

Ferroptosis: The Potential Target in Heart Failure with Preserved Ejection Fraction
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
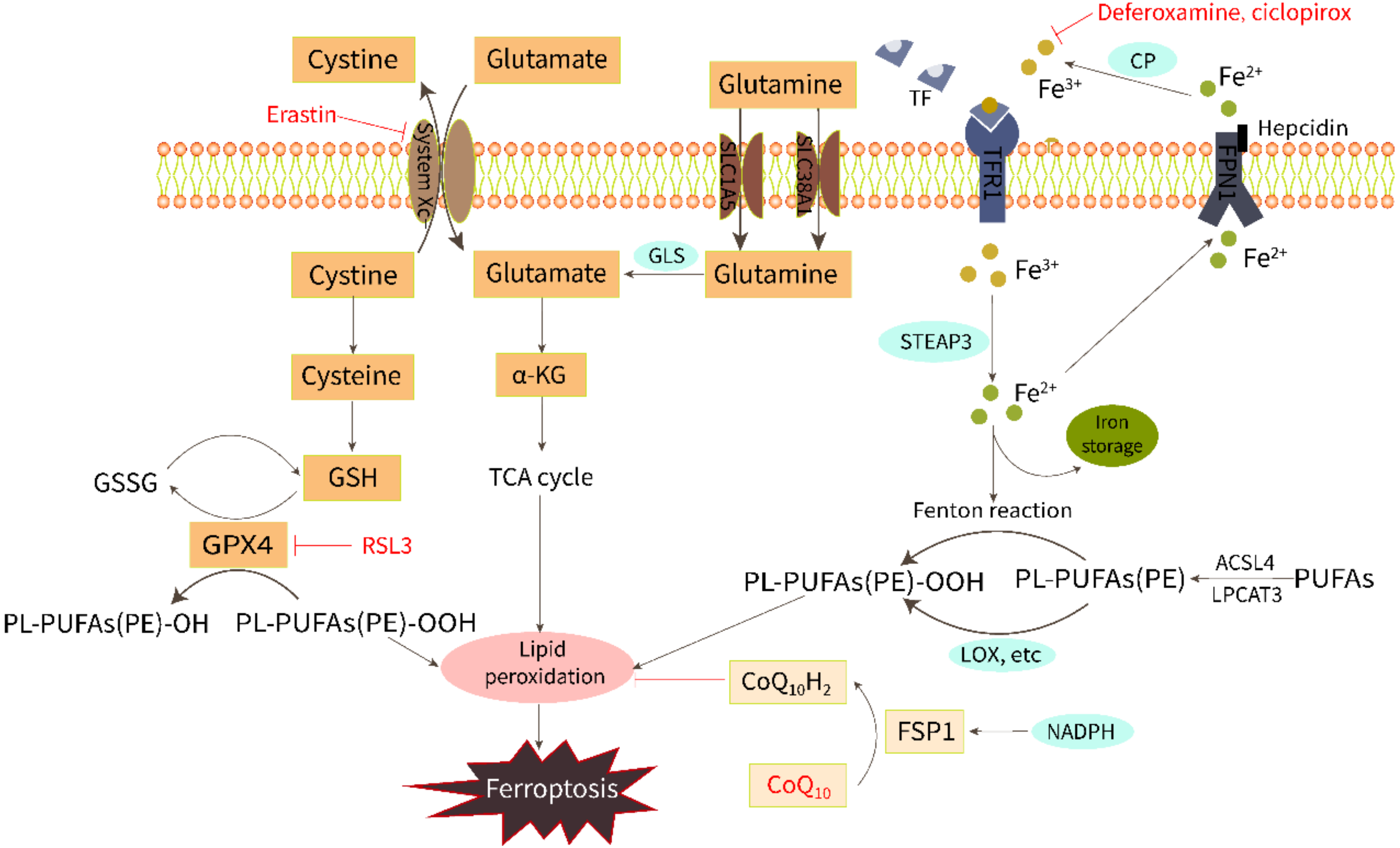
2. Biochemical Regulation of Ferroptosis
2.1. Iron Overload
2.2. Lipid Peroxidation
2.3. Amino Acid Metabolism
3. The Comorbidity–Inflammation Paradigm in HFpEF Is Closely Related to the Regulatory Mechanism of Ferroptosis
3.1. The Comorbidity–Inflammation Paradigm in HFpEF
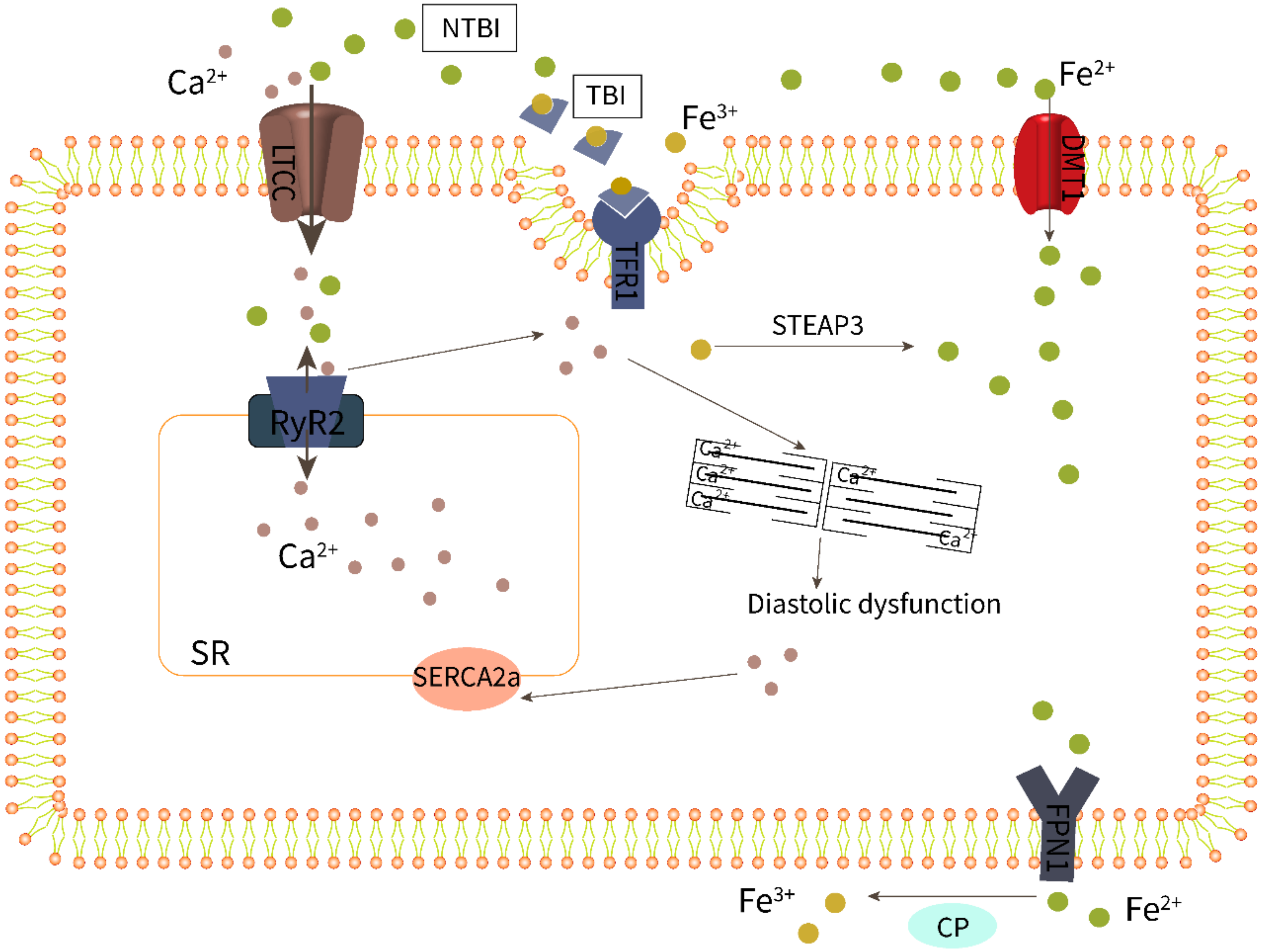
3.2. Iron Overload Leads to Endothelial Dysfunction
3.3. Macrophage Iron Overload Is Involved in Myocardial Inflammation and Fibrosis
3.4. Cardiomyocyte Iron Overload Impairs Excitation–Contraction Coupling
3.5. Relationship between LPO and HFpEF
4. Research Progress of Anti-Ferroptosis in HFpEF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. Heart failure with preserved ejection fraction: Present status and future directions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Ovchinnikov, A.G.; Arefieva, T.I.; Potekhina, A.V.; Filatova, A.Y.; Ageev, F.T.; Boytsov, S. The Molecular and Cellular Mechanisms Associated with a Microvascular Inflammation in the Pathogenesis of Heart Failure with Preserved Ejection Fraction. Acta Nat. 2020, 12, 40–51. [Google Scholar] [CrossRef]
- Leonardi, B.; Margossian, R.; Colan, S.D.; Powell, A.J. Relationship of magnetic resonance imaging estimation of myocardial iron to left ventricular systolic and diastolic function in thalassemia. JACC Cardiovasc. Imaging 2008, 1, 572–578. [Google Scholar] [CrossRef]
- Kremastinos, D.T.; Farmakis, D.; Aessopos, A.; Hahalis, G.; Hamodraka, E.; Tsiapras, D.; Keren, A. Beta-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circ. Heart Fail. 2010, 3, 451–458. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Muller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, Q.; Chen, F.; Pang, J.; Pan, C.; Xu, F.; Chen, Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 2021, 9, 742483. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Li, N.; Wang, W.; Zhou, H.; Wu, Q.; Duan, M.; Liu, C.; Wu, H.; Deng, W.; Shen, D.; Tang, Q. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 2020, 160, 303–318. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- DuBrock, H.M.; AbouEzzeddine, O.F.; Redfield, M.M. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS ONE 2018, 13, e0201836. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Orlenko, A.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Spires, T.E.; Yarde, M.; Wang, Z.; Seiffert, D.A.; et al. Multiple Plasma Biomarkers for Risk Stratification in Patients With Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 75, 1281–1295. [Google Scholar] [CrossRef]
- Sanders-van Wijk, S.; Tromp, J.; Beussink-Nelson, L.; Hage, C.; Svedlund, S.; Saraste, A.; Swat, S.A.; Sanchez, C.; Njoroge, J.; Tan, R.-S.; et al. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure With Preserved Ejection Fraction: Results From the PROMIS-HFpEF Study. Circulation 2020, 142, 2029–2044. [Google Scholar] [CrossRef]
- Hage, C.; Michaelsson, E.; Kull, B.; Miliotis, T.; Svedlund, S.; Linde, C.; Donal, E.; Daubert, J.; Gan, L.; Lund, L.H. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. 2020, 7, 1534–1546. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E.B. Macrophages in Heart Failure with Reduced versus Preserved Ejection Fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis The Heart Failure With Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Marques, V.B.; Nunes, D.O.; Stefanon, I.; Dos Santos, L. Chronic iron overload induces functional and structural vascular changes in small resistance arteries via NADPH oxidase-dependent O2(-) production. Toxicol. Lett. 2017, 279, 43–52. [Google Scholar] [CrossRef]
- Marques, V.B.; Nascimento, T.B.; Ribeiro, R.F., Jr.; Broseghini-Filho, G.B.; Rossi, E.M.; Graceli, J.B.; dos Santos, L. Chronic iron overload in rats increases vascular reactivity by increasing oxidative stress and reducing nitric oxide bioavailability. Life Sci. 2015, 143, 89–97. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5, e132964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Locati, M.; Gammella, E.; Invernizzi, P.; Cairo, G. Iron levels in polarized macrophages: Regulation of immunity and autoimmunity. Autoimmun. Rev. 2012, 11, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.-P.; Liu, Z.-J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harrington, L.; Trebicka, E.; Shi, H.N.; Kagan, J.C.; Hong, C.C.; Lin, H.Y.; Babitt, J.L.; Cherayil, B.J. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J. Clin. Investig. 2009, 119, 3322–3328. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Haschka, D.; Dichtl, S.; Sonnweber, T.; Theurl, I.; Theurl, M.; Lindner, E.; Demetz, E.; Aßhoff, M.; et al. Lipocalin-2 ensures host defense against Salmonella Typhimurium by controlling macrophage iron homeostasis and immune response. Eur. J. Immunol. 2015, 45, 3073–3086. [Google Scholar] [CrossRef]
- Nai, A.; Rubio, A.; Campanella, A.; Gourbeyre, O.; Artuso, I.; Bordini, J.; Gineste, A.; Latour, C.; Besson-Fournier, C.; Lin, H.Y.; et al. Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 2016, 127, 2327–2336. [Google Scholar] [CrossRef]
- Santos, M.M.; de Sousa, M.; Rademakers, L.H.P.M.; Clevers, H.; Marx, J.; Schilham, M.W. Iron overload and heart fibrosis in mice deficient for both beta 2-microglobulin and Rag1. Am. J. Pathol. 2000, 157, 1883–1892. [Google Scholar] [CrossRef]
- Pepe, A.; Positano, V.; Capra, M.; Maggio, A.; Pinto, C.L.; Spasiano, A.; Forni, G.; Derchi, G.; Favilli, B.; Rossi, G.; et al. Myocardial scarring by delayed enhancement cardiovascular magnetic resonance in thalassaemia major. Heart 2009, 95, 1688–1693. [Google Scholar] [CrossRef]
- Ishizaka, N.; Aizawa, T.; Yamazaki, I.; Usui, S.-I.; Mori, I.; Kurokawa, K.; Tang, S.-S.; Ingelfinger, J.R.; Ohno, M.; Nagai, R. Abnormal iron deposition in renal cells in the rat with chronic angiotensin II administration. Lab. Investig. 2002, 82, 87–96. [Google Scholar] [CrossRef]
- Ishizaka, N.; Saito, K.; Mitani, H.; Yamazaki, I.; Sata, M.; Usui, S.-I.; Mori, I.; Ohno, M.; Nagai, R. Iron overload augments angiotensin II-induced cardiac fibrosis and promotes neointima formation. Circulation 2002, 106, 1840–1846. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Chen, Y.Y.; Chang, Y.Z.; Duan, X.L.; Ho, K.P.; Jiang, D.H.; Wang, K.; Qian, Z.M. Post-transcriptional expression of DMT1 in the heart of rat. J. Cell. Physiol. 2003, 196, 124–130. [Google Scholar] [CrossRef]
- Liu, Y.; Parkes, J.G.; Templeton, D.A. Differential accumulation of non-transferrin-bound iron by cardiac myocytes and fibroblasts. J. Mol. Cell. Cardiol. 2003, 35, 505–514. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Sun, H.; Trivieri, M.G.; E Koch, S.; Dawood, F.; Ackerley, C.; Yazdanpanah, M.; Wilson, G.J.; Schwartz, A.; Liu, P.P.; et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 2003, 9, 1187–1194. [Google Scholar] [CrossRef]
- Tsushima, R.G.; Wickenden, A.D.; Bouchard, R.A.; Oudit, G.Y.; Liu, P.P.; Backx, P.H. Modulation of iron uptake in heart by L-type Ca2+ channel modifiers: Possible implications in iron overload. Circ. Res. 1999, 84, 1302–1309. [Google Scholar] [CrossRef]
- Runte, K.E.; Bell, S.P.; Selby, D.E.; Häußler, T.N.; Ashikaga, T.; LeWinter, M.M.; Palmer, B.M.; Meyer, M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium From Patients With Hypertensive Heart Disease and Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2017, 10, e004311. [Google Scholar] [CrossRef]
- Selby, D.E.; Palmer, B.M.; LeWinter, M.M.; Meyer, M. Tachycardia-induced diastolic dysfunction and resting tone in myocardium from patients with a normal ejection fraction. J. Am. Coll. Cardiol. 2011, 58, 147–154. [Google Scholar] [CrossRef]
- Adeniran, I.; MacIver, D.H.; Hancox, J.C.; Zhang, H.G. Abnormal calcium homeostasis in heart failure with preserved ejection fraction is related to both reduced contractile function and incomplete relaxation: An electromechanically detailed biophysical modeling study. Front. Physiol. 2015, 6, 78. [Google Scholar] [CrossRef]
- Durland, L. Distinguishing HF with reduced and preserved ejection fraction at the level of individual cardiomyocytes: Implications for therapeutic development. J. Physiol. 2021, 599, 1027–1029. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct features of calcium handling and beta-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef]
- Rouhana, S.; Farah, C.; Roy, J.; Finan, A.; de Araujo, G.R.; Bideaux, P.; Scheuermann, V.; Saliba, Y.; Reboul, C.; Cazorla, O.; et al. Early calcium handling imbalance in pressure overload-induced heart failure with nearly normal left ventricular ejection fraction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 230–242. [Google Scholar] [CrossRef]
- Warbrick, I.; Rabkin, S.W. Effect of the peptides Relaxin, Neuregulin, Ghrelin and Glucagon-like peptide-1, on cardiomyocyte factors involved in the molecular mechanisms leading to diastolic dysfunction and/or heart failure with preserved ejection fraction. Peptides 2019, 111, 33–41. [Google Scholar] [CrossRef]
- Nunez, M.T.; Hidalgo, C. Noxious Iron-Calcium Connections in Neurodegeneration. Front. Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Gao, Z.; Li, H. Association of cardiac injury with iron-increased oxidative and nitrative modifications of the SERCA2a isoform of sarcoplasmic reticulum Ca(2+)-ATPase in diabetic rats. Biochimie 2016, 127, 144–152. [Google Scholar] [CrossRef]
- Houser, S.R. Role of RyR2 Phosphorylation in Heart Failure and Arrhythmias: Protein Kinase A-Mediated Hyperphosphorylation of the Ryanodine Receptor at Serine 2808 Does Not Alter Cardiac. Circ. Res. 2014, 114, 1320–1327. [Google Scholar] [CrossRef]
- Qin, F.; Siwik, D.A.; Lancel, S.; Zhang, J.; Kuster, G.M.; Luptak, I.; Wang, L.; Tong, X.; Kang, Y.J.; Cohen, R.A.; et al. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J. Am. Heart Assoc. 2013, 2, e000184. [Google Scholar] [CrossRef]
- Wongjaikam, S.; Kumfu, S.; Khamseekaew, J.; Chattipakorn, S.C.; Chattipakorn, N. Restoring the impaired cardiac calcium homeostasis and cardiac function in iron overload rats by the combined deferiprone and N-acetyl cysteine. Sci. Rep. 2017, 7, 44460. [Google Scholar] [CrossRef]
- Reddy, Y.N.V.; Andersen, M.J.; Obokata, M.; Koepp, K.E.; Kane, G.C.; Melenovsky, V.; Olson, T.P.; Borlaug, B.A. Arterial Stiffening with Exercise in Patients with Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 136–148. [Google Scholar] [CrossRef]
- Lim, G.B. New mouse model reveals nitrosative stress as a novel driver of HFpEF. Nat. Rev. Cardiol. 2019, 16, 383. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef]
- Pop, C.; Stefan, M.G.; Muntean, D.M.; Stoicescu, L.; Gal, A.F.; Kiss, B.; Morgovan, C.; Loghin, F.; Rochette, L.; Lauzier, B.; et al. Protective Effects of a Discontinuous Treatment with Alpha-Lipoic Acid in Obesity-Related Heart Failure with Preserved Ejection Fraction, in Rats. Antioxidants 2020, 9, 1073. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Velez, C.R.; Garcia-Castineiras, S.; Mendoza-Ramos, E.; Hernandez-Lopez, E. Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am. Heart J. 1996, 131, 146–152. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Benderdour, M.; Charron, G.; deBlois, D.; Comte, B.; Des Rosiers, C. Cardiac mitochondrial NADP(+)-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation—An event that precedes hypertrophy development. J. Biol. Chem. 2003, 278, 45154–45159. [Google Scholar] [CrossRef]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43. [Google Scholar] [CrossRef]
- Ma, S.; He, L.L.; Zhang, G.R.; Zuo, Q.J.; Wang, Z.L.; Zhai, J.; Zhang, T.; Wang, Y.; Ma, H.; Guo, Y. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn Schmiedeberg’s Arch. Pharmacol. 2022, 395, 945–962. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef]
- Paneni, F.; Sciarretta, S.; Costantino, S. Tackling myocardial oxidative stress with empagliflozin: Are we big enough to fight heart failure with preserved ejection fraction? Cardiovasc. Res. 2021, 117, 343–345. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Adarsh, K.; Kaur, H.; Mohan, V. Coenzyme Q10 (CoQ10) in isolated diastolic heart failure in hypertrophic cardiomyopathy (HCM). Biofactors 2008, 32, 145–149. [Google Scholar] [CrossRef]
- Samuel, T.Y.; Hasin, T.; Gotsman, I.; Weitzman, T.; Ben Ivgi, F.; Dadon, Z.; Asher, E.; Amir, O.; Glikson, M.; Alcalai, R.; et al. Coenzyme Q10 in the Treatment of Heart Failure with Preserved Ejection Fraction: A Prospective, Randomized, Double-Blind, Placebo-Controlled Trial. Drugs R&D 2021, 22, 25–33. [Google Scholar] [CrossRef]
- Sobirin, M.A.; Herry, Y.; Sofia, S.N.; Uddin, I.; Rifqi, S.; Tsutsui, H. Effects of coenzyme Q10 supplementation on diastolic function in patients with heart failure with preserved ejection fraction. Drug Discov. Ther. 2019, 13, 38–46. [Google Scholar] [CrossRef]
- Lopez-Lluch, G.; del Pozo-Cruz, J.; Sanchez-Cuesta, A.; Cortes-Rodriguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Ouchi, N.; Shibata, R.; Walsh, K. Adiponectin actions in the cardiovascular system. Cardiovasc. Res. 2007, 74, 11–18. [Google Scholar] [CrossRef]
- Sam, F.; Duhaney, T.A.S.; Sato, K.; Wilson, R.M.; Ohashi, K.; Sono-Romanelli, S.; Higuchi, A.; De Silva, D.S.; Qin, F.; Walsh, K.; et al. Adiponectin Deficiency, Diastolic Dysfunction, and Diastolic Heart Failure. Endocrinology 2010, 151, 322–331. [Google Scholar] [CrossRef]
- Essick, E.E.; Wilson, R.M.; Pimentel, D.R.; Shimano, M.; Baid, S.; Ouchi, N.; Sam, F. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS ONE 2013, 8, e68697. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.L.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Bayeva, M.; Khechaduri, A.; Puig, S.; Chang, H.-C.; Patial, S.; Blackshear, P.J.; Ardehali, H. mTOR Regulates Cellular Iron Homeostasis through Tristetraprolin. Cell Metab. 2012, 16, 645–657. [Google Scholar] [CrossRef]
- Sato, T.; Chang, H.C.; Bayeva, M.; Shapiro, J.S.; Ramos-Alonso, L.; Kouzu, H.; Jiang, X.; Liu, T.; Yar, S.; Sawicki, K.T.; et al. mRNA-binding protein tristetraprolin is essential for cardiac response to iron deficiency by regulating mitochondrial function. Proc. Natl. Acad. Sci. USA 2018, 115, E6291–E6300. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Ravassa, S.; López, B.; Moreno, M.; Beaumont, J.; José, G.S.; Querejeta, R.; Bayés-Genís, A.; Díez, J. Myocardial Remodeling in Hypertension. Hypertension 2018, 72, 549–558. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, C.; Gao, Z.; Chen, H.; Li, K.; Wang, L.; Zheng, Y.; Li, C.; Zhang, H.; Gong, M.; et al. SLC7A11/xCT Prevents Cardiac Hypertrophy by Inhibiting Ferroptosis. Cardiovasc. Drugs Ther. 2022, 36, 437–447. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Voors, A.A.; de Boer, R.A.; Solomon, S.D.; van Veldhuisen, D.J. Heart failure with preserved ejection fraction: From mechanisms to therapies. Eur. Heart J. 2018, 39, 2780–2792. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, E895–E1032. [Google Scholar] [CrossRef]
- Kremastinos, D.T.; Tsiapras, D.P.; Kostopoulou, A.G.; Hamodraka, E.S.; Chaidaroglou, A.S.; Kapsali, E.D. NT-proBNP levels and diastolic dysfunction in beta-thalassaemia major patients. Eur. J. Heart Fail. 2007, 9, 531–536. [Google Scholar] [CrossRef]
- Balkan, C.; Tuluce, S.Y.; Basol, G.; Tuluce, K.; Ay, Y.; Karapinar, D.Y.; Gurgun, C.; Bayindir, O.; Kavakli, K. Relation between NT-proBNP levels, iron overload, and early stage of myocardial dysfunction in β-thalassemia major patients. Echocardiography 2012, 29, 318–325. [Google Scholar] [CrossRef]
- Silvilairat, S.; Charoenkwan, P.; Saekho, S.; Tantiworawit, A.; Srichairatanakool, S. Early detection of ventricular dysfunction by tissue Doppler echocardiography related to cardiac iron overload in patients with thalassemia. Int. J. Cardiovasc. Imaging 2021, 37, 91–98. [Google Scholar] [CrossRef]
- Anderson, L.J.; Holden, S.; Davis, B.; Prescott, E.; Charrier, C.; Bunce, N.; Firmin, D.; Wonke, B.; Porter, J.; Walker, J.M.; et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur. Heart J. 2001, 22, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Westwood, M.A.; Wonke, B.; Maceira, A.M.; Prescott, E.; Walker, J.M.; Porter, J.; Pennell, D.J. Left ventricular diastolic function compared with T2* cardiovascular magnetic resonance for early detection of myocardial iron overload in thalassemia major. J. Magn. Reson. Imaging 2005, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kondur, A.K.; Li, T.; Vaitkevicius, P.; Afonso, L. Quantification of myocardial iron overload by cardiovascular magnetic resonance imaging T2* and review of the literature. Clin. Cardiol. 2009, 32, E55–E59. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283. [Google Scholar] [CrossRef]
- Jankowska, E.A.; von Haehling, S.; Anker, S.D.; Macdougall, I.C.; Ponikowski, P. Iron deficiency and heart failure: Diagnostic dilemmas and therapeutic perspectives. Eur. Heart J. 2013, 34, 816–829. [Google Scholar] [CrossRef]
- Okonko, D.O.; Mandal, A.K.; Missouris, C.G.; Poole-Wilson, P.A. Disordered iron homeostasis in chronic heart failure: Prevalence, predictors, and relation to anemia, exercise capacity, and survival. J. Am. Coll. Cardiol. 2011, 58, 1241–1251. [Google Scholar] [CrossRef]
- Kremastinos, D.T.; Farmakis, D. Iron overload cardiomyopathy in clinical practice. Circulation 2011, 124, 2253–2263. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Zhao, Z.; Zhou, X.; Yan, Y.; Shi, L.; Chen, J.; Fu, B.; Mao, J. Ferroptosis: The Potential Target in Heart Failure with Preserved Ejection Fraction. Cells 2022, 11, 2842. https://doi.org/10.3390/cells11182842
Li Q, Zhao Z, Zhou X, Yan Y, Shi L, Chen J, Fu B, Mao J. Ferroptosis: The Potential Target in Heart Failure with Preserved Ejection Fraction. Cells. 2022; 11(18):2842. https://doi.org/10.3390/cells11182842
Chicago/Turabian StyleLi, Qing, Zhiqiang Zhao, Xia Zhou, Yuting Yan, Lusi Shi, Jiafan Chen, Baohui Fu, and Jingyuan Mao. 2022. "Ferroptosis: The Potential Target in Heart Failure with Preserved Ejection Fraction" Cells 11, no. 18: 2842. https://doi.org/10.3390/cells11182842