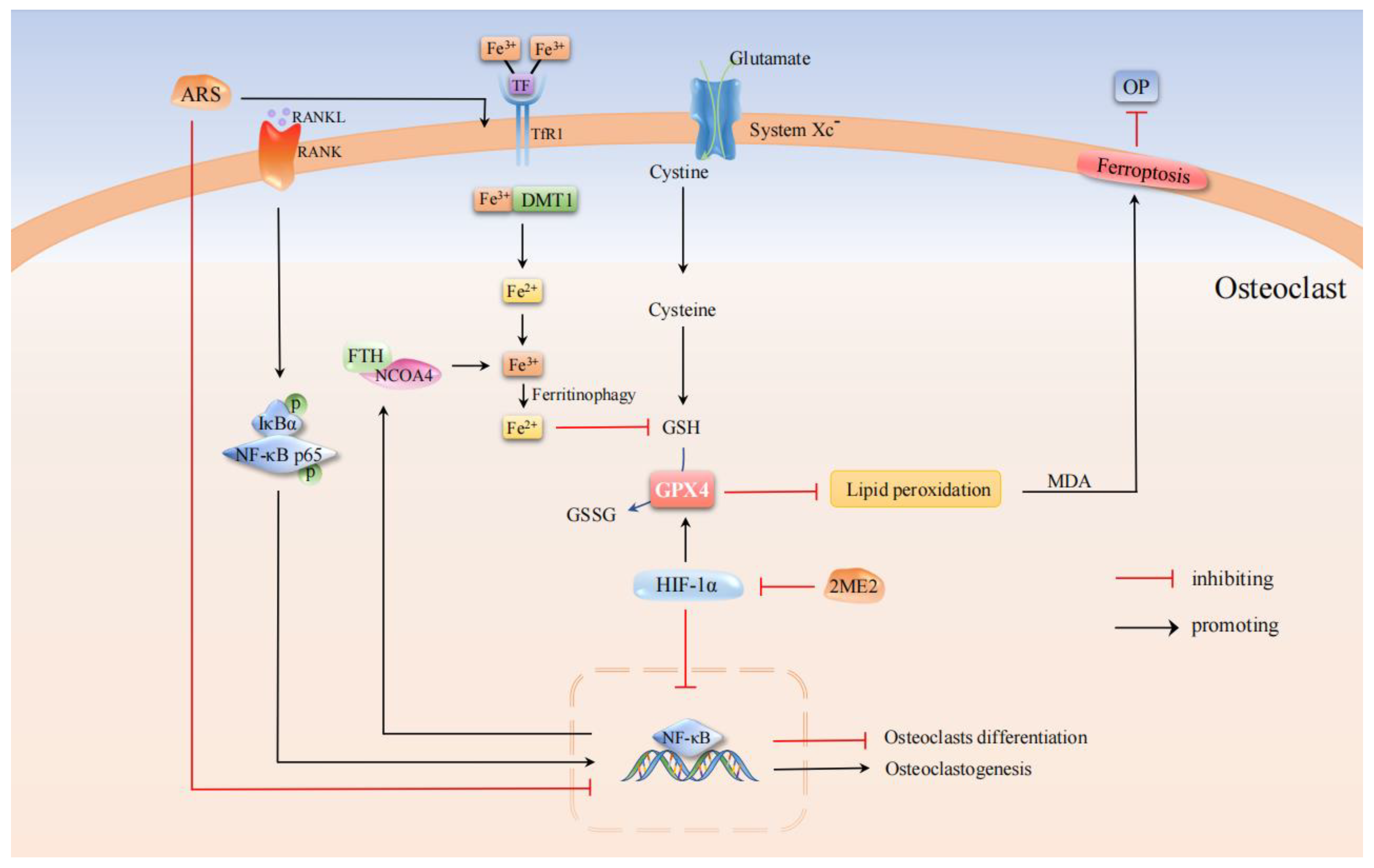
3.2. OP-Associated Osteoclasts with Ferroptosis
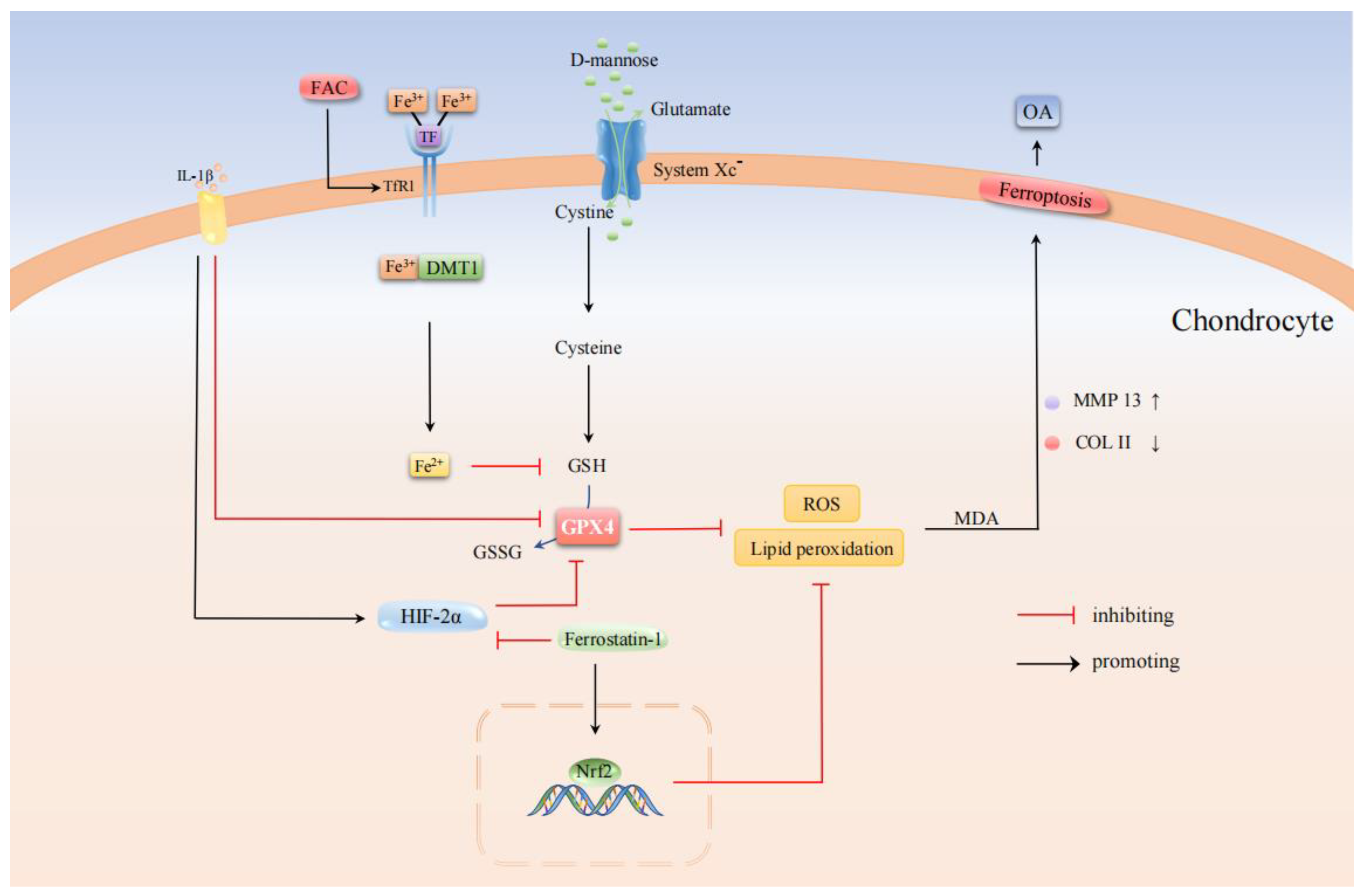
4. Osteoarthritis
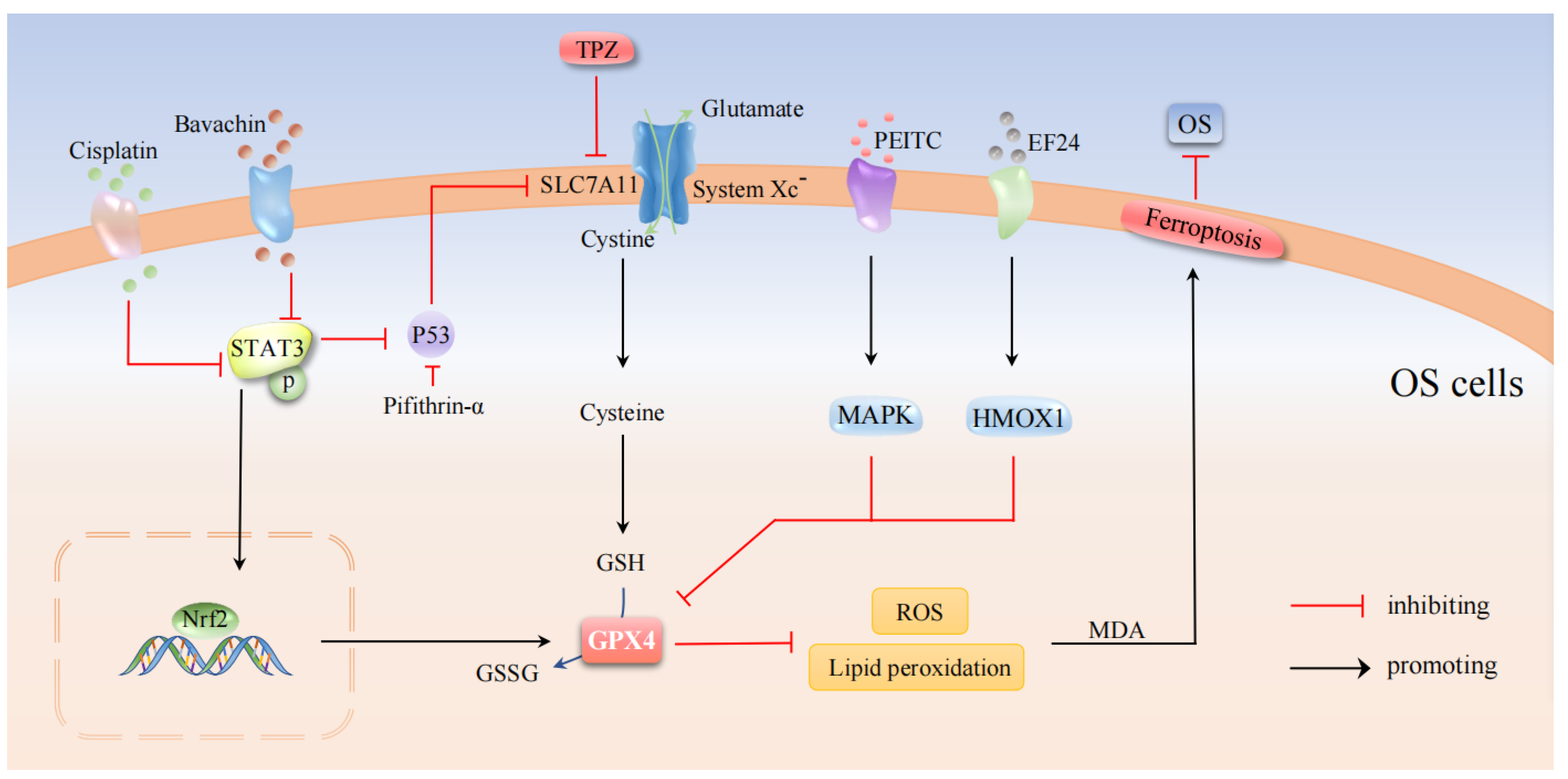
5. Osteosarcoma
6. Conclusions and Prospects
- Research on activators and inhibitors regulating key targets of ferroptosis.
- Research on interventions (DNA repair proteins, flavonoids, antineoplastic agents, etc.) of ferroptosis signaling pathways in major bone-associated cells.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012, 19, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Bergmann, A. Autophagy and cell death: No longer at odds. Cell 2007, 131, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [Green Version]
- Artyukhova, M.A.; Tyurina, Y.Y.; Chu, C.T.; Zharikova, T.M.; Bayir, H.; Kagan, V.E.; Timashev, P.S. Interrogating Parkinson’s disease associated redox targets: Potential application of CRISPR editing. Free Radic. Biol. Med. 2019, 144, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xie, Y.; Kang, R.; Hou, W.; Sun, X.; Epperly, M.W.; Greenberger, J.S.; Tang, D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem. Biophys. Res. Commun. 2016, 480, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid. Med. Cell. Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, Y.; Sun, W.; Zhang, Z.; Liu, J.; Yang, W.; Yuan, W.; Yi, Y.; Wang, J.; Liu, J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2alpha-dependent manner. Cell Prolif. 2021, 54, e13134. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhen, C.; Liu, J.; Shang, P. beta-Phenethyl Isothiocyanate Induces Cell Death in Human Osteosarcoma through Altering Iron Metabolism, Disturbing the Redox Balance, and Activating the MAPK Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5021983. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, B.; Xiong, Q.; Feng, Y.; Du, H. Erastininduced ferroptosis causes physiological and pathological changes in healthy tissues of mice. Mol. Med. Rep. 2021, 24, 713. [Google Scholar] [CrossRef]
- Travlos, G.S. Normal structure, function, and histology of the bone marrow. Toxicol. Pathol. 2006, 34, 548–565. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Salvesen, G. Regulated cell death: Signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell. Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Berhane, H.; Shinde, A.; Kalash, R.; Xu, K.; Epperly, M.W.; Goff, J.; Franicola, D.; Zhang, X.; Dixon, T.; Shields, D.; et al. Amelioration of radiation-induced oral cavity mucositis and distant bone marrow suppression in fanconi anemia Fancd2-/- (FVB/N) mice by intraoral GS-nitroxide JP4-039. Radiat. Res. 2014, 182, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Epperly, M.W.; Fisher, R.; Zhang, X.; Shields, D.; Hou, W.; Wang, H.; Li, S.; Wipf, P.; Parmar, K.; et al. Amelioration of Head and Neck Radiation-Induced Mucositis and Distant Marrow Suppression in Fanca(−/−) and Fancg(−/−) Mice by Intraoral Administration of GS-Nitroxide (JP4-039). Radiat. Res. 2018, 189, 560–578. [Google Scholar] [CrossRef]
- Epperly, M.W.; Fisher, R.; Zhang, X.; Hou, W.; Shields, D.; Wipf, P.; Wang, H.; Thermozier, S.; Greenberger, J.S. Fanconi Anemia Mouse Genotype-specific Mitigation of Total Body Irradiation by GS-Nitroxide JP4-039. In Vivo 2020, 34, 33–38. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E. Epidemiology, etiology, and diagnosis of osteoporosis. Am. J. Obstet. Gynecol. 2006, 194, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T. Osteoporosis--concept, classification and epidemiology. Nihon Rinsho 1994, 52, 2275–2280. [Google Scholar] [PubMed]
- Park-Min, K.H. Mechanisms involved in normal and pathological osteoclastogenesis. Cell. Mol. Life Sci. 2018, 75, 2519–2528. [Google Scholar] [CrossRef]
- Zhang, J.; Qiu, X.; Xi, K.; Hu, W.; Pei, H.; Nie, J.; Wang, Z.; Ding, J.; Shang, P.; Li, B.; et al. Therapeutic ionizing radiation induced bone loss: A review of in vivo and in vitro findings. Connect. Tissue Res. 2018, 59, 509–522. [Google Scholar] [CrossRef]
- Zhang, J.J.; Liu, W.Q.; Peng, J.J.; Ma, Q.L.; Peng, J.; Luo, X.J. miR-21-5p/203a-3p promote ox-LDL-induced endothelial cell senescence through down-regulation of mitochondrial fission protein Drp1. Mech. Ageing Dev. 2017, 164, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Colunga, M.G.; Weidner, H.; Vujic Spasic, M.; Hofbauer, L.C.; Baschant, U.; Rauner, M. Shaping the bone through iron and iron-related proteins. Semin. Hematol. 2021, 58, 188–200. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, W.L.; Meng, H.Z.; Cai, Z.Y.; Yang, M.W. Regulation of DMT1 on autophagy and apoptosis in osteoblast. Int. J. Med. Sci. 2017, 14, 275–283. [Google Scholar] [CrossRef]
- Meng, H.Z.; Zhang, W.L.; Liu, F.; Yang, M.W. Advanced Glycation End Products Affect Osteoblast Proliferation and Function by Modulating Autophagy Via the Receptor of Advanced Glycation End Products/Raf Protein/Mitogen-activated Protein Kinase/Extracellular Signal-regulated Kinase Kinase/Extracellular Signal-regulated Kinase (RAGE/Raf/MEK/ERK) Pathway. J. Biol. Chem. 2015, 290, 28189–28199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.L.; Meng, H.Z.; Yang, M.W. Regulation of DMT1 on Bone Microstructure in Type 2 Diabetes. Int. J. Med. Sci. 2015, 12, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, H.; Sun, J.; Zheng, T.; Zhao, P.; Li, H.; Yang, M. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis Via Mitophagy in Type 2 Diabetic Osteoporosis. Biol. Trace Elem. Res. 2022, 200, 298–307. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sang, W.; Zhong, Y.; Xue, S.; Yang, M.; Wang, C.; Lu, H.; Huan, R.; Mao, X.; Zhu, L.; et al. CoCrMo-Nanoparticles induced peri-implant osteolysis by promoting osteoblast ferroptosis via regulating Nrf2-ARE signalling pathway. Cell Prolif. 2021, 54, e13142. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Tang, C.; Yan, G. Ferritinophagy and ferroptosis in cardiovascular disease: Mechanisms and potential applications. Biomed Pharm. 2021, 141, 111872. [Google Scholar] [CrossRef]
- Arnett, T.R.; Orriss, I.R. Metabolic properties of the osteoclast. Bone 2018, 115, 25–30. [Google Scholar] [CrossRef]
- Ishii, K.A.; Fumoto, T.; Iwai, K.; Takeshita, S.; Ito, M.; Shimohata, N.; Aburatani, H.; Taketani, S.; Lelliott, C.J.; Vidal-Puig, A.; et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat. Med. 2009, 15, 259–266. [Google Scholar] [CrossRef]
- Ni, S.; Yuan, Y.; Qian, Z.; Zhong, Z.; Lv, T.; Kuang, Y.; Yu, B. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic. Biol. Med. 2021, 169, 271–282. [Google Scholar] [CrossRef]
- Zhang, J. The osteoprotective effects of artemisinin compounds and the possible mechanisms associated with intracellular iron: A review of in vivo and in vitro studies. Environ. Toxicol. Pharmacol. 2020, 76, 103358. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Ramos, E.; Branco, J. Osteoarthritis. Acta Med. Port. 2015, 28, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.E.; Scanzello, C.; Felson, D.T.; Au, M.B.; Goggins, J.; Plunkett, A.; Misra, D. Correlation between senescence-associated secretory phenotypes factors in synovial fluid and serum and structural changes in osteoarthritis. Eur. J. Rheumatol. 2020, 7, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Bian, Q.; Wang, Y.J.; Liu, S.F.; Li, Y.P. Osteoarthritis: Genetic factors, animal models, mechanisms, and therapies. Front. Biosci. 2012, 4, 74–100. [Google Scholar] [CrossRef]
- Murata, K.; Yoshitomi, H.; Tanida, S.; Ishikawa, M.; Nishitani, K.; Ito, H.; Nakamura, T. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 2010, 12, R86. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Nims, R.J.; Dicks, A.; Wu, C.L.; Meulenbelt, I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018, 71–72, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef]
- Yao, X.; Sun, K.; Yu, S.; Luo, J.; Guo, J.; Lin, J.; Wang, G.; Guo, Z.; Ye, Y.; Guo, F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J. Orthop. Translat. 2021, 27, 33–43. [Google Scholar] [CrossRef]
- Saito, T.; Kawaguchi, H. HIF-2alpha as a possible therapeutic target of osteoarthritis. Osteoarthr. Cartil. 2010, 18, 1552–1556. [Google Scholar] [CrossRef]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2alpha activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Investig. 2021, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 18. [Google Scholar] [CrossRef]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef]
- Luo, H.; Zhang, R. Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the Xc-/GPX4 axis. Exp. Ther. Med. 2021, 21, 72. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef]
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Faisham, W.I.; Mat Saad, A.Z.; Alsaigh, L.N.; Nor Azman, M.Z.; Kamarul Imran, M.; Biswal, B.M.; Bhavaraju, V.M.; Salzihan, M.S.; Hasnan, J.; Ezane, A.M.; et al. Prognostic factors and survival rate of osteosarcoma: A single-institution study. Asia Pac. J. Clin. Oncol. 2017, 13, e104–e110. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Van Tine, B.A. Pathogenesis and Current Treatment of Osteosarcoma: Perspectives for Future Therapies. J. Clin. Med. 2021, 10, 1182. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Gao, S.; Zhan, L.; Yang, Z.; Shi, R.; Li, H.; Xia, Z.; Yuan, S.; Wu, Q.P.; Wang, T.; Yao, S. Remote Limb Ischaemic Postconditioning Protects Against Myocardial Ischaemia/Reperfusion Injury in Mice: Activation of JAK/STAT3-Mediated Nrf2-Antioxidant Signalling. Cell. Physiol. Biochem. 2017, 43, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.; Lim, C. Anticancer activity of flavonoids accompanied by redox state modulation and the potential for a chemotherapeutic strategy. Food Sci. Biotechnol. 2021, 30, 321–340. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, X.; Zou, L.; Lei, M.; Feng, J.; Hu, Z. Bavachin Induces Ferroptosis through the STAT3/P53/SLC7A11 Axis in Osteosarcoma Cells. Oxid. Med. Cell. Longev. 2021, 2021, 1783485. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Kushlinskii, N.E.; Fridman, M.V.; Braga, E.A. Molecular Mechanisms and microRNAs in Osteosarcoma Pathogenesis. Biochemistry 2016, 81, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, P.; Kim, J.S. Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers. Molecules 2018, 23, 2983. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Li, J.; Peng, Z.H.; Sheinin, Y.; Zhou, J.; Oupicky, D. Tumor-Penetrating Nanoparticles for Enhanced Anticancer Activity of Combined Photodynamic and Hypoxia-Activated Therapy. ACS Nano 2017, 11, 2227–2238. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gong, M.; Deng, Z.; Liu, H.; Chang, Y.; Yang, Z.; Cai, L. Tirapazamine suppress osteosarcoma cells in part through SLC7A11 mediated ferroptosis. Biochem. Biophys. Res. Commun. 2021, 567, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Cai, J.; Armstrong, J.; Herold, M.; Lu, Y.J.; Sun, A.; Snyder, J.P.; Liotta, D.C.; Jones, D.P.; Shoji, M. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs 2005, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of EF24, a Novel Curcumin Analog: A Review. Front. Oncol. 2018, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, S.; Shi, L.; Zhang, S.; Ren, J.; Yao, S.; Yun, D.; Huang, L.; Wang, J.; Li, W.; et al. Design, synthesis, and evaluation of asymmetric EF24 analogues as potential anti-cancer agents for lung cancer. Eur. J. Med. Chem. 2017, 125, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, X.; Zhang, C.; Yang, T.; Deng, Z.; Song, Y.; Huang, L.; Li, F.; Li, Q.; Lin, S.; et al. EF24 induces ferroptosis in osteosarcoma cells through HMOX1. Biomed Pharmacother. 2021, 136, 111202. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Hua, W.; Tian, L.; Zhou, X.; Wang, D.; Yang, Y.; Ni, G. Molecular Mechanism of Ferroptosis in Orthopedic Diseases. Cells 2022, 11, 2979. https://doi.org/10.3390/cells11192979
Gao L, Hua W, Tian L, Zhou X, Wang D, Yang Y, Ni G. Molecular Mechanism of Ferroptosis in Orthopedic Diseases. Cells. 2022; 11(19):2979. https://doi.org/10.3390/cells11192979
Chicago/Turabian StyleGao, Lu, Weizhong Hua, Lixiang Tian, Xuchang Zhou, Dongxue Wang, Yajing Yang, and Guoxin Ni. 2022. "Molecular Mechanism of Ferroptosis in Orthopedic Diseases" Cells 11, no. 19: 2979. https://doi.org/10.3390/cells11192979