
Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review

 , , ,
, , ,
Abstract
:1. Introduction
2. NAFLD Epidemiology and Pathogenesis
3. Endothelial Cells in the Pathogenesis of NAFLD
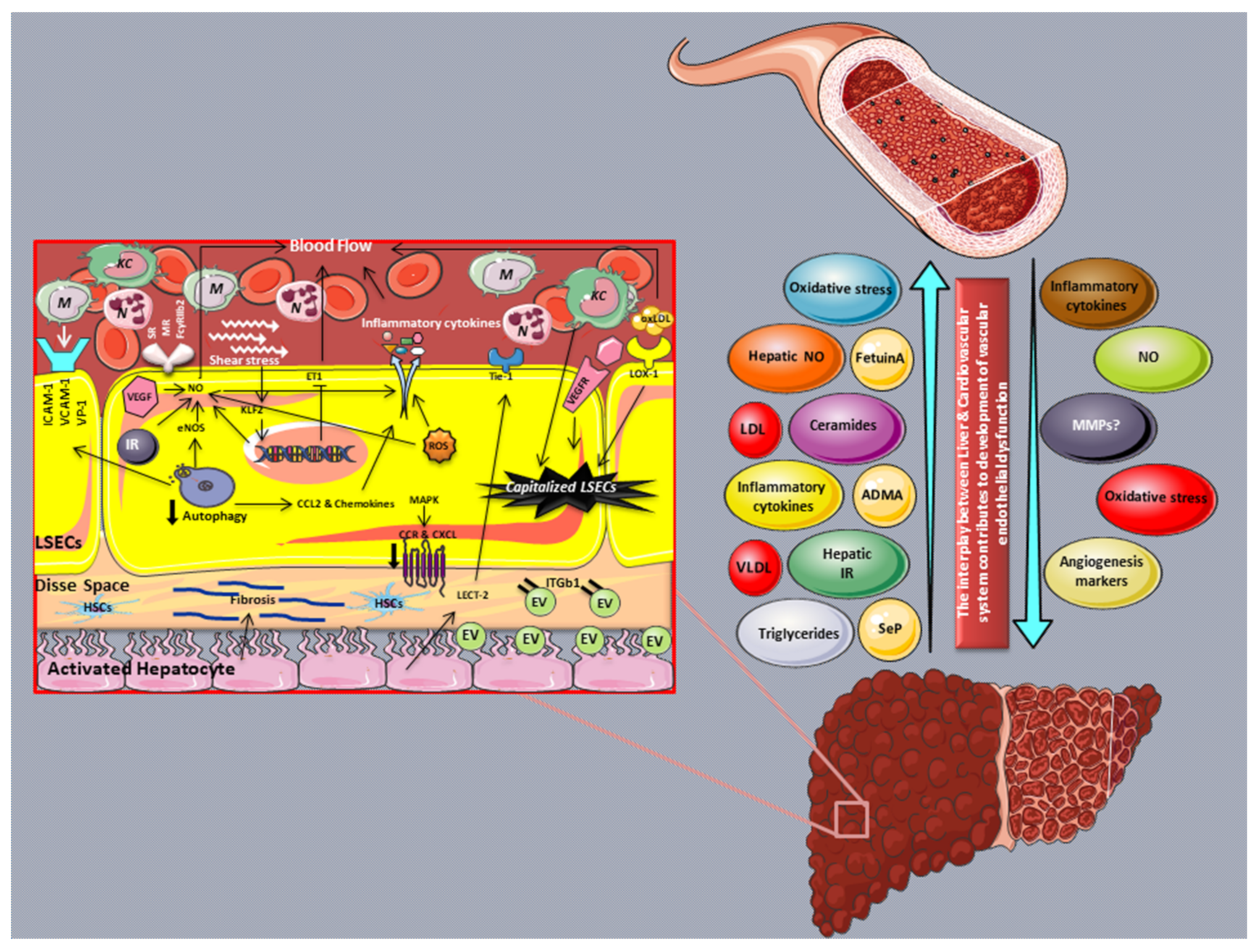
3.1. LSECs and Their Role in the Regulation of Blood Flow and Hepatic Microcirculation
3.2. Capillarization of LSECs in NAFLD
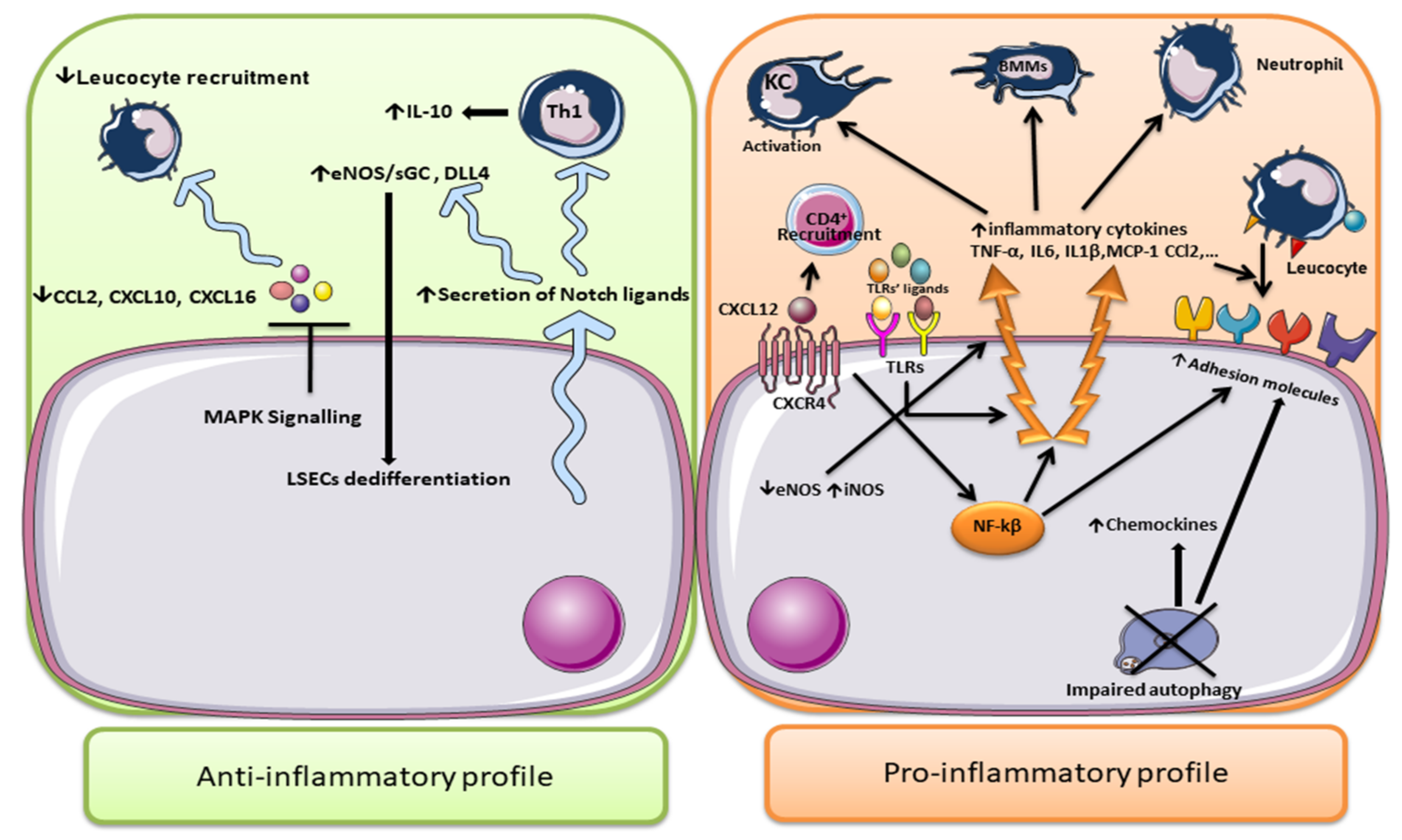
3.3. LSECs in the Regulation of Inflammation in NAFLD
3.4. LSECs in NAFLD-Related HCC
3.5. Novel Markers of LSEC Dysfunction
4. Endothelial Cells as Therapeutic Targets for the Treatment of NAFLD
Targeting LSECs in Experimental Studies
5. Vascular Endothelial Dysfunction and NAFLD: Human Studies
Liver-Secreted Molecules Leading to Vascular Endothelial Dysfunction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.K.; Peng, Z.G. Targeting Liver Sinusoidal Endothelial Cells: An Attractive Therapeutic Strategy to Control Inflammation in Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2021, 12, 655557. [Google Scholar] [CrossRef] [PubMed]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar] [PubMed]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 Inhibitors in NAFLD: Expanding Their Role beyond Diabetes and Cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar] [CrossRef]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Dela Cruz, R.; Theise, N.; Nieto, N. High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernandez-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef]
- Pasarin, M.; La Mura, V.; Gracia-Sancho, J.; Garcia-Caldero, H.; Rodriguez-Vilarrupla, A.; Garcia-Pagan, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 2012, 7, e32785. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar]
- Francque, S.; Laleman, W.; Verbeke, L.; Van Steenkiste, C.; Casteleyn, C.; Kwanten, W.; Van Dyck, C.; D’Hondt, M.; Ramon, A.; Vermeulen, W.; et al. Increased intrahepatic resistance in severe steatosis: Endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab. Investig. 2012, 92, 1428–1439. [Google Scholar] [CrossRef]
- Ozturk, K.; Uygun, A.; Guler, A.K.; Demirci, H.; Ozdemir, C.; Cakir, M.; Sakin, Y.S.; Turker, T.; Sari, S.; Demirbas, S.; et al. Nonalcoholic fatty liver disease is an independent risk factor for atherosclerosis in young adult men. Atherosclerosis 2015, 240, 380–386. [Google Scholar] [CrossRef]
- Bugianesi, E.; Leone, N.; Vanni, E.; Marchesini, G.; Brunello, F.; Carucci, P.; Musso, A.; De Paolis, P.; Capussotti, L.; Salizzoni, M.; et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002, 123, 134–140. [Google Scholar] [CrossRef]
- Hui, J.M.; Kench, J.G.; Chitturi, S.; Sud, A.; Farrell, G.C.; Byth, K.; Hall, P.; Khan, M.; George, J. Long-term outcomes of cirrhosis in nonalcoholic steatohepatitis compared with hepatitis C. Hepatology 2003, 38, 420–427. [Google Scholar] [CrossRef]
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, Y.J.; You, Y.H.; Moon, M.K.; Yoon, K.H.; Ahn, Y.B.; Ko, S.H. Effect of sodium-glucose cotransporter 2 inhibitor, empagliflozin, and alpha-glucosidase inhibitor, voglibose, on hepatic steatosis in an animal model of type 2 diabetes. J. Cell. Biochem. 2019, 120, 8534–8546. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Mahady, S.E.; George, J. Exercise and diet in the management of nonalcoholic fatty liver disease. Metabolism 2016, 65, 1172–1182. [Google Scholar] [CrossRef]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE((−/−)) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, K.; Kong, A.; Zhou, Y.; Chen, D.; Gu, J.; Shi, H. Dysregulation of autophagy acts as a pathogenic mechanism of non-alcoholic fatty liver disease (NAFLD) induced by common environmental pollutants. Ecotoxicol. Environ. Saf. 2021, 217, 112256. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Yang, J.; Zhang, H.; Li, X. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J. Hepatol. 2014, 60, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, J.; Man, K.; Li, X.; Du, J.; Chu, E.S.; Go, M.Y.; Sung, J.J.; Yu, J. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J. Hepatol. 2016, 64, 160–170. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Lee, Y.A.; Friedman, S.L. Inflammatory and fibrotic mechanisms in NAFLD-Implications for new treatment strategies. J. Intern. Med. 2022, 291, 11–31. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111S, 154170. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and increased risk of cardiovascular disease: Clinical associations, pathophysiological mechanisms and pharmacological implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef]
- Velarde-Ruiz Velasco, J.A.; Garcia-Jimenez, E.S.; Garcia-Zermeno, K.R.; Morel-Cerda, E.C.; Aldana-Ledesma, J.M.; Castro-Narro, G.E.; Cerpa-Cruz, S.; Tapia-Calderon, D.K.; Mercado-Jauregui, L.A.; Contreras-Omana, R. Extrahepatic complications of non-alcoholic fatty liver disease: Its impact beyond the liver. Rev. Gastroenterol. Mex. 2019, 84, 472–481. [Google Scholar] [CrossRef]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef]
- Ogresta, D.; Mrzljak, A.; Cigrovski Berkovic, M.; Bilic-Curcic, I.; Stojsavljevic-Shapeski, S.; Virovic-Jukic, L. Coagulation and Endothelial Dysfunction Associated with NAFLD: Current Status and Therapeutic Implications. J. Clin. Transl. Hepatol. 2022, 10, 339–355. [Google Scholar] [CrossRef]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Junior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Pi, X.; Xie, L.; Patterson, C. Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ. Res. 2018, 123, 477–494. [Google Scholar] [CrossRef]
- McCuskey, R.S. The hepatic microvascular system in health and its response to toxicants. Anat. Rec. 2008, 291, 661–671. [Google Scholar] [CrossRef]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Sorensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrod, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [CrossRef]
- Maslak, E.; Gregorius, A.; Chlopicki, S. Liver sinusoidal endothelial cells (LSECs) function and NAFLD; NO-based therapy targeted to the liver. Pharmacol. Rep. 2015, 67, 689–694. [Google Scholar] [CrossRef]
- Schierwagen, R.; Uschner, F.E.; Magdaleno, F.; Klein, S.; Trebicka, J. Rationale for the use of statins in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G407–G412. [Google Scholar] [CrossRef]
- Schleicher, J.; Guthke, R.; Dahmen, U.; Dirsch, O.; Holzhuetter, H.G.; Schuster, S. A theoretical study of lipid accumulation in the liver-implications for nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2014, 1841, 62–69. [Google Scholar] [CrossRef]
- Farrell, G.C.; Teoh, N.C.; McCuskey, R.S. Hepatic microcirculation in fatty liver disease. Anat. Rec. 2008, 291, 684–692. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, R.S.; Nishida, J.; Eguchi, H.; McDonnell, D.; Baker, G.L.; Ekataksin, W.; Krasovich, M.A.; Rudi, V.; Seitz, H.K.; Urbaschek, B.; et al. Role of endotoxin in the hepatic microvascular inflammatory response to ethanol. J. Gastroenterol. Hepatol. 1995, 10 (Suppl. 1), S18–S23. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, S.; Yang, W.; Winslet, M.C.; Seifalian, A.M. The role of nitric oxide in the modulation of hepatic microcirculation and tissue oxygenation in an experimental model of hepatic steatosis. Microvasc. Res. 2005, 70, 129–136. [Google Scholar] [CrossRef]
- Pereira, E.; Silvares, R.R.; Flores, E.E.I.; Rodrigues, K.L.; Daliry, A. Pyridoxamine improves metabolic and microcirculatory complications associated with nonalcoholic fatty liver disease. Microcirculation 2020, 27, e12603. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.; Silvares, R.R.; Flores, E.E.I.; Rodrigues, K.L.; Ramos, I.P.; da Silva, I.J.; Machado, M.P.; Miranda, R.A.; Pazos-Moura, C.C.; Goncalves-de-Albuquerque, C.F.; et al. Hepatic microvascular dysfunction and increased advanced glycation end products are components of non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0179654. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.K.; Zhang, X.Y.; Zimmermann, A.; Davis, G.; Wheatley, A.M. Effect of ischemia-reperfusion injury on the microcirculation of the steatotic liver of the Zucker rat. Transplantation 2001, 72, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Rosenstengel, S.; Stoeppeler, S.; Bahde, R.; Spiegel, H.U.; Palmes, D. Type of steatosis influences microcirculation and fibrogenesis in different rat strains. J. Investig. Surg. 2011, 24, 273–282. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ito, Y.; Wijeweera, J.; Liu, J.; Malle, E.; Farhood, A.; McCuskey, R.S.; Jaeschke, H. Reduced inflammatory response and increased microcirculatory disturbances during hepatic ischemia-reperfusion injury in steatotic livers of ob/ob mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1385–G1395. [Google Scholar] [CrossRef]
- Teoh, N.C.; Williams, J.; Hartley, J.; Yu, J.; McCuskey, R.S.; Farrell, G.C. Short-term therapy with peroxisome proliferation-activator receptor-alpha agonist Wy-14,643 protects murine fatty liver against ischemia-reperfusion injury. Hepatology 2010, 51, 996–1006. [Google Scholar] [CrossRef]
- McCuskey, R.S.; Ito, Y.; Robertson, G.R.; McCuskey, M.K.; Perry, M.; Farrell, G.C. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology 2004, 40, 386–393. [Google Scholar] [CrossRef]
- Seifalian, A.M.; Piasecki, C.; Agarwal, A.; Davidson, B.R. The effect of graded steatosis on flow in the hepatic parenchymal microcirculation. Transplantation 1999, 68, 780–784. [Google Scholar] [CrossRef]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A., Jr.; et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Investig. 2006, 116, 49–58. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Li, N.; Pan, D.Y.; Miao, Q.; Ma, G.F.; Liu, Y.M.; Tseng, Y.J.; Li, F.; Xu, L.L.; Chen, S.Y. Kruppel-like factor 2 inhibit the angiogenesis of cultured human liver sinusoidal endothelial cells through the ERK1/2 signaling pathway. Biochem. Biophys. Res. Commun. 2015, 464, 1241–1247. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Garcia-Caldero, H.; Hide, D.; Marrone, G.; Guixe-Muntet, S.; Peralta, C.; Garcia-Pagan, J.C.; Abraldes, J.G.; Bosch, J. Simvastatin maintains function and viability of steatotic rat livers procured for transplantation. J. Hepatol. 2013, 58, 1140–1146. [Google Scholar] [CrossRef]
- Marrone, G.; Maeso-Diaz, R.; Garcia-Cardena, G.; Abraldes, J.G.; Garcia-Pagan, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef]
- Bravo, M.; Raurell, I.; Barbera, A.; Hide, D.; Gil, M.; Estrella, F.; Salcedo, M.T.; Augustin, S.; Genesca, J.; Martell, M. Synergic effect of atorvastatin and ambrisentan on sinusoidal and hemodynamic alterations in a rat model of NASH. Dis. Model Mech. 2021, 14, dmm048884. [Google Scholar] [CrossRef]
- Zhang, D.; Utsumi, T.; Huang, H.C.; Gao, L.; Sangwung, P.; Chung, C.; Shibao, K.; Okamoto, K.; Yamaguchi, K.; Groszmann, R.J.; et al. Reticulon 4B (Nogo-B) is a novel regulator of hepatic fibrosis. Hepatology 2011, 53, 1306–1315. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [CrossRef]
- Rockey, D.C.; Chung, J.J. Regulation of inducible nitric oxide synthase in hepatic sinusoidal endothelial cells. Am. J. Physiol. 1996, 271, G260–G267. [Google Scholar] [CrossRef]
- Leung, T.M.; Fung, M.L.; Liong, E.C.; Lau, T.Y.; Nanji, A.A.; Tipoe, G.L. Role of nitric oxide in the regulation of fibrogenic factors in experimental liver fibrosis in mice. Histol. Histopathol. 2011, 26, 201–211. [Google Scholar] [CrossRef]
- Leung, T.M.; Tipoe, G.L.; Liong, E.C.; Lau, T.Y.; Fung, M.L.; Nanji, A.A. Endothelial nitric oxide synthase is a critical factor in experimental liver fibrosis. Int. J. Exp. Pathol. 2008, 89, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Lavina, B.; Rodriguez-Vilarrupla, A.; Garcia-Caldero, H.; Fernandez, M.; Bosch, J.; Garcia-Pagan, J.C. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology 2008, 47, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Pasarin, M.; Abraldes, J.G.; Liguori, E.; Kok, B.; La Mura, V. Intrahepatic vascular changes in non-alcoholic fatty liver disease: Potential role of insulin-resistance and endothelial dysfunction. World J. Gastroenterol. 2017, 23, 6777–6787. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Paredes, F.J.; Hernandez Mesa, G.; Morales Arraez, D.; Marcelino Reyes, R.; Abrante, B.; Diaz-Flores, F.; Salido, E.; Quintero, E.; Hernandez-Guerra, M. Contribution of Cyclooxygenase End Products and Oxidative Stress to Intrahepatic Endothelial Dysfunction in Early Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0156650. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Herrnberger, L.; Hennig, R.; Kremer, W.; Hellerbrand, C.; Goepferich, A.; Kalbitzer, H.R.; Tamm, E.R. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PLoS ONE 2014, 9, e115005. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef]
- Peng, Q.; Zhang, Q.; Xiao, W.; Shao, M.; Fan, Q.; Zhang, H.; Zou, Y.; Li, X.; Xu, W.; Mo, Z.; et al. Protective effects of Sapindus mukorossi Gaertn against fatty liver disease induced by high fat diet in rats. Biochem. Biophys. Res. Commun. 2014, 450, 685–691. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, A.; Xi, L.; Yang, J.; Zhou, W.; Wang, Y.; Liang, S.; Yu, W.; Wang, Y.; Zhu, J. RETRACTION: Leukocyte cell-derived chemotaxin 2 affects nonalcoholic fatty liver disease. J. Endocrinol. 2020, 246, Z1. [Google Scholar] [CrossRef]
- Okumura, A.; Unoki-Kubota, H.; Matsushita, Y.; Shiga, T.; Moriyoshi, Y.; Yamagoe, S.; Kaburagi, Y. Increased serum leukocyte cell-derived chemotaxin 2 (LECT2) levels in obesity and fatty liver. Biosci. Trends 2013, 7, 276–283. [Google Scholar] [CrossRef]
- Hwang, H.J.; Jung, T.W.; Hong, H.C.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Choi, D.S.; Baik, S.H.; Yoo, H.J. LECT2 induces atherosclerotic inflammatory reaction via CD209 receptor-mediated JNK phosphorylation in human endothelial cells. Metabolism 2015, 64, 1175–1182. [Google Scholar] [CrossRef]
- Xu, M.; Xu, H.H.; Lin, Y.; Sun, X.; Wang, L.J.; Fang, Z.P.; Su, X.H.; Liang, X.J.; Hu, Y.; Liu, Z.M.; et al. LECT2, a Ligand for Tie1, Plays a Crucial Role in Liver Fibrogenesis. Cell 2019, 178, 1478–1492.e1420. [Google Scholar] [CrossRef]
- Plaza, A.; Naranjo, V.; Blonda, A.M.; Cano, V.; Gonzalez-Martin, C.; Gil-Ortega, M.; Ruiz-Gayo, M.; Merino, B. Inflammatory stress and altered angiogenesis evoked by very high-fat diets in mouse liver. Endocrinol. Diabetes Nutr. 2019, 66, 434–442. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G757–G763. [Google Scholar] [CrossRef]
- Su, T.; Yang, Y.; Lai, S.; Jeong, J.; Jung, Y.; McConnell, M.; Utsumi, T.; Iwakiri, Y. Single-Cell Transcriptomics Reveals Zone-Specific Alterations of Liver Sinusoidal Endothelial Cells in Cirrhosis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1139–1161. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver sinusoidal endothelial cells and liver regeneration. J. Clin. Investig. 2013, 123, 1861–1866. [Google Scholar] [CrossRef]
- Garg, M.; Kaur, S.; Banik, A.; Kumar, V.; Rastogi, A.; Sarin, S.K.; Mukhopadhyay, A.; Trehanpati, N. Bone marrow endothelial progenitor cells activate hepatic stellate cells and aggravate carbon tetrachloride induced liver fibrosis in mice via paracrine factors. Cell Prolif. 2017, 50, e12355. [Google Scholar] [CrossRef]
- Ankoma-Sey, V.; Wang, Y.; Dai, Z. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology 2000, 31, 141–148. [Google Scholar] [CrossRef]
- Novo, E.; Cannito, S.; Zamara, E.; Valfre di Bonzo, L.; Caligiuri, A.; Cravanzola, C.; Compagnone, A.; Colombatto, S.; Marra, F.; Pinzani, M.; et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am. J. Pathol. 2007, 170, 1942–1953. [Google Scholar] [CrossRef]
- Peyter, A.C.; Armengaud, J.B.; Guillot, E.; Yzydorczyk, C. Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 6667. [Google Scholar] [CrossRef]
- Kaur, S.; Tripathi, D.; Dongre, K.; Garg, V.; Rooge, S.; Mukopadhyay, A.; Sakhuja, P.; Sarin, S.K. Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident liver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J. Hepatol. 2012, 57, 1193–1198. [Google Scholar] [CrossRef]
- Lian, J.; Lu, Y.; Xu, P.; Ai, A.; Zhou, G.; Liu, W.; Cao, Y.; Zhang, W.J. Prevention of liver fibrosis by intrasplenic injection of high-density cultured bone marrow cells in a rat chronic liver injury model. PLoS ONE 2014, 9, e103603. [Google Scholar] [CrossRef]
- Liu, F.; Fei, R.; Rao, H.Y.; Cong, X.; Ha, M.H.; Wei, L. The effects of endothelial progenitor cell transplantation in carbon tetrachloride induced hepatic fibrosis rats. Zhonghua Gan Zang Bing Za Zhi 2007, 15, 589–592. [Google Scholar]
- Lan, L.; Liu, R.; Qin, L.Y.; Cheng, P.; Liu, B.W.; Zhang, B.Y.; Ding, S.Z.; Li, X.L. Transplantation of bone marrow-derived endothelial progenitor cells and hepatocyte stem cells from liver fibrosis rats ameliorates liver fibrosis. World J. Gastroenterol. 2018, 24, 237–247. [Google Scholar] [CrossRef]
- Gutierrez-Grobe, Y.; Gavilanes-Espinar, J.G.; Masso-Rojas, F.A.; Sanchez-Valle, V.; Paez-Arenas, A.; Ponciano-Rodriguez, G.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Metabolic syndrome and nonalcoholic fatty liver disease. The role of endothelial progenitor cells. Ann. Hepatol. 2013, 12, 908–914. [Google Scholar] [CrossRef]
- Chiang, C.H.; Huang, P.H.; Chung, F.P.; Chen, Z.Y.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS ONE 2012, 7, e31799. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, Y.; Tsuchiya, K.; Komiya, C.; Shiba, K.; Shimazu, N.; Yamaguchi, S.; Deushi, M.; Osaka, M.; Inoue, K.; Sato, Y.; et al. Roles for Cell-Cell Adhesion and Contact in Obesity-Induced Hepatic Myeloid Cell Accumulation and Glucose Intolerance. Cell Rep. 2017, 18, 2766–2779. [Google Scholar] [CrossRef]
- Bae, C.R.; Zhang, H.; Kwon, Y.G. The endothelial dysfunction blocker CU06-1004 ameliorates choline-deficient L-amino acid diet-induced non-alcoholic steatohepatitis in mice. PLoS ONE 2020, 15, e0243497. [Google Scholar] [CrossRef]
- Drescher, H.K.; Schippers, A.; Rosenhain, S.; Gremse, F.; Bongiovanni, L.; Bruin, A.; Eswaran, S.; Gallage, S.U.; Pfister, D.; Szydlowska, M.; et al. L-Selectin/CD62L is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men. Cells 2020, 9, 1106. [Google Scholar] [CrossRef]
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Chernavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef]
- Zhou, Y.; Cao, H.B.; Li, W.J.; Zhao, L. The CXCL12 (SDF-1)/CXCR4 chemokine axis: Oncogenic properties, molecular targeting, and synthetic and natural product CXCR4 inhibitors for cancer therapy. Chin. J. Nat. Med. 2018, 16, 801–810. [Google Scholar] [CrossRef]
- Boujedidi, H.; Robert, O.; Bignon, A.; Cassard-Doulcier, A.M.; Renoud, M.L.; Gary-Gouy, H.; Hemon, P.; Tharinger, H.; Prevot, S.; Bachelerie, F.; et al. CXCR4 dysfunction in non-alcoholic steatohepatitis in mice and patients. Clin. Sci. 2015, 128, 257–267. [Google Scholar] [CrossRef]
- Li, Y.; Li, N.; Liu, J.; An, X. Gr-1(high)Ly6G(+)Myeloid-derived suppressor cells and their role in a murine model of non-alcoholic steatohepatitis. Am. J. Transl. Res. 2020, 12, 2827–2842. [Google Scholar]
- Neumann, K.; Erben, U.; Kruse, N.; Wechsung, K.; Schumann, M.; Klugewitz, K.; Scheffold, A.; Kuhl, A.A. Chemokine Transfer by Liver Sinusoidal Endothelial Cells Contributes to the Recruitment of CD4+ T Cells into the Murine Liver. PLoS ONE 2015, 10, e0123867. [Google Scholar] [CrossRef]
- Wang, S.; Gao, S.; Li, Y.; Qian, X.; Luan, J.; Lv, X. Emerging Importance of Chemokine Receptor CXCR4 and Its Ligand in Liver Disease. Front Cell Dev. Biol. 2021, 9, 716842. [Google Scholar] [CrossRef]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef]
- Knolle, P.A.; Wohlleber, D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol. Immunol. 2016, 13, 347–353. [Google Scholar] [CrossRef]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The Role of Sinusoidal Endothelial Cells in the Axis of Inflammation and Cancer Within the Liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Knolle, P.A.; Schmitt, E.; Jin, S.; Germann, T.; Duchmann, R.; Hegenbarth, S.; Gerken, G.; Lohse, A.W. Induction of cytokine production in naive CD4(+) T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology 1999, 116, 1428–1440. [Google Scholar] [CrossRef]
- Nakamoto, N.; Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef]
- Furuta, K.; Guo, Q.; Hirsova, P.; Ibrahim, S.H. Emerging Roles of Liver Sinusoidal Endothelial Cells in Nonalcoholic Steatohepatitis. Biology 2020, 9, 395. [Google Scholar] [CrossRef]
- Martin-Armas, M.; Simon-Santamaria, J.; Pettersen, I.; Moens, U.; Smedsrod, B.; Sveinbjornsson, B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J. Hepatol. 2006, 44, 939–946. [Google Scholar] [CrossRef]
- Sutter, A.G.; Palanisamy, A.P.; Lench, J.H.; Esckilsen, S.; Geng, T.; Lewin, D.N.; Cowart, L.A.; Chavin, K.D. Dietary Saturated Fat Promotes Development of Hepatic Inflammation Through Toll-Like Receptor 4 in Mice. J. Cell. Biochem. 2016, 117, 1613–1621. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Shabgah, A.G.; Norouzi, F.; Hedayati-Moghadam, M.; Soleimani, D.; Pahlavani, N.; Navashenaq, J.G. A comprehensive review of long non-coding RNAs in the pathogenesis and development of non-alcoholic fatty liver disease. Nutr. Metab. 2021, 18, 22. [Google Scholar] [CrossRef]
- Tripathi, D.M.; Rohilla, S.; Kaur, I.; Siddiqui, H.; Rawal, P.; Juneja, P.; Kumar, V.; Kumari, A.; Naidu, V.G.M.; Ramakrishna, S.; et al. Immunonano-Lipocarrier-Mediated Liver Sinusoidal Endothelial Cell-Specific RUNX1 Inhibition Impedes Immune Cell Infiltration and Hepatic Inflammation in Murine Model of NASH. Int. J. Mol. Sci. 2021, 22, 8489. [Google Scholar] [CrossRef]
- Kaur, S.; Rawal, P.; Siddiqui, H.; Rohilla, S.; Sharma, S.; Tripathi, D.M.; Baweja, S.; Hassan, M.; Vlaic, S.; Guthke, R.; et al. Increased Expression of RUNX1 in Liver Correlates with NASH Activity Score in Patients with Non-Alcoholic Steatohepatitis (NASH). Cells 2019, 8, 1277. [Google Scholar] [CrossRef]
- Marcher, A.B.; Bendixen, S.M.; Terkelsen, M.K.; Hohmann, S.S.; Hansen, M.H.; Larsen, B.D.; Mandrup, S.; Dimke, H.; Detlefsen, S.; Ravnskjaer, K. Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci. Rep. 2019, 9, 2324. [Google Scholar] [CrossRef]
- McMahan, R.H.; Porsche, C.E.; Edwards, M.G.; Rosen, H.R. Free Fatty Acids Differentially Downregulate Chemokines in Liver Sinusoidal Endothelial Cells: Insights into Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0159217. [Google Scholar] [CrossRef]
- Neumann, K.; Rudolph, C.; Neumann, C.; Janke, M.; Amsen, D.; Scheffold, A. Liver sinusoidal endothelial cells induce immunosuppressive IL-10-producing Th1 cells via the Notch pathway. Eur. J. Immunol. 2015, 45, 2008–2016. [Google Scholar] [CrossRef]
- Chen, L.; Gu, T.; Li, B.; Li, F.; Ma, Z.; Zhang, Q.; Cai, X.; Lu, L. Delta-like ligand 4/DLL4 regulates the capillarization of liver sinusoidal endothelial cell and liver fibrogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1663–1675. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.M.; Kyrou, I.; Nasiri-Ansari, N.; Kaltsas, G.; Papavassiliou, A.G.; Kassi, E.; Randeva, H.S. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr. Obes. Rep. 2021, 10, 134–161. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Biquard, L.; Lasselin, J.; Kheloufi, M.; Tanguy, M.; Vion, A.C.; Merian, J.; Colnot, N.; Loyer, X.; Tedgui, A.; et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J. Hepatol. 2020, 72, 528–538. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Camprecios, G.; Suarez-Herrera, N.; Montironi, C.; Guixe-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagan, J.C.; Hernandez-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Sorensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrod, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef]
- Smedsrod, B.; Le Couteur, D.; Ikejima, K.; Jaeschke, H.; Kawada, N.; Naito, M.; Knolle, P.; Nagy, L.; Senoo, H.; Vidal-Vanaclocha, F.; et al. Hepatic sinusoidal cells in health and disease: Update from the 14th International Symposium. Liver Int. 2009, 29, 490–501. [Google Scholar] [CrossRef]
- Bhandari, S.; Larsen, A.K.; McCourt, P.; Smedsrod, B.; Sorensen, K.K. The Scavenger Function of Liver Sinusoidal Endothelial Cells in Health and Disease. Front. Physiol. 2021, 12, 757469. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yokoyama, H.; Matsuura, T.; Fujiwara, Y. Fc gamma RIIb expression levels in human liver sinusoidal endothelial cells during progression of non-alcoholic fatty liver disease. PLoS ONE 2019, 14, e0211543. [Google Scholar] [CrossRef]
- Milner, K.L.; van der Poorten, D.; Xu, A.; Bugianesi, E.; Kench, J.G.; Lam, K.S.; Chisholm, D.J.; George, J. Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1926–1934. [Google Scholar] [CrossRef]
- Laouirem, S.; Sannier, A.; Norkowski, E.; Cauchy, F.; Doblas, S.; Rautou, P.E.; Albuquerque, M.; Garteiser, P.; Sognigbe, L.; Raffenne, J.; et al. Endothelial fatty liver binding protein 4: A new targetable mediator in hepatocellular carcinoma related to metabolic syndrome. Oncogene 2019, 38, 3033–3046. [Google Scholar] [CrossRef]
- Verhulst, S.; van Os, E.A.; De Smet, V.; Eysackers, N.; Mannaerts, I.; van Grunsven, L.A. Gene Signatures Detect Damaged Liver Sinusoidal Endothelial Cells in Chronic Liver Diseases. Front. Med. 2021, 8, 750044. [Google Scholar] [CrossRef]
- Carlessi, R.; Denisenko, E.; Boslem, E.; Koehn-Gaone, J.; Main, N.; Abu Bakar, N.D.B.; Shirolkar, G.D.; Jones, M.; Poppe, D.; Dwyer, B.J.; et al. Single Nucleus RNA Sequencing of Pre-Malignant Liver Reveals Disease-Associated Hepatocyte State with HCC Prognostic Potential. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Olsavszky, V.; Sticht, C.; Schmid, C.D.; Winkler, M.; Wohlfeil, S.A.; Olsavszky, A.; Schledzewski, K.; Geraud, C.; Goerdt, S.; Leibing, T.; et al. Exploring the transcriptomic network of multi-ligand scavenger receptor Stabilin-1- and Stabilin-2-deficient liver sinusoidal endothelial cells. Gene 2021, 768, 145284. [Google Scholar] [CrossRef]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef]
- Elvevold, K.; Smedsrod, B.; Martinez, I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G391–G400. [Google Scholar] [CrossRef]
- Wu, X.; Shu, L.; Zhang, Z.; Li, J.; Zong, J.; Cheong, L.Y.; Ye, D.; Lam, K.S.L.; Song, E.; Wang, C.; et al. Adipocyte Fatty Acid Binding Protein Promotes the Onset and Progression of Liver Fibrosis via Mediating the Crosstalk between Liver Sinusoidal Endothelial Cells and Hepatic Stellate Cells. Adv. Sci. 2021, 8, e2003721. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Xiu, A.Y.; Meng, D.X.; Wang, S.N.; Zhang, C.Q. Carvedilol attenuates carbon tetrachloride-induced liver fibrosis and hepatic sinusoidal capillarization in mice. Drug Des. Devel. Ther. 2019, 13, 2667–2676. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Z.; Qian, L.; Lin, Q.; Zhang, C.; Shao, J.; Zhang, F.; Zheng, S. Tetramethylpyrazine attenuates carbon tetrachloride-caused liver injury and fibrogenesis and reduces hepatic angiogenesis in rats. Biomed. Pharmacother. 2017, 86, 521–530. [Google Scholar] [CrossRef]
- Bravo, M.; Raurell, I.; Hide, D.; Fernandez-Iglesias, A.; Gil, M.; Barbera, A.; Salcedo, M.T.; Augustin, S.; Genesca, J.; Martell, M. Restoration of liver sinusoidal cell phenotypes by statins improves portal hypertension and histology in rats with NASH. Sci. Rep. 2019, 9, 20183. [Google Scholar] [CrossRef]
- Yu, Z.; Guo, J.; Liu, Y.; Wang, M.; Liu, Z.; Gao, Y.; Huang, L. Nano delivery of simvastatin targets liver sinusoidal endothelial cells to remodel tumor microenvironment for hepatocellular carcinoma. J. Nanobiotechnol. 2022, 20, 9. [Google Scholar] [CrossRef]
- Pereira, E.; Araujo, B.P.; Rodrigues, K.L.; Silvares, R.R.; Martins, C.S.M.; Flores, E.E.I.; Fernandes-Santos, C.; Daliry, A. Simvastatin Improves Microcirculatory Function in Nonalcoholic Fatty Liver Disease and Downregulates Oxidative and ALE-RAGE Stress. Nutrients 2022, 14, 716. [Google Scholar] [CrossRef]
- Dekker, R.J.; van Soest, S.; Fontijn, R.D.; Salamanca, S.; de Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef]
- Siasos, G.; Tousoulis, D.; Siasou, Z.; Stefanadis, C.; Papavassiliou, A.G. Shear stress, protein kinases and atherosclerosis. Curr. Med. Chem. 2007, 14, 1567–1572. [Google Scholar] [CrossRef]
- Russo, L.; Gracia-Sancho, J.; Garcia-Caldero, H.; Marrone, G.; Garcia-Pagan, J.C.; Garcia-Cardena, G.; Bosch, J. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology 2012, 55, 921–930. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef]
- Ming, X.F.; Viswambharan, H.; Barandier, C.; Ruffieux, J.; Kaibuchi, K.; Rusconi, S.; Yang, Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol. Cell Biol. 2002, 22, 8467–8477. [Google Scholar] [CrossRef]
- Morales-Ruiz, M.; Cejudo-Martin, P.; Fernandez-Varo, G.; Tugues, S.; Ros, J.; Angeli, P.; Rivera, F.; Arroyo, V.; Rodes, J.; Sessa, W.C.; et al. Transduction of the liver with activated Akt normalizes portal pressure in cirrhotic rats. Gastroenterology 2003, 125, 522–531. [Google Scholar] [CrossRef]
- Rodriguez, S.; Raurell, I.; Torres-Arauz, M.; Garcia-Lezana, T.; Genesca, J.; Martell, M. A Nitric Oxide-Donating Statin Decreases Portal Pressure with a Better Toxicity Profile than Conventional Statins in Cirrhotic Rats. Sci. Rep. 2017, 7, 40461. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, C.; Zhou, J.; Zhen, Z.; Wang, Y.; Shen, C. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS ONE 2013, 8, e76538. [Google Scholar] [CrossRef] [PubMed]
- Trocha, M.; Merwid-Lad, A.; Szuba, A.; Chlebda, E.; Piesniewska, M.; Sozanski, T.; Szelag, A. Effect of simvastatin on nitric oxide synthases (eNOS, iNOS) and arginine and its derivatives (ADMA, SDMA) in ischemia/reperfusion injury in rat liver. Pharmacol. Rep. 2010, 62, 343–351. [Google Scholar] [CrossRef]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Huang, J.; Rockey, D.C. Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein betagamma subunit signaling to protein jinase B/Akt. J. Biol. Chem. 2003, 278, 49929–49935. [Google Scholar] [CrossRef] [PubMed]
- Furuta, K.; Guo, Q.; Pavelko, K.D.; Lee, J.H.; Robertson, K.D.; Nakao, Y.; Melek, J.; Shah, V.H.; Hirsova, P.; Ibrahim, S.H. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J. Clin. Investig. 2021, 131, e143690. [Google Scholar] [CrossRef]
- Nati, M.; Chung, K.J.; Chavakis, T. The Role of Innate Immune Cells in Nonalcoholic Fatty Liver Disease. J. Innate Immun. 2022, 14, 31–41. [Google Scholar] [CrossRef]
- Guo, Q.; Furuta, K.; Lucien, F.; Gutierrez Sanchez, L.H.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef]
- Geraud, C.; Mogler, C.; Runge, A.; Evdokimov, K.; Lu, S.; Schledzewski, K.; Arnold, B.; Hammerling, G.; Koch, P.S.; Breuhahn, K.; et al. Endothelial transdifferentiation in hepatocellular carcinoma: Loss of Stabilin-2 expression in peri-tumourous liver correlates with increased survival. Liver Int. 2013, 33, 1428–1440. [Google Scholar] [CrossRef]
- Tardif, J.C.; McMurray, J.J.; Klug, E.; Small, R.; Schumi, J.; Choi, J.; Cooper, J.; Scott, R.; Lewis, E.F.; L’Allier, P.L.; et al. Effects of succinobucol (AGI-1067) after an acute coronary syndrome: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 371, 1761–1768. [Google Scholar] [CrossRef]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Zambon, A.; Gervois, P.; Pauletto, P.; Fruchart, J.C.; Staels, B. Modulation of hepatic inflammatory risk markers of cardiovascular diseases by PPAR-alpha activators: Clinical and experimental evidence. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 977–986. [Google Scholar] [CrossRef]
- Kostadinova, R.; Wahli, W.; Michalik, L. PPARs in diseases: Control mechanisms of inflammation. Curr. Med. Chem. 2005, 12, 2995–3009. [Google Scholar] [CrossRef]
- Dela Pena, A.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Sempoux, C.; dela Pena, A.; Horsmans, Y. Curcumin inhibits NF-kappaB activation and reduces the severity of experimental steatohepatitis in mice. J. Hepatol. 2004, 41, 926–934. [Google Scholar] [CrossRef]
- Cai, W.; Duan, X.M.; Liu, Y.; Yu, J.; Tang, Y.L.; Liu, Z.L.; Jiang, S.; Zhang, C.P.; Liu, J.Y.; Xu, J.X. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. Biomed. Res. Int. 2017, 2017, 4391920. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Yin, X.M. Role of High-Mobility Group Box-1 in Liver Pathogenesis. Int. J. Mol. Sci. 2019, 20, 5314. [Google Scholar] [CrossRef]
- Ibrahim, S.H. Sinusoidal endotheliopathy in nonalcoholic steatohepatitis: Therapeutic implications. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G67–G74. [Google Scholar] [CrossRef]
- Lefere, S.; Van de Velde, F.; Hoorens, A.; Raevens, S.; Van Campenhout, S.; Vandierendonck, A.; Neyt, S.; Vandeghinste, B.; Vanhove, C.; Debbaut, C.; et al. Angiopoietin-2 Promotes Pathological Angiogenesis and Is a Therapeutic Target in Murine Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1087–1104. [Google Scholar] [CrossRef]
- Orlandi, P.; Solini, A.; Banchi, M.; Brunetto, M.R.; Cioni, D.; Ghiadoni, L.; Bocci, G. Antiangiogenic Drugs in NASH: Evidence of a Possible New Therapeutic Approach. Pharmaceuticals 2021, 14, 995. [Google Scholar] [CrossRef]
- Coulon, S.; Legry, V.; Heindryckx, F.; Van Steenkiste, C.; Casteleyn, C.; Olievier, K.; Libbrecht, L.; Carmeliet, P.; Jonckx, B.; Stassen, J.M.; et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 2013, 57, 1793–1805. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Liu, R.S.; Lee, P.C.; Yeh, Y.C.; Huang, Y.T.; Lee, W.P.; Lee, K.C.; Hsieh, Y.C.; Lee, F.Y.; Tan, T.W.; et al. Anti-VEGFR agents ameliorate hepatic venous dysregulation/microcirculatory dysfunction, splanchnic venous pooling and ascites of NASH-cirrhotic rat. Liver Int. 2014, 34, 521–534. [Google Scholar] [CrossRef]
- Rai, R.P.; Liu, Y.; Iyer, S.S.; Liu, S.; Gupta, B.; Desai, C.; Kumar, P.; Smith, T.; Singhi, A.D.; Nusrat, A.; et al. Blocking integrin alpha4beta7-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J. Hepatol. 2020, 73, 1013–1022. [Google Scholar] [CrossRef]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar] [CrossRef]
- Mendt, M.; Cardier, J.E. Stromal-derived factor-1 and its receptor, CXCR4, are constitutively expressed by mouse liver sinusoidal endothelial cells: Implications for the regulation of hematopoietic cell migration to the liver during extramedullary hematopoiesis. Stem Cells Dev. 2012, 21, 2142–2151. [Google Scholar] [CrossRef]
- Chow, L.N.; Schreiner, P.; Ng, B.Y.; Lo, B.; Hughes, M.R.; Scott, R.W.; Gusti, V.; Lecour, S.; Simonson, E.; Manisali, I.; et al. Impact of a CXCL12/CXCR4 Antagonist in Bleomycin (BLM) Induced Pulmonary Fibrosis and Carbon Tetrachloride (CCl4) Induced Hepatic Fibrosis in Mice. PLoS ONE 2016, 11, e0151765. [Google Scholar] [CrossRef]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef]
- Moosavian, S.A.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The Emerging Role of Nanomedicine in the Management of Nonalcoholic Fatty Liver Disease: A State-of-the-Art Review. Bioinorg. Chem. Appl. 2021, 2021, 4041415. [Google Scholar] [CrossRef]
- Salunkhe, S.A.; Chitkara, D.; Mahato, R.I.; Mittal, A. Lipid based nanocarriers for effective drug delivery and treatment of diabetes associated liver fibrosis. Adv. Drug Deliv. Rev. 2021, 173, 394–415. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Xu, G.; Yao, P. Liver-targeted delivery of insulin-loaded nanoparticles via enterohepatic circulation of bile acids. Drug Deliv. 2018, 25, 1224–1233. [Google Scholar] [CrossRef]
- Thomas, R.G.; Moon, M.J.; Kim, J.H.; Lee, J.H.; Jeong, Y.Y. Effectiveness of Losartan-Loaded Hyaluronic Acid (HA) Micelles for the Reduction of Advanced Hepatic Fibrosis in C3H/HeN Mice Model. PLoS ONE 2015, 10, e0145512. [Google Scholar] [CrossRef]
- Jimenez Calvente, C.; Sehgal, A.; Popov, Y.; Kim, Y.O.; Zevallos, V.; Sahin, U.; Diken, M.; Schuppan, D. Specific hepatic delivery of procollagen alpha1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology 2015, 62, 1285–1297. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chiang, T.; Liu, C.H.; Chern, G.G.; Lin, T.T.; Gao, D.Y.; Chen, Y. Delivery of siRNA Using CXCR4-targeted Nanoparticles Modulates Tumor Microenvironment and Achieves a Potent Antitumor Response in Liver Cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; McCourt, P.A.G.; Le Couteur, D.G.; Cogger, V.C. Novel targets for delaying aging: The importance of the liver and advances in drug delivery. Adv. Drug Deliv. Rev. 2018, 135, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Grice, J.E.; Zhu, Y.; Liu, D.; Sanchez, W.Y.; Li, Z.; Crawford, D.H.; Le Couteur, D.G.; Cogger, V.C.; Liu, X.; et al. Intravital multiphoton imaging of the selective uptake of water-dispersible quantum dots into sinusoidal liver cells. Small 2015, 11, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Bargheer, D.; Giemsa, A.; Freund, B.; Heine, M.; Waurisch, C.; Stachowski, G.M.; Hickey, S.G.; Eychmuller, A.; Heeren, J.; Nielsen, P. The distribution and degradation of radiolabeled superparamagnetic iron oxide nanoparticles and quantum dots in mice. Beilstein J. Nanotechnol. 2015, 6, 111–123. [Google Scholar] [CrossRef]
- Lao, Y.; Li, Y.; Zhang, P.; Shao, Q.; Lin, W.; Qiu, B.; Lv, Y.; Tang, L.; Su, S.; Zhang, H.; et al. Targeting Endothelial Erk1/2-Akt Axis as a Regeneration Strategy to Bypass Fibrosis during Chronic Liver Injury in Mice. Mol. Ther. 2018, 26, 2779–2797. [Google Scholar] [CrossRef]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a mediator of non-alcoholic Fatty liver disease and associated atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef]
- Gaudio, E.; Nobili, V.; Franchitto, A.; Onori, P.; Carpino, G. Nonalcoholic fatty liver disease and atherosclerosis. Intern Emerg. Med. 2012, 7 (Suppl. 3), S297–S305. [Google Scholar] [CrossRef]
- Dogru, T.; Genc, H.; Tapan, S.; Aslan, F.; Ercin, C.N.; Ors, F.; Kara, M.; Sertoglu, E.; Karslioglu, Y.; Bagci, S.; et al. Plasma fetuin-A is associated with endothelial dysfunction and subclinical atherosclerosis in subjects with nonalcoholic fatty liver disease. Clin. Endocrinol. 2013, 78, 712–717. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Kozakova, M.; Hojlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; Balkau, B.; Investigators, R. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009, 49, 1537–1544. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kim, D.; Kim, H.J.; Kang, J.H.; Chung, S.J.; Park, M.J.; Kim, Y.S.; Kim, C.H.; Choi, S.H.; Kim, W.; et al. The relation between non-alcoholic fatty liver disease and the risk of coronary heart disease in Koreans. Am. J. Gastroenterol. 2009, 104, 1953–1960. [Google Scholar] [CrossRef]
- Luo, J.; Xu, L.; Li, J.; Zhao, S. Nonalcoholic fatty liver disease as a potential risk factor of cardiovascular disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 193–199. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: Implications for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1155–1161. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. [Google Scholar] [CrossRef]
- Janes, F.; Cifu, A.; Pessa, M.E.; Domenis, R.; Gigli, G.L.; Sanvilli, N.; Nilo, A.; Garbo, R.; Curcio, F.; Giacomello, R.; et al. ADMA as a possible marker of endothelial damage. A study in young asymptomatic patients with cerebral small vessel disease. Sci. Rep. 2019, 9, 14207. [Google Scholar] [CrossRef]
- Al-Hamoudi, W.; Alsadoon, A.; Hassanian, M.; Alkhalidi, H.; Abdo, A.; Nour, M.; Halwani, R.; Sanai, F.; Alsharaabi, A.; Alswat, K.; et al. Endothelial dysfunction in nonalcoholic steatohepatitis with low cardiac disease risk. Sci. Rep. 2020, 10, 8825. [Google Scholar] [CrossRef]
- Arinc, H.; Sarli, B.; Baktir, A.O.; Saglam, H.; Demirci, E.; Dogan, Y.; Kurtul, S.; Karaman, H.; Erden, A.; Karaman, A. Serum gamma glutamyl transferase and alanine transaminase concentrations predict endothelial dysfunction in patients with non-alcoholic steatohepatitis. Ups. J. Med. Sci. 2013, 118, 228–234. [Google Scholar] [CrossRef]
- Cetindagli, I.; Kara, M.; Tanoglu, A.; Ozalper, V.; Aribal, S.; Hancerli, Y.; Unal, M.; Ozari, O.; Hira, S.; Kaplan, M.; et al. Evaluation of endothelial dysfunction in patients with nonalcoholic fatty liver disease: Association of selenoprotein P with carotid intima-media thickness and endothelium-dependent vasodilation. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 516–524. [Google Scholar] [CrossRef]
- Colak, Y.; Senates, E.; Yesil, A.; Yilmaz, Y.; Ozturk, O.; Doganay, L.; Coskunpinar, E.; Kahraman, O.T.; Mesci, B.; Ulasoglu, C.; et al. Assessment of endothelial function in patients with nonalcoholic fatty liver disease. Endocrine 2013, 43, 100–107. [Google Scholar] [CrossRef]
- Dogru, T.; Genc, H.; Tapan, S.; Ercin, C.N.; Ors, F.; Aslan, F.; Kara, M.; Sertoglu, E.; Bagci, S.; Kurt, I.; et al. Elevated asymmetric dimethylarginine in plasma: An early marker for endothelial dysfunction in non-alcoholic fatty liver disease? Diabetes Res. Clin. Pract. 2012, 96, 47–52. [Google Scholar] [CrossRef]
- Gurel, H.; Genc, H.; Celebi, G.; Sertoglu, E.; Cicek, A.F.; Kayadibi, H.; Ercin, C.N.; Dogru, T. Plasma pentraxin-3 is associated with endothelial dysfunction in non-alcoholic fatty liver disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4305–4312. [Google Scholar]
- Jose, N.; Vasant, P.K.; Kulirankal, K.G. Study of Endothelial Dysfunction in Patients with Non-alcoholic Fatty Liver Disease. Cureus 2021, 13, e20515. [Google Scholar] [CrossRef]
- Kasumov, T.; Edmison, J.M.; Dasarathy, S.; Bennett, C.; Lopez, R.; Kalhan, S.C. Plasma levels of asymmetric dimethylarginine in patients with biopsy-proven nonalcoholic fatty liver disease. Metabolism 2011, 60, 776–781. [Google Scholar] [CrossRef]
- Kucukazman, M.; Ata, N.; Yavuz, B.; Dal, K.; Sen, O.; Deveci, O.S.; Agladioglu, K.; Yeniova, A.O.; Nazligul, Y.; Ertugrul, D.T. Evaluation of early atherosclerosis markers in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2013, 25, 147–151. [Google Scholar] [CrossRef]
- Loffredo, L.; Baratta, F.; Ludovica, P.; Battaglia, S.; Carnevale, R.; Nocella, C.; Novo, M.; Pannitteri, G.; Ceci, F.; Angelico, F.; et al. Effects of dark chocolate on endothelial function in patients with non-alcoholic steatohepatitis. Nutr. Metab. Cardiovasc. Dis. 2017, 28, 143–149. [Google Scholar] [CrossRef]
- Long, M.T.; Wang, N.; Larson, M.G.; Mitchell, G.F.; Palmisano, J.; Vasan, R.S.; Hoffmann, U.; Speliotes, E.K.; Vita, J.A.; Benjamin, E.J.; et al. Nonalcoholic fatty liver disease and vascular function: Cross-sectional analysis in the Framingham heart study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1284–1291. [Google Scholar] [CrossRef]
- Nahandi, M.Z.; Khoshbaten, M.; Ramazanzadeh, E.; Abbaszadeh, L.; Javadrashid, R.; Shirazi, K.M.; Gholami, N. Effect of non-alcoholic fatty liver disease on carotid artery intima-media thickness as a risk factor for atherosclerosis. Gastroenterol. Hepatol. Bed. Bench 2014, 7, 55–62. [Google Scholar]
- Narayan, J.; Das, H.S.; Nath, P.; Singh, A.; Mishra, D.; Padhi, P.K.; Singh, S.P. Endothelial Dysfunction, a Marker of Atherosclerosis, Is Independent of Metabolic Syndrome in NAFLD Patients. Int. J. Hepatol. 2020, 2020, 1825142. [Google Scholar] [CrossRef]
- Perticone, M.; Cimellaro, A.; Maio, R.; Caroleo, B.; Sciacqua, A.; Sesti, G.; Perticone, F. Additive Effect of Non-Alcoholic Fatty Liver Disease on Metabolic Syndrome-Related Endothelial Dysfunction in Hypertensive Patients. Int. J. Mol. Sci. 2016, 17, 456. [Google Scholar] [CrossRef]
- Pinarbasi, B.; Demir, K.; Oflaz, H.; Ahishali, E.; Akyuz, F.; Elitok, A.; Cimen, A.O.; Golcuk, E.; Gulluoglu, M.; Issever, H.; et al. Measurement of the coronary flow velocity reserve in patients with non-alcoholic fatty liver disease. Turk. J. Gastroenterol. 2012, 23, 720–726. [Google Scholar] [CrossRef]
- Pugh, C.J.; Spring, V.S.; Kemp, G.J.; Richardson, P.; Shojaee-Moradie, F.; Umpleby, A.M.; Green, D.J.; Cable, N.T.; Jones, H.; Cuthbertson, D.J. Exercise training reverses endothelial dysfunction in nonalcoholic fatty liver disease. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1298–H1306. [Google Scholar] [CrossRef] [PubMed]
- Sapmaz, F.; Uzman, M.; Basyigit, S.; Ozkan, S.; Yavuz, B.; Yeniova, A.; Kefeli, A.; Asilturk, Z.; Nazligul, Y. Steatosis Grade is the Most Important Risk Factor for Development of Endothelial Dysfunction in NAFLD. Medicine 2016, 95, e3280. [Google Scholar] [CrossRef] [PubMed]
- Sayki Arslan, M.; Turhan, S.; Dincer, I.; Mizrak, D.; Corapcioglu, D.; Idilman, R. A potential link between endothelial function, cardiovascular risk, and metabolic syndrome in patients with Non-alcoholic fatty liver disease. Diabetol. Metab. Syndr. 2014, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Sciacqua, A.; Perticone, M.; Miceli, S.; Laino, I.; Tassone, E.J.; Grembiale, R.D.; Andreozzi, F.; Sesti, G.; Perticone, F. Endothelial dysfunction and non-alcoholic liver steatosis in hypertensive patients. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Senturk, O.; Kocaman, O.; Hulagu, S.; Sahin, T.; Aygun, C.; Konduk, T.; Celebi, A. Endothelial dysfunction in Turkish patients with non-alcoholic fatty liver disease. Intern. Med. J. 2008, 38, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Fatima, J.; Chaudhary, S.; Ali, M.; Mishra, I. A Study of Endothelial Dysfunction in Patients of Non-Alcoholic Fatty Liver Disease. J. Assoc. Physicians India 2017, 65, 18–22. [Google Scholar] [PubMed]
- Thakur, M.L.; Sharma, S.; Kumar, A.; Bhatt, S.P.; Luthra, K.; Guleria, R.; Pandey, R.M.; Vikram, N.K. Nonalcoholic fatty liver disease is associated with subclinical atherosclerosis independent of obesity and metabolic syndrome in Asian Indians. Atherosclerosis 2012, 223, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased arterial stiffness and impaired endothelial function in nonalcoholic Fatty liver disease: A pilot study. Am. J. Hypertens. 2010, 23, 1183–1189. [Google Scholar] [CrossRef]
- Weghuber, D.; Roden, M.; Franz, C.; Chmelik, M.; Torabia, S.; Nowotny, P.; Gruber, S.; Waldhausl, W.; Klingler, A.; Bieglmayer, C.; et al. Vascular function in obese children with non-alcoholic fatty liver disease. Int. J. Pediatr. Obes. 2011, 6, 120–127. [Google Scholar] [CrossRef]
- Zhou, S.L.; Li, C.; Huang, J.J.; Xiong, Q.; Li, Q.Z.; Peng, B.K.; Tang, Z. The relationship between endoplasmic reticulum stress and liver function, insulin resistance and vascular endothelial function in patients with non-alcoholic fatty liver disease. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11707–11715. [Google Scholar] [CrossRef]
- Yu, X.Y.; Li, Y.; Liu, T.; Wang, R.T. Association of whole blood viscosity with non-alcoholic fatty liver disease. Clin. Hemorheol. Microcirc. 2015, 62, 335–343. [Google Scholar] [CrossRef]
- Baldini, F.; Khalil, M.; Serale, N.; Voci, A.; Portincasa, P.; Vergani, L. Extent and features of liver steatosis in vitro pave the way to endothelial dysfunction without physical cell-to-cell contact. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 3522–3532. [Google Scholar] [CrossRef]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar] [CrossRef]
- Boon, J.; Hoy, A.J.; Stark, R.; Brown, R.D.; Meex, R.C.; Henstridge, D.C.; Schenk, S.; Meikle, P.J.; Horowitz, J.F.; Kingwell, B.A.; et al. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 2013, 62, 401–410. [Google Scholar] [CrossRef]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef]
- Yang, S.J.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: Implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metab. 2011, 96, E1325–E1329. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ref. | Animal Model | Treatment | Markers of NAFLD | Markers of Endothelial Dysfunction | Outcome after Therapy |
|---|---|---|---|---|---|
| [62] | foz/foz mice under HFD/chew diet | PPAR-alpha agonist (Wy-14,643) (10 days) | ALT Hepatic necrosis area Inflammation (IL-1a, TNF-a, IL-12) | Adhesion molecules (VCAM-1) Microvascular narrowing | ⬇ ALT ⬇ Adhesion molecules ⬇ Inflammation ⬇ Fibrosis ⬇ Microvascular narrowing |
| [69] | Male Sprague Dawley rats under HFGFD | Atorvastatin and/or Ambrisentan (2 weeks) | ALT Contractile response in HSCs NA Score | Portal hypertension eNOS | Combination therapy normalizes liver hemodynamics and reverses HSC procontractile and profibrogenic profile |
| [152] | Sprague Dawley rats under HFGFD for 8 weeks | Simvastatin or Atorvastatin (2 weeks) | NA Score Lipid droplet HSC activation | Portal pressure CD32b/CD11b ratio Contractile phenotype | Reverses NASH histology features ⬇ LSEC differentiation ⬇ LSEC capillarization |
| [153] | C57BL/6 mice received ip CCl4 injection 2/W for 4 weeks | Simvastatin by tail vein injection and simvastatin-free drug daily (5 days) | HSC activation | CD31 KLF2-NO signaling CXCL16 | Restores the quiescence of activated HSCs Alleviates LSEC capillarization Induces NKT recruitment into HCC microenvironment through CXCL16 |
| [154] | C57BL/6 mice under HFHC diet | Simvastatin For 5 weeks | ALT and AST Serum and hepatic TG and TC NA score Hepatic fibrosis | Microcirculatory dysfunction Microvascular blood flow Leukocyte recruitment | Restores the endothelium-dependent vasodilatory response and blood flow Restores antioxidant enzymatic activity ⬇ Lipid peroxidation |
| [162] | Wistar rats under HFD models | Simvastatin (4 weeks) | ALT and AST Fibrosis α-SMA and Collagen I TC and TG | iNOS eNOS | Improves NASH-related fibrosis by increasing ⬆ eNOS ⬇ iNOS |
| [158] | Male Sprague Dawley rats underwent BDL | Atorvastatin (7 days) | ALT and AST HSC activation | eNOS Portal pressure | ⬆ eNOS ⬆ NO/PKG pathway activation ⬇ Portal pressure |
| [165] | C57BL6/J mice under FFC and chew diets and endothelial-cell-specific Vcam1 knockout mice | VCAM-1-neutralizing Ab or MLK3 inhibitor | ALT NA score Fibrosis | Adhesion molecules such as VCAM-1 Inflammatory markers such as IL-1b, CCR2, TNF-a, and CD68 | ⬇ Inflammation ⬇ Improved NA score ⬇ Fibrosis |
| [174] | C57BL6/J mice under MCD treatment | Curcumin (NF-kappaB inhibitor) (up to 4 weeks) | ALT NA Score Lipoperoxides, Pro-inflammatory markers such as COX-2, MCP-1, and CINC | Adhesion molecules such as ICAM-1 | ⬇ Inflammation ⬇ Liver histological features |
| [6] | C57BL6/J mice under MCD treatment | Anti-HMGB1 antibody | ALT NA score Fibrosis | CD31 | Prevents liver fibrosis |
| [180] | C57BL6/J mice and db/db mice under MCD | Anti-VEGFR2 antibody (2 weeks) | ALT and AST HSC activation Fibrosis | Inflammation, CD105 | Prevents liver fibrosis ⬇ Inflammation ⬇ Disrupted vascular architecture |
| [178] | C57BL6/J mice under MCD, C57BL6/J mice injected with strepto-zotocin (STZ), and C57BL6/J mice under WD | Ang-2/Tie2 interaction inhibiting peptibody L1–10 | ALT and AST Inflammation, MCP-1, CXCL2, and CCR2 Fibrosis | Adhesion molecules ICAM-1 and VCAM-1 CD34 Ang-2 | Reverses NASH ⬇ HCC progression ⬇ Inflammation ⬇ Liver histological features ⬇ Fibrosis ⬇ iNOS |
| [184] | LSECs isolated from C57BL6/J mice | AMD3100 (CXCR4 antagonist) | HSC migration | CXCR-4 expression under treatment with inflammatory cytokines PECAM-1 | ⬇ Inflammation ⬇ CXCR4 ⬇ CXCl12 ⬇HSC migration |
| [196] | C57BL/6J mice under CCl4 and LSECs isolated from C57BL/6J mice under CCl4 | NP carrying honokiol or Ad-CA-MEK1 Ad-DN Akt | HSC activation HSC senescence | LSEC profibrotic phenotype NO/ROS | promotes Erk1/2 activity ⬇ Liver fibrosis ⬆ Enhances liver regeneration |
| Ref. | Study Population (nr) | Method of NAFLD Diagnosis | Method of Endothelial Dysfunction Diagnosis | Outcome |
|---|---|---|---|---|
| [229] | 39 NASH vs. 13 NAFL vs. 28 healthy controls | U/S, LB | FMD | FMD levels lower in NASH than all and lower in NAFL than controls |
| [226] | 15 NASH vs. 17 NAFLD vs. 16 healthy controls | U/S, LB | FMD | Lower FMD in NASH but not in NAFLD |
| [225] | 20 NAFLD with arterial hypertension vs. 20 hypertensive controls | U/S | FBF | FBF significantly reduced in NAFLD patients |
| [212] | 70 NAFLD vs. 70 healthy controls | LB | PTX-3, ADMA | Higher PTX-3 and ADMA serum levels in patients with NAFLD, higher PTX-3 but not ADMA in NASH |
| [224] | 100 NAFLD vs. 45 healthy controls | Biochemical, radiological and/or histological criteria | ADMA, FMD | No statistically significant differences |
| [227] | 32 NAFLD vs. 16 healthy controls | U/S | FMD | Lower % of FMD change |
| [220] | 93 MS+/NAFLD+ vs. 78 MS+/NAFLD− vs. 101 MS−/NAFLD− | FLI | Vasodilating response of FBF to acetylcholine | Worse response to vasodilation in MS+/NAFLD+ and MS+/NAFLD− patients |
| [210] | 51 NAFLD vs. 21 healthy controls | LB | ADMA, FMD, CIMT | Higher CIMT and lower FMD in patients with NAFLD, no difference in ADMA |
| [216] | 19 NASH vs. 19 NAFL vs. 19 healthy controls | U/S, LB | FMD | FMD levels lower in NASH than all and lower in NAFL than controls |
| [211] | 24 NASH vs. 23 borderline NASH vs. 20 NAFL | LB | ADMA, CIMT | Higher ADMA in NAFLD, no differences in CIMT when adjusted for metabolic parameters and insulin sensitivity |
| [230] | 23 NAFLD vs. 28 healthy controls | LB | PWV, CIMT, FMD | Higher PWV and CIMT and lower FMD in NAFLD |
| [231] | 14 obese children with NAFLD vs. 14 obese children | MR spectroscopy | FMD | No difference |
| [214] | 24 nondiabetic NASH vs. 11 nondiabetic NAFL vs. 25 healthy controls | LB | ADMA | Higher in NAFLD, no difference when adjusted for insulin resistance |
| [215] | 117 NAFLD vs. 44 healthy controls | U/S | FMD, CIMT | Lower FMD in NAFLD correlated with U/S staging, higher CIMT in NAFLD, not correlated with U/S staging |
| [221] | 12 NASH vs. 12 NAFLD vs. 28 healthy controls | LB | APDV, AMDV, CFVR, CIMT | 2 min after dipyridamole infusion, APDV, AMDV, and CFVR were lower in patients with NAFLD, no differences between NAFL and NASH patients |
| [228] | 40 nondiabetic NAFLD vs. 40 healthy controls | U/S | FMD, CIMT | Higher CIMT and lower FMD in NAFLD patients |
| [199] | 115 NAFLD (50 NASH, 35 borderline NASH, 30 NAFL) vs. 74 healthy controls | LB | Fetuin-A, ADMA, adiponectin, CIMT | Higher ADMA, CIMT, and fetuin-A and lower adiponectin serum levels in NAFLD patients. No difference when the findings were adjusted according to the BMI, glucose, lipids, and HOMA-IR index |
| [208] | 50 NASH vs. 30 healthy controls | LB | CIMT, FMD | Lower FMD and higher CIMT in NASH patients |
| [222] | 34 obese NAFLD vs. 20 obese controls | Proton MR spectroscopy | FMD | Lower in NAFLD |
| [218] | 49 NAFLD + DM vs. 50 NAFLD vs. 52 healthy controls | U/S | CIMT | Higher median CIMT in NAFLD patients when compared with controls irrespective of DM when adjusted for confounders |
| [217] | 350 NAFLD vs. 1934 controls with no cardiovascular disease | CAP | FMD, PAT ratio, PWV | Lower FMD and PAT ratio and higher PWV in NAFLD patients. Only PAT ratio statistically significant after adjusting for mean arterial pressure |
| [12] | 39 young men with NASH vs. 22 young men with NAFL vs. 41 young healthy control men | LB | PWV, FMD, CIMT | Lower FMD and higher PWV in NAFLD vs. controls, no difference between NAFL and NASH; higher CIMT in NASH vs. NAFL and controls |
| [223] | 176 NAFLD vs. 90 non-NAFLD controls | U/S | FMD | Lower FMD in NAFLD patients, especially in patients with grade 3 steatosis |
| [209] | 93 NAFLD vs. 37 healthy controls | U/S, LB | FMD, CIMT | Lower FMD in NAFLD, no statistically significant difference in CIMT |
| [207] | 42 NASH vs. 47 NAFL vs. 50 healthy controls | LB | FMD | Lower FMD in NAFLD, especially in NASH |
| [219] | 126 NAFLD (58 with MS, 68 without MS) vs. 31 CHB | U/S | FMD, CIMT | Lower FMD in NAFLD independent of MS; higher CIMT in patients with MS |
| [232] | 95 NAFLD vs. 90 obese controls | C/T | FMD, NMD | Lower FMD in NAFLD, no differences in NMD |
| [213] | 25 NAFLD vs. 25 healthy controls | U/S | FMD | Lower FMD in patients with NAFLD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.-M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. https://doi.org/10.3390/cells11162511
Nasiri-Ansari N, Androutsakos T, Flessa C-M, Kyrou I, Siasos G, Randeva HS, Kassi E, Papavassiliou AG. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells. 2022; 11(16):2511. https://doi.org/10.3390/cells11162511
Chicago/Turabian StyleNasiri-Ansari, Narjes, Theodoros Androutsakos, Christina-Maria Flessa, Ioannis Kyrou, Gerasimos Siasos, Harpal S. Randeva, Eva Kassi, and Athanasios G. Papavassiliou. 2022. "Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review" Cells 11, no. 16: 2511. https://doi.org/10.3390/cells11162511