
Ferroptosis: A New Development Trend in Periodontitis


{kind=link}
{kind=link}
Abstract
:1. Introduction
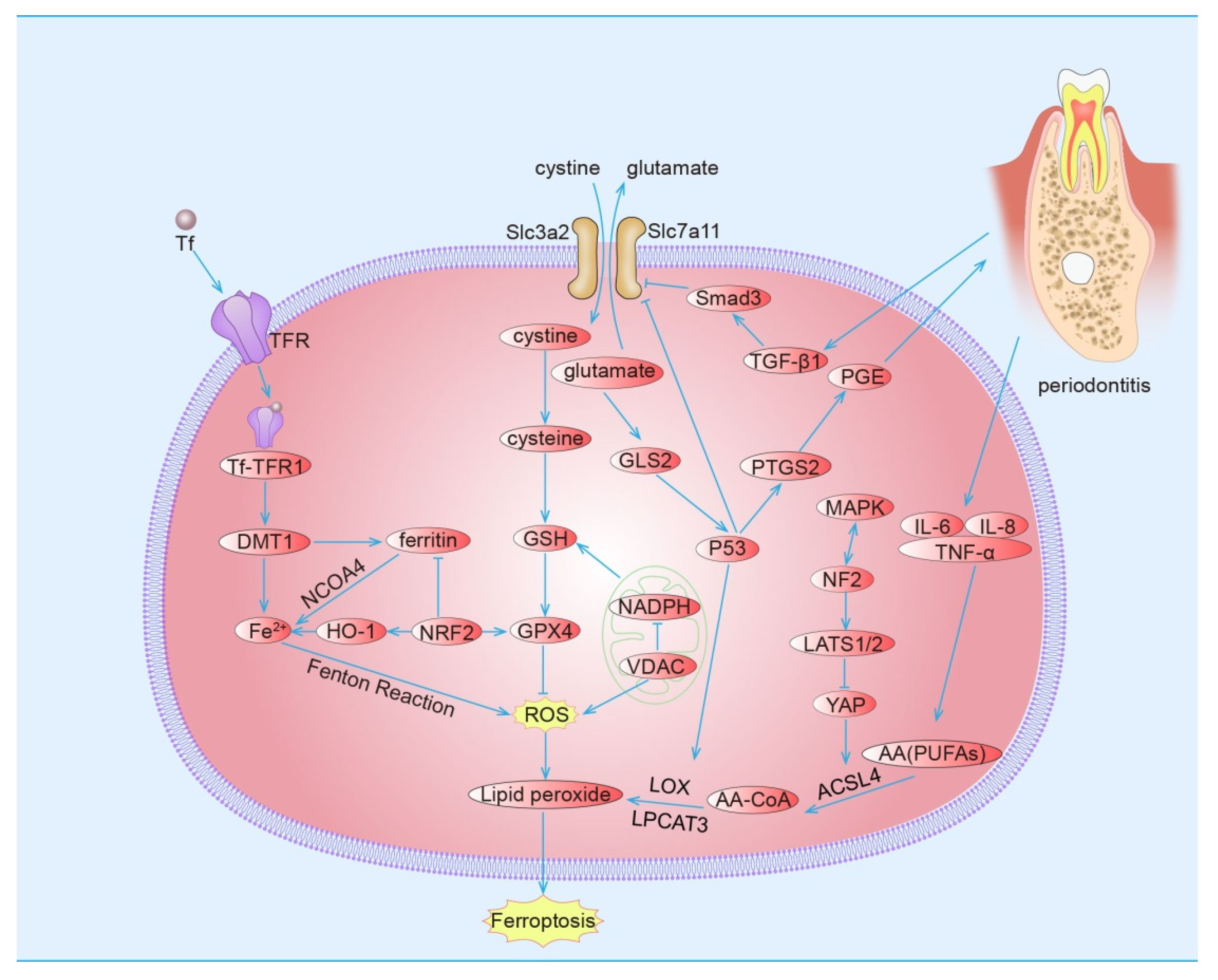
2. Ferroptosis
2.1. Biological Function of Iron
2.2. Characteristics of Ferroptosis
2.3. Pathways of Ferroptosis
2.3.1. Induction of Ferroptosis by Inhibiting the Cystine/Glutamate Transport Receptor (System Xc−)
2.3.2. Induction of Ferroptosis by Voltage-Dependent Anion Channel (VDAC)
2.3.3. Induction of Ferroptosis by Ferritin Phagocytosis
2.3.4. Induction of Ferroptosis by the P53 Pathway
2.3.5. Induction of Ferroptosis by the MAPK Pathway
2.3.6. Inhibiting Ferroptosis by the Hippo Pathway
2.3.7. Inhibiting Ferroptosis by Nuclear Factor Erythroid 2-Related Factor 2 (NRF2)
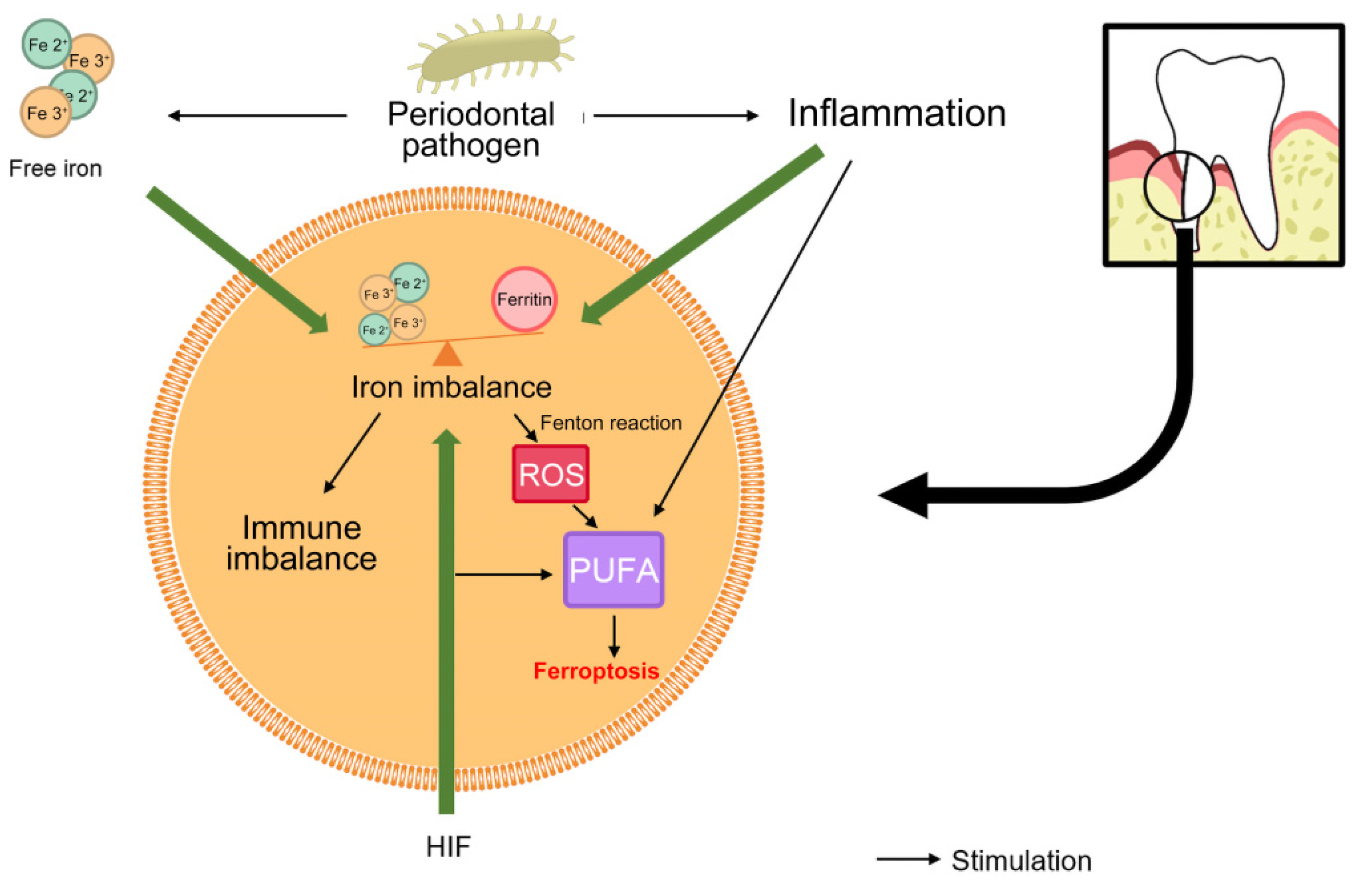
3. Ferroptosis and Periodontitis
3.1. Iron Overload
3.2. Lipid Peroxidation
3.3. Ferritin Phagocytosis
3.4. MAPK Pathway
3.5. P53 Pathway
3.6. Transforming Growth Factor β
4. Clinical Application and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, X.; Fang, X.; Wang, F.; Min, J. The role of ferroptosis in chronic diseases. J. Zhejiang Univ. Med. Sci. 2020, 49, 44–57. [Google Scholar] [CrossRef]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y. The Protective Effects of Cryptochlorogenic Acid on β-Cells Function in Diabetes in vivo and vitro via Inhibition of Ferroptosis. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1921–1931. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin—A peptide hormone at the interface of innate immunity and iron metabolism. Curr. Top. Microbiol. Immunol. 2006, 306, 183–198. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G199–G203. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B. Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef]
- Hoeft, K.; Bloch, D.B.; Graw, J.A.; Malhotra, R.; Ichinose, F.; Bagchi, A. Iron Loading Exaggerates the Inflammatory Response to the Toll-like Receptor 4 Ligand Lipopolysaccharide by Altering Mitochondrial Homeostasis. Anesthesiology 2017, 127, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Boyer, E.; Le Gall-David, S.; Martin, B.; Fong, S.B.; Loréal, O.; Deugnier, Y.; Bonnaure-Mallet, M.; Meuric, V. Increased transferrin saturation is associated with subgingival microbiota dysbiosis and severe periodontitis in genetic haemochromatosis. Sci. Rep. 2018, 8, 15532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.N.; Yang, Y.Z.; Feng, Y.Z. Serum and salivary ferritin and Hepcidin levels in patients with chronic periodontitis and type 2 diabetes mellitus. BMC Oral Health 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Bains, V.K.; Bains, R. The antioxidant master glutathione and periodontal health. Dent. Res. J. 2015, 12, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.M.; Brock, G.R.; Matthews, J.B.; Chapple, I.L. Crevicular fluid glutathione levels in periodontitis and the effect of non-surgical therapy. J. Clin. Periodontol. 2010, 37, 17–23. [Google Scholar] [CrossRef]
- Oliveira, F.; Rocha, S.; Fernandes, R. Iron metabolism: From health to disease. J. Clin. Lab. Anal. 2014, 28, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Frazer, D.M.; Anderson, G.J. The regulation of iron transport. Biofactors 2014, 40, 206–214. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.; Kowdley, K.V. Review article: The iron overload syndromes. Aliment. Pharmacol. Ther. 2012, 35, 876–893. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559s–1566s. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, P.A.; Reed, G.H. The ubiquity of iron. ACS Chem. Biol. 2012, 7, 1477–1481. [Google Scholar] [CrossRef]
- Chew, K.C.; Ang, E.-T.; Tai, Y.K.; Tsang, F.; Lo, S.Q.; Ong, E.; Ong, W.-Y.; Shen, H.-M.; Lim, K.-L.; Dawson, V.L. Enhanced autophagy from chronic toxicity of iron and mutant A53T α-synuclein: Implications for neuronal cell death in Parkinson disease. J. Biol. Chem. 2011, 286, 33380–33389. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Lin, P.P.; Lin, C.Y.; Lin, C.H.; Huang, C.H.; Huang, Y.J.; Lane, H.Y. Decreased mRNA expression for the two subunits of system xc(-), SLC3A2 and SLC7A11, in WBC in patients with schizophrenia: Evidence in support of the hypo-glutamatergic hypothesis of schizophrenia. J. Psychiatr. Res. 2016, 72, 58–63. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Skonieczna, M.; Cieslar-Pobuda, A.; Saenko, Y.; Foksinski, M.; Olinski, R.; Rzeszowska-Wolny, J.; Wiechec, E. The impact of DIDS-induced inhibition of voltage-dependent anion channels (VDAC) on cellular response of lymphoblastoid cells to ionizing radiation. Med. Chem. 2017, 13, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N. VDAC-Tubulin, an Anti-Warburg Pro-Oxidant Switch. Front. Oncol. 2017, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemasters, J.J. Evolution of voltage-dependent anion channel function: From molecular sieve to governator to actuator of ferroptosis. Front. Oncol. 2017, 7, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 89–100. [Google Scholar]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [Green Version]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026146. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid metabolism and ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, B.; Ahmad, K.; Roos, J.; Lehmann, C.; Chiba, T.; Ulrich-Rückert, S.; Smeenk, L.; van Heeringen, S.; Maier, T.J.; Groner, B.; et al. 5-Lipoxygenase is a direct p53 target gene in humans. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 1003–1016. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Hattori, K.; Ishikawa, H.; Sakauchi, C.; Takayanagi, S.; Naguro, I.; Ichijo, H. Cold stress-induced ferroptosis involves the ASK 1-p38 pathway. EMBO Rep. 2017, 18, 2067–2078. [Google Scholar] [CrossRef]
- Tang, Q.; Bai, L.; Zou, Z.; Meng, P.; Xia, Y.; Cheng, S.; Mu, S.; Zhou, J.; Wang, X.; Qin, X.; et al. Ferroptosis is newly characterized form of neuronal cell death in response to arsenite exposure. Neurotoxicology 2018, 67, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhen, C.; Liu, J.; Shang, P. β-Phenethyl isothiocyanate induces cell death in human osteosarcoma through altering iron metabolism, disturbing the redox balance, and activating the MAPK signaling pathway. Oxidative Med. Cell. Longev. 2020, 2020, 5021983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Han, W.; Qin, A.; Wang, Z.; Xu, J.; Qian, Y. The emerging role of Hippo signaling pathway in regulating osteoclast formation. J. Cell. Physiol. 2018, 233, 4606–4617. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, C.; Yang, L.; Zeng, Z.; Zeng, M.; Jiang, W.; Liu, L.; Zhao, M. Role of Nrf2/HO-1 signal axis in the mechanisms for oxidative stress-relevant diseases. Zhong Nan Da Xue Xue Bao Yi Xue Ban = J. Cent. South Univ. Med. Sci. 2019, 44, 74–80. [Google Scholar] [CrossRef]
- Gou, Z.; Su, X.; Hu, X.; Zhou, Y.; Huang, L.; Fan, Y.; Li, J.; Lu, L. Melatonin improves hypoxic-ischemic brain damage through the Akt/Nrf2/Gpx4 signaling pathway. Brain Res. Bull. 2020, 163, 40–48. [Google Scholar] [CrossRef]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.-T.; Yen, C.-J.; Chang, C.-C.; Huang, K.-T.; Chen, K.-H.; Zhang, R.-Y.; Lee, P.-Y.; Miaw, S.-C.; Huang, J.-W.; Chiang, C.-K. Ferritin heavy chain mediates the protective effect of heme oxygenase-1 against oxidative stress. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 2506–2517. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef] [Green Version]
- Costa, S.A.; Ribeiro, C.C.C.; Moreira, A.R.O.; Carvalho Souza, S.F. High serum iron markers are associated with periodontitis in post-menopausal women: A population-based study (NHANES III). J. Clin. Periodontol. 2022, 49, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. The role of crevicular fluid iron in periodontal disease. J. Periodontol. 1985, 56, 22–27. [Google Scholar] [CrossRef]
- Liu, L.Y.; McGregor, N.; Wong, B.K.; Butt, H.; Darby, I.B. The association between clinical periodontal parameters and free haem concentration within the gingival crevicular fluid: A pilot study. J. Periodontal Res. 2016, 51, 86–94. [Google Scholar] [CrossRef]
- Olczak, T.; Simpson, W.; Liu, X.; Genco, C.A. Iron and heme utilization in Porphyromonas gingivalis. FEMS Microbiol. Rev. 2005, 29, 119–144. [Google Scholar] [CrossRef] [Green Version]
- Ratnayake, D.B.; Wai, S.N.; Shi, Y.; Amako, K.; Nakayama, H.; Nakayama, K. Ferritin from the obligate anaerobe Porphyromonas gingivalis: Purification, gene cloning and mutant studiesThe GenBank accession number for the sequence reported in this paper is AB016086. Microbiology 2000, 146, 1119–1127. [Google Scholar] [CrossRef]
- Lewis, J.P. Metal uptake in host-pathogen interactions: Role of iron in Porphyromonas gingivalis interactions with host organisms. Periodontology 2000 2010, 52, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afacan, B.; Öztürk, V.; Paşalı, Ç.; Bozkurt, E.; Köse, T.; Emingil, G. Gingival crevicular fluid and salivary HIF-1α, VEGF, and TNF-α levels in periodontal health and disease. J. Periodontol. 2019, 90, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, R.C.; Costa, A.d.L.L.; Freitas, R.d.A.; Bezerra, B.A.d.A.; Santos, B.R.M.d.; Pinto, L.P.; Gurgel, B.C.d.V. Immunoexpression of HIF-1α and VEGF in periodontal disease and healthy gingival tissues. Braz. Dent. J. 2016, 27, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takedachi, M.; Yamamoto, S.; Kawasaki, K.; Shimomura, J.; Murata, M.; Morimoto, C.; Hirai, A.; Kawakami, K.; Bhongsatiern, P.; Iwayama, T.; et al. Reciprocal role of PLAP-1 in HIF-1α-mediated responses to hypoxia. J. Periodontal Res. 2022, 57, 470–478. [Google Scholar] [CrossRef]
- Ezzeddini, R.; Taghikhani, M.; Salek Farrokhi, A.; Somi, M.H.; Samadi, N.; Esfahani, A.; Rasaee, M.J. Downregulation of fatty acid oxidation by involvement of HIF-1α and PPARγ in human gastric adenocarcinoma and related clinical significance. J. Physiol. Biochem. 2021, 77, 249–260. [Google Scholar] [CrossRef]
- Ramirez-Tortosa, M.C.; Quiles, J.L.; Battino, M.; Granados, S.; Morillo, J.M.; Bompadre, S.; Newman, H.N.; Bullon, P. Periodontitis is associated with altered plasma fatty acids and cardiovascular risk markers. Nutr. Metab. Cardiovasc. Dis. NMCD 2010, 20, 133–139. [Google Scholar] [CrossRef]
- Zaloga, G.P. Narrative review of n-3 polyunsaturated fatty acid supplementation upon immune functions, resolution molecules and lipid peroxidation. Nutrients 2021, 13, 662. [Google Scholar] [CrossRef]
- Kantarci, A.; Oyaizu, K.; Van Dyke, T.E. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: Findings from localized aggressive periodontitis. J. Periodontol. 2003, 74, 66–75. [Google Scholar] [CrossRef]
- Hirschfeld, J.; White, P.C.; Milward, M.R.; Cooper, P.R.; Chapple, I.L.C. Modulation of Neutrophil Extracellular Trap and Reactive Oxygen Species Release by Periodontal Bacteria. Infect. Immun. 2017, 85, e00297-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Andrukhov, O.; Rausch-Fan, X. Oxidative Stress and Antioxidant System in Periodontitis. Front. Physiol. 2017, 8, 910. [Google Scholar] [CrossRef]
- Borges Jr, I.; Moreira, E.A.M.; De Oliveira, T.B.; da Silva, M.B.S.; Fröde, T.S. Proinflammatory and oxidative stress markers in patients with periodontal disease. Mediat. Inflamm. 2007, 2007, 45794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, H.E.; Lindhe, J. Conditions and pathological features of rapidly destructive, experimental periodontitis in dogs. J. Periodontol. 1980, 51, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Chathoth, K.; Martin, B.; Cornelis, P.; Yvenou, S.; Bonnaure-Mallet, M.; Baysse, C. The events that may contribute to subgingival dysbiosis: A focus on the interplay between iron, sulfide and oxygen. FEMS Microbiol. Lett. 2020, 367, fnaa100. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.A.; Moreira, A.R.O.; Costa, C.P.S.; Carvalho Souza, S.d.F. Iron overload and periodontal status in patients with sickle cell anaemia: A case series. J. Clin. Periodontol. 2020, 47, 668–675. [Google Scholar] [CrossRef]
- Guo, W.; Zhao, Y.; Li, H.; Lei, L. NCOA4-mediated ferritinophagy promoted inflammatory responses in periodontitis. J. Periodontal Res. 2021, 56, 523–534. [Google Scholar] [CrossRef]
- Leung, K.-P.; Folk, S.P. Effects of porphyrins and inorganic iron on the growth of Prevotella intermedia. FEMS Microbiol. Lett. 2002, 209, 15–21. [Google Scholar] [CrossRef]
- DeCarlo, A.A.; Paramaesvaran, M.; Yun, P.L.; Collyer, C.; Hunter, N. Porphyrin-mediated binding to hemoglobin by the HA2 domain of cysteine proteinases (gingipains) and hemagglutinins from the periodontal pathogen Porphyromonas gingivalis. J. Bacteriol. 1999, 181, 3784–3791. [Google Scholar] [CrossRef] [Green Version]
- Goulet, V.; Britigan, B.; Nakayama, K.; Grenier, D. Cleavage of human transferrin by Porphyromonas gingivalis gingipains promotes growth and formation of hydroxyl radicals. Infect. Immun. 2004, 72, 4351–4356. [Google Scholar] [CrossRef] [Green Version]
- Guan, S.M.; Nagata, H.; Shizukuishi, S.; Wu, J.Z. Degradation of human hemoglobin by Prevotella intermedia. Anaerobe 2006, 12, 279–282. [Google Scholar] [CrossRef]
- Ke, J.Y.; Cen, W.J.; Zhou, X.Z.; Li, Y.R.; Kong, W.D.; Jiang, J.W. Iron overload induces apoptosis of murine preosteoblast cells via ROS and inhibition of AKT pathway. Oral Dis. 2017, 23, 784–794. [Google Scholar] [CrossRef]
- Wu, D.; Lin, Z.; Zhang, S.; Cao, F.; Liang, D.; Zhou, X. Decreased hemoglobin concentration and iron metabolism disorder in periodontitis: Systematic review and meta-analysis. Front. Physiol. 2020, 10, 1620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Chen, L.; Gao, X.; Shen, S.; Sheng, W.; Min, J.; Wang, F. The role of iron homeostasis in remodeling immune function and regulating inflammatory disease. Sci. Bull. 2021, 66, 1806–1816. [Google Scholar] [CrossRef]
- Han, Y.; Huang, W.; Meng, H.; Zhan, Y.; Hou, J. Pro-inflammatory cytokine interleukin-6-induced hepcidin, a key mediator of periodontitis-related anemia of inflammation. J. Periodontal Res. 2021, 56, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.A.M.; Casanovas, R.C.; Rodrigues, V.P.; Pereira, A.F.V.; Ferreira, T.C.A.; Nascimento, F.; Nascimento, J.R.D.; Gomes-Filho, I.S.; Bastos, M.G.; Pereira, A.L.A. The effect of nonsurgical periodontal therapy on hepcidin and on inflammatory and iron marker levels. Braz. Oral Res. 2019, 33, e055. [Google Scholar] [CrossRef] [Green Version]
- Akalin, F.A.; Baltacioğlu, E.; Alver, A.; Karabulut, E. Lipid peroxidation levels and total oxidant status in serum, saliva and gingival crevicular fluid in patients with chronic periodontitis. J. Clin. Periodontol. 2007, 34, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Inonu, E.; Hakki, S.S.; Kayis, S.A.; Nielsen, F.H. The Association Between Some Macro and Trace Elements in Saliva and Periodontal Status. Biol. Trace Elem. Res. 2020, 197, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, M.; Li, Z.; Sa, R.; Chu, Q.; Zhang, Q.; Zhang, H.; Tang, W.; Zhang, M.; Yin, H. Mass spectrometry-based metabolomic profiling identifies alterations in salivary redox status and fatty acid metabolism in response to inflammation and oxidative stress in periodontal disease. Free Radic. Biol. Med. 2014, 70, 223–232. [Google Scholar] [CrossRef]
- Gaur, S.; Agnihotri, R. Trace Mineral Micronutrients and Chronic Periodontitis-a Review. Biol. Trace Elem. Res. 2017, 176, 225–238. [Google Scholar] [CrossRef]
- Fentoğlu, Ö.; Kırzıoğlu, F.Y.; Bulut, M.T.; Kumbul Doğuç, D.; Kulaç, E.; Önder, C.; Günhan, M. Evaluation of lipid peroxidation and oxidative DNA damage in patients with periodontitis and hyperlipidemia. J. Periodontol. 2015, 86, 682–688. [Google Scholar] [CrossRef]
- Toczewska, J.; Maciejczyk, M.; Konopka, T.; Zalewska, A. Total oxidant and antioxidant capacity of gingival crevicular fluid and saliva in patients with periodontitis: Review and clinical study. Antioxidants 2020, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiao, X.; Zhang, C.; Hou, J.; Qi, S. Long non-coding RNA LINC00616 promotes ferroptosis of periodontal ligament stem cells via the microRNA-370/transferrin receptor axis. Bioengineered 2022, 13, 13070–13081. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Aust, S.D. The role of iron in the initiation of lipid peroxidation. Chem. Phys. Lipids 1987, 44, 191–208. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrin, G.L.; Strauss, F.J.; Benfatti, C.A.M.; Maia, L.C.; Gruber, R. Effects of short-chain fatty acids on human oral epithelial cells and the potential impact on periodontal disease: A systematic review of in vitro studies. Int. J. Mol. Sci. 2020, 21, 4895. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Meng, H.; Gao, X.; Feng, L.; Xu, L. Analysis of short chain fatty acids in gingival crevicular fluid of patients with aggressive periodontitis. Zhonghua Kou Qiang Yi Xue Za Zhi = Zhonghua Kouqiang Yixue Zazhi = Chin. J. Stomatol. 2008, 43, 664–667. [Google Scholar]
- Zhao, Y.; Li, J.; Guo, W.; Li, H.; Lei, L. Periodontitis-level butyrate-induced ferroptosis in periodontal ligament fibroblasts by activation of ferritinophagy. Cell Death Discov. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.Z.; Hu, J.X.; Wu, H.; Li, Y.L.; Chen, H.L.; Bai, H.; Hai, C.X. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic. Biol. Med. 2011, 51, 539–551. [Google Scholar] [CrossRef]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [Green Version]
- Dörner, M.H.; Silverstone, A.; Nishiya, K.; de Sostoa, A.; Munn, G.; de Sousa, M. Ferritin synthesis by human T lymphocytes. Science 1980, 209, 1019–1021. [Google Scholar] [CrossRef]
- Huang, W.; Zhan, Y.; Zheng, Y.; Han, Y.; Hu, W.; Hou, J. Up-regulated ferritin in periodontitis promotes inflammatory cytokine expression in human periodontal ligament cells through transferrin receptor via ERK/P38 MAPK pathways. Clin. Sci. 2019, 133, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.; Mertens, C.; Meier, J.K.; Fuhrmann, D.C.; Brüne, B.; Jung, M. Strategies to Interfere with Tumor Metabolism through the Interplay of Innate and Adaptive Immunity. Cells 2019, 8, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adonogianaki, E.; Mooney, J.; Kinane, D.F. The ability of gingival crevicular fluid acute phase proteins to distinguish healthy, gingivitis and periodontitis sites. J. Clin. Periodontol. 1992, 19, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Medikeri, R.S.; Lele, S.V.; Mali, P.P.; Jain, P.M.; Darawade, D.A.; Medikeri, M.R. Effect of Camphylobacter rectus on Serum Iron and Transferrin- In-Vivo Findings. J. Clin. Diagn. Res. JCDR 2015, 9, ZC26–ZC30. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, H.; Yao, G.; Qiao, P.; Li, L.; Wu, S. Therapeutic potential of iron chelators on osteoporosis and their cellular mechanisms. Biomed. Pharmacother. 2021, 137, 111380. [Google Scholar] [CrossRef]
- Memmert, S.; Gölz, L.; Pütz, P.; Jäger, A.; Deschner, J.; Appel, T.; Baumgarten, G.; Rath-Deschner, B.; Frede, S.; Götz, W. Regulation of p53 under hypoxic and inflammatory conditions in periodontium. Clin. Oral Investig. 2016, 20, 1781–1789. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [Green Version]
- Sommakia, S.; Baker, O.J. Regulation of inflammation by lipid mediators in oral diseases. Oral Dis. 2017, 23, 576–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, W.D.; Kim, S.K.; Moon, D.H.; Lee, S.J. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 406. [Google Scholar] [CrossRef] [PubMed]
- Groenink, J.; Walgreen-Weterings, E.; Nazmi, K.; Bolscher, J.G.; Veerman, E.C.; van Winkelhoff, A.J.; Nieuw Amerongen, A.V. Salivary lactoferrin and low-Mr mucin MG2 in Actinobacillus actinomycetemcomitans-associated periodontitis. J. Clin. Periodontol. 1999, 26, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Ghallab, N.A.; Hamdy, E.; Shaker, O.G. Malondialdehyde, superoxide dismutase and melatonin levels in gingival crevicular fluid of aggressive and chronic periodontitis patients. Aust. Dent. J. 2016, 61, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-Y.; Meng, X.; Wang, Y.-R.; Wang, Q.-Q.; He, X.; Sun, X.-Y.; Cheng, N.; Zhang, L. PRDX6 alleviates lipopolysaccharide-induced inflammation and ferroptosis in periodontitis. Acta Odontol. Scand. 2022, 80, 535–546. [Google Scholar] [CrossRef]
- Chung, J.H.; Kim, Y.S.; Noh, K.; Lee, Y.M.; Chang, S.W.; Kim, E.C. Deferoxamine promotes osteoblastic differentiation in human periodontal ligament cells via the nuclear factor erythroid 2-related factor-mediated antioxidant signaling pathway. J. Periodontal Res. 2014, 49, 563–573. [Google Scholar] [CrossRef]
- Grenier, D.; Huot, M.P.; Mayrand, D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 2000, 44, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Jin, H.; Da, J.; Zhang, K.; Liu, L.; Guo, Y.; Zhang, W.; Geng, Y.; Liu, X.; Zhang, J. Role of ferroptosis-related genes in periodontitis based on integrated bioinformatics analysis. PLoS ONE 2022, 17, e0271202. [Google Scholar] [CrossRef]
- Greabu, M.; Giampieri, F.; Imre, M.M.; Mohora, M.; Totan, A.; Pituru, S.M.; Ionescu, E. Autophagy, One of the Main Steps in Periodontitis Pathogenesis and Evolution. Molecules 2020, 25, 4338. [Google Scholar] [CrossRef]
- Xu, G.; Li, X.; Zhu, Z.; Wang, H.; Bai, X. Iron Overload Induces Apoptosis and Cytoprotective Autophagy Regulated by ROS Generation in Mc3t3-E1 Cells. Biol. Trace Elem. Res. 2021, 199, 3781–3792. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Ma, S.; Deng, J.; Jiang, X.; Ma, F.; Li, Z. Ferroptosis: A New Development Trend in Periodontitis. Cells 2022, 11, 3349. https://doi.org/10.3390/cells11213349
Chen K, Ma S, Deng J, Jiang X, Ma F, Li Z. Ferroptosis: A New Development Trend in Periodontitis. Cells. 2022; 11(21):3349. https://doi.org/10.3390/cells11213349
Chicago/Turabian StyleChen, Kexiao, Shuyuan Ma, Jianwen Deng, Xinrong Jiang, Fengyu Ma, and Zejian Li. 2022. "Ferroptosis: A New Development Trend in Periodontitis" Cells 11, no. 21: 3349. https://doi.org/10.3390/cells11213349