
The Role of Endothelial Cells in the Onset, Development and Modulation of Vein Graft Disease

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
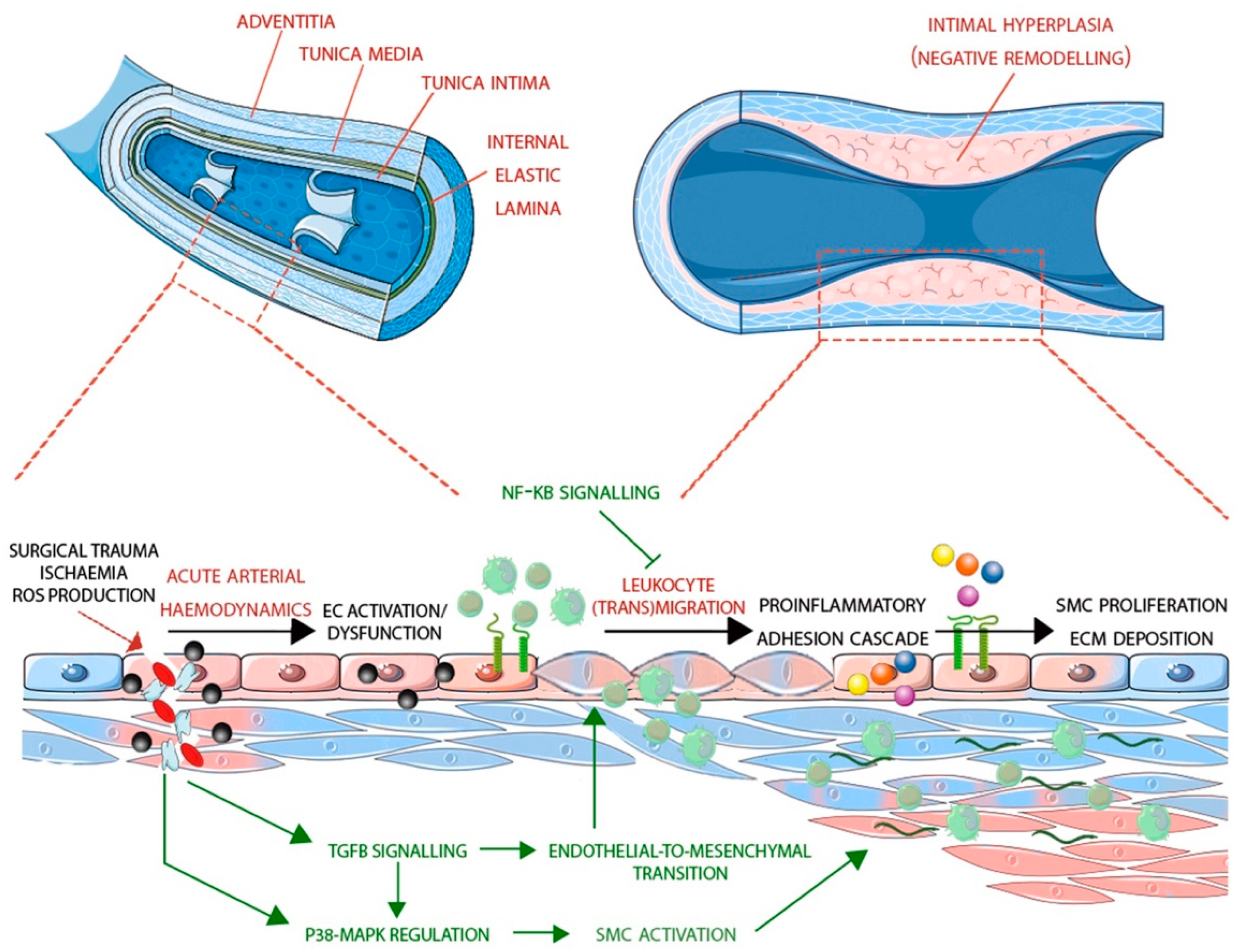
2. Differences between Arteries and Veins
3. Endothelial Cells Function in Vascular Homeostasis and Inflammation
4. The Role of Endothelial Cells in Vein Graft Disease
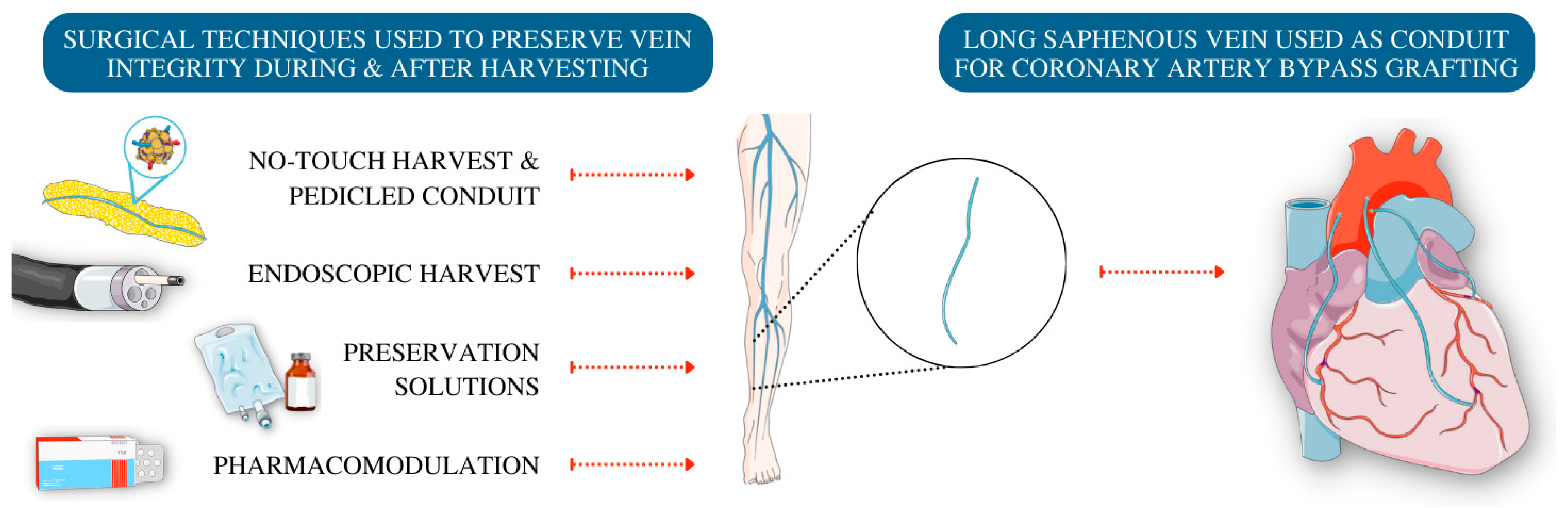
5. Techniques to Preserve Endothelial Cells in Vein Grafts
5.1. Harvesting Techniques
5.2. Storage Conditions
5.3. Pharma-Modulation
5.3.1. Aspirin
5.3.2. Statins
5.3.3. ACE Inhibitors/AngII Receptor Antagonists
5.4. Novel Approaches to Endothelial Preservation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Golledge, J.; Turner, R.; Harley, S.; Powell, J. Development of an in vitro model to study the response of saphenous vein endothelium to pulsatile arterial flow and circumferential deformation. Eur. J. Vasc. Endovasc. Surg. 1997, 13, 605–612. [Google Scholar] [CrossRef]
- Kockx, M.M.; Cambier, B.A.; Bortier, H.E.; De Meyer, G.R.; Van Cauwelaert, P.A. The modulation of smooth muscle cell phenotype is an early event in human aorto-coronary saphenous vein grafts. Virchows Arch. A 1992, 420, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Luong, L.A.; Chaudhury, H.; Ruud, O.; Punjabi, P.P.; Anderson, J.R.; Mullholand, J.W.; Clements, A.T.; Krams, R.; Foin, N.; et al. Dexamethasone Arterializes Venous Endothelial Cells by Inducing Mitogen-Activated Protein Kinase Phosphatase-1. Circulation 2011, 123, 524–532. [Google Scholar] [CrossRef]
- Ungogo, M.A. Targeting Smad-Mediated TGFβ Pathway in Coronary Artery Bypass Graft. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 119–130. [Google Scholar] [CrossRef] [PubMed]
- West, N.E.; Guzik, T.J.; Black, E.; Channon, K.M. Enhanced superoxide production in experimental venous bypass graft intimal hyperplasia: Role of NAD (P) H oxidase. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 189–194. [Google Scholar] [CrossRef]
- Sun, Q.; Kawamura, T.; Masutani, K.; Peng, X.; Sun, Q.; Stolz, D.B.; Pribis, J.P.; Billiar, T.R.; Sun, X.; Bermudez, C.A. Oral intake of hydrogen-rich water inhibits intimal hyperplasia in arterialized vein grafts in rats. Cardiovasc. Res. 2012, 94, 144–153. [Google Scholar] [CrossRef]
- Weaver, H.; Shukla, N.; Ellinsworth, D.; Jeremy, J.Y. Oxidative stress and vein graft failure: A focus on NADH oxidase, nitric oxide and eicosanoids. Curr. Opin. Pharmacol. 2012, 12, 160–165. [Google Scholar] [CrossRef]
- Lawrie, G.M.; Weilbacher, D.E.; Henry, P.D. Endothelium-dependent relaxation in human saphenous vein grafts: Effects of preparation and clinicopathologic correlations. J. Thorac. Cardiovasc. Surg. 1990, 100, 612–620. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Mehta, J.; Li, D. Identification and autoregulation of receptor for OX-LDL in cultured human coronary artery endothelial cells. Biochem. Biophys. Res. Commun. 1998, 248, 511–514. [Google Scholar] [CrossRef]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Moriwaki, H.; Murase, T.; Sawamura, T.; Masaki, T.; Hashimoto, N.; Kita, T. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999, 99, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, Y.; Philips, M.; Sawamura, T.; Mehta, J. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ. Res. 1999, 84, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Morawietz, H.; Rueckschloss, U.; Niemann, B.; Duerrschmidt, N.; Galle, J.; Hakim, K.; Zerkowski, H.-R.; Sawamura, T.; Holtz, J. Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 1999, 100, 899–902. [Google Scholar] [CrossRef]
- Chen, H.; Li, D.; Sawamura, T.; Inoue, K.; Mehta, J.L. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: Modulation by losartan. Biochem. Biophys. Res. Commun. 2000, 276, 1100–1104. [Google Scholar] [CrossRef]
- British Heart Foundation. BHF UK Facts and Figures. 2022. Available online: https://www.bhf.org.uk/what-we-do/news-from-the-bhf/contact-the-press-office/facts-and-figures#:~:text=Heart%20and%20circulatory%20diseases%20cause,men%20and%203.6%20million%20women (accessed on 25 September 2022).
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef]
- Gupta, K.K.; Ali, S.; Sanghera, R.S. Pharmacological options in atherosclerosis: A review of the existing evidence. Cardiol. Ther. 2019, 8, 5–20. [Google Scholar] [CrossRef]
- Neumann, F.-J.; Sousa-Uva, M.; Ahlsson, A.; Alfonso, F.; Banning, A.P.; Benedetto, U.; Byrne, R.A.; Collet, J.-P.; Falk, V.; Head, S.J.; et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur. Heart J. 2018, 40, 87–165. [Google Scholar] [CrossRef]
- Spadaccio, C.; Benedetto, U. Coronary artery bypass grafting (CABG) vs. percutaneous coronary intervention (PCI) in the treatment of multivessel coronary disease: Quo vadis?—A review of the evidences on coronary artery disease. Ann. Cardiothorac. Surg. 2018, 7, 506–515. [Google Scholar] [CrossRef]
- Bello, S.O.; Peng, E.W.; Sarkar, P.K. Conduits for coronary artery bypass surgery: The quest for second best. J. Cardiovasc. Med. 2011, 12, 411–421. [Google Scholar] [CrossRef]
- Gaudino, M.; Antoniades, C.; Benedetto, U.; Deb, S.; Di Franco, A.; Di Giammarco, G.; Fremes, S.; Glineur, D.; Grau, J.; He, G.-W. Mechanisms, consequences, and prevention of coronary graft failure. Circulation 2017, 136, 1749–1764. [Google Scholar] [CrossRef]
- Sabik, J.F., III. Understanding saphenous vein graft patency. Am. Heart Assoc. 2011, 124, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Guida, G.; Ward, A.O.; Bruno, V.D.; George, S.J.; Caputo, M.; Angelini, G.D.; Zakkar, M. Saphenous vein graft disease, pathophysiology, prevention, and treatment. A review of the literature. J. Card. Surg. 2020, 35, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.O.; Caputo, M.; Angelini, G.D.; George, S.J.; Zakkar, M. Activation and inflammation of the venous endothelium in vein graft disease. Atherosclerosis 2017, 265, 266–274. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, P.A. Arterial versus venous endothelial cells. Cell Tissue Res. 2009, 335, 5–16. [Google Scholar]
- Florey, L. The endothelial cell. Br. Med. J. 1966, 2, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Wang, Y.; Li, Y.; LeMaire, S.A.; Shen, Y.H. New technologies with increased precision improve understanding of endothelial cell heterogeneity in cardiovascular health and disease. Front. Cell Dev. Biol. 2021, 9, 679995. [Google Scholar] [CrossRef] [PubMed]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid shear stress sensing by the endothelial layer. Front. Physiol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Paszkowiak, J.J.; Dardik, A. Arterial wall shear stress: Observations from the bench to the bedside. Vasc. Endovasc. Surg. 2003, 37, 47–57. [Google Scholar] [CrossRef]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef]
- McQueen, L.W.; Ladak, S.S.; Zakkar, M. Acute shear stress and vein graft disease. Int. J. Biochem. Cell Biol. 2022, 144, 106173. [Google Scholar] [CrossRef]
- Zakkar, M.; Angelini, G.D.; Emanueli, C. Regulation of vascular endothelium inflammatory signalling by shear stress. Curr. Vasc. Pharmacol. 2016, 14, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Muñoz-Cánoves, P.; Yang, P.-C.; Brunelli, S. Mesenchymal Transitions in Development and Disease; Hindawi: London, UK, 2016. [Google Scholar]
- Moonen, J.-R.A.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; Van Kooten, T.G.; Van Luyn, M.J.; Zeebregts, C.J.; Krenning, G. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Van Varik, B.J.; Rennenberg, R.J.; Reutelingsperger, C.P.; Kroon, A.A.; de Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular endothelial cell biology: An update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Gori, T. Endothelial function: A short guide for the interventional cardiologist. Int. J. Mol. Sci. 2018, 19, 3838. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.; Dora, K. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, J.; Banga, J.; Moncada, S.; Palmer, R.; de Groot, P.; Sixma, J. Nitric oxide functions as an inhibitor of platelet adhesion under flow conditions. Circulation 1992, 85, 2284–2290. [Google Scholar] [CrossRef]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial–vascular smooth muscle cells interactions in atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.; Geczy, C. Coagulation and the expression of cell-mediated immunity. Immunol. Cell Biol. 1987, 65, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J. 2006, 273, 4377–4389. [Google Scholar] [CrossRef]
- Spertini, O. Regulation of leukocyte migration by adhesion molecules. Schweiz. Med. Wochenschr. 1996, 126, 1926–1934. [Google Scholar] [PubMed]
- Smith, E.M. Neuropeptides as Signal Molecules in Common with Leukocytes and the Hypothalamic–Pituitary–Adrenal Axis; Elsevier: Amsterdam, The Netherlands, 2008; pp. 3–14. [Google Scholar]
- Jung, U.; Ley, K. Regulation of E-selectin, P-selectin, and intercellular adhesion molecule 1 expression in mouse cremaster muscle vasculature. Microcirculation 1997, 4, 311–319. [Google Scholar] [CrossRef]
- Ley, K.; Tedder, T.F. Leukocyte interactions with vascular endothelium. New insights into selectin-mediated attachment and rolling. J. Immunol. 1995, 155, 525–528. [Google Scholar]
- Ley, K. Molecular mechanisms of leukocyte recruitment in the inflammatory process. Cardiovasc. Res. 1996, 32, 733–742. [Google Scholar] [CrossRef]
- Furie, M.B.; Tancinco, M.; Smith, C.W. Monoclonal antibodies to leukocyte integrins CD11a/CD18 and CD11b/CD18 or intercellular adhesion molecule-1 inhibit chemoattractant-stimulated neutrophil transendothelial migration in vitro. Blood 1991, 78, 2089–2097. [Google Scholar] [CrossRef]
- Mandell, K.J.; Parkos, C.A. The JAM family of proteins. Adv. Drug Deliv. Rev. 2005, 57, 857–867. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal. 2014, 20, 887–898. [Google Scholar] [CrossRef]
- Jerkic, M.; Letarte, M. Contribution of oxidative stress to endothelial dysfunction in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 6, 34. [Google Scholar] [CrossRef]
- Westerband, A.; Crouse, D.; Richter, L.C.; Aguirre, M.L.; Wixon, C.C.; James, D.C.; Mills, J.L.; Hunter, G.C.; Heimark, R.L. Vein adaptation to arterialization in an experimental model. J. Vasc. Surg. 2001, 33, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.O.; Angelini, G.D.; Caputo, M.; Evans, P.C.; Johnson, J.L.; Suleiman, M.S.; Tulloh, R.M.; George, S.J.; Zakkar, M. NF-κB inhibition prevents acute shear stress-induced inflammation in the saphenous vein graft endothelium. Sci. Rep. 2020, 10, 15133. [Google Scholar] [CrossRef]
- Wan, S.; George, S.; Berry, C.; Baker, A. Vein graft failure: Current clinical practice and potential for gene therapeutics. Gene Ther. 2012, 19, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, Q. New mouse model of vein bypass graft atherosclerosis. Heart Lung Circ. 2002, 11, 182–188. [Google Scholar] [CrossRef]
- Tseng, C.-N.; Karlöf, E.; Chang, Y.-T.; Lengquist, M.; Rotzius, P.; Berggren, P.-O.; Hedin, U.; Eriksson, E.E. Contribution of endothelial injury and inflammation in early phase to vein graft failure: The causal factors impact on the development of intimal hyperplasia in murine models. PLoS ONE 2014, 9, e98904. [Google Scholar] [CrossRef]
- Shintani, T.; Sawa, Y.; Takahashi, T.; Matsumiya, G.; Matsuura, N.; Miyamoto, Y.; Matsuda, H. Intraoperative transfection of vein grafts with the NFκB decoy in a canine aortocoronary bypass model: A strategy to attenuate intimal hyperplasia. Ann. Thorac. Surg. 2002, 74, 1132–1137. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Wang, J.-T.; Fan, Q.-X.; Geng, J.-G. Andrographolide inhibits NF-κB activation and attenuates neointimal hyperplasia in arterial restenosis. Cell Res. 2007, 17, 933–941. [Google Scholar] [CrossRef]
- Miyake, T.; Aoki, M.; Shiraya, S.; Tanemoto, K.; Ogihara, T.; Kaneda, Y.; Morishita, R. Inhibitory effects of NFκB decoy oligodeoxynucleotides on neointimal hyperplasia in a rabbit vein graft model. J. Mol. Cell. Cardiol. 2006, 41, 431–440. [Google Scholar] [CrossRef]
- Saunders, P.C.; Pintucci, G.; Bizekis, C.S.; Sharony, R.; Hyman, K.M.; Saponara, F.; Baumann, F.G.; Grossi, E.A.; Colvin, S.B.; Mignatti, P. Vein graft arterialization causes differential activation of mitogen-activated protein kinases. J. Thorac. Cardiovasc. Surg. 2004, 127, 1276–1284. [Google Scholar] [CrossRef]
- Gulkarov, I.; Bohmann, K.; Cinnante, K.M.; Pirelli, L.; Yu, P.-J.; Grau, J.B.; Pintucci, G.; Galloway, A.C.; Mignatti, P. Topical mitogen-activated protein kinases inhibition reduces intimal hyperplasia in arterialized vein grafts. J. Surg. Res. 2009, 154, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Wong, J.; Luo, H.; Hsiang, Y.N.; van Breemen, C.; Okon, E.B. Arterialization of a vein graft promotes cell cycle progression through Akt and p38 mitogen-activated protein kinase pathways: Impact of the preparation procedure. Can. J. Cardiol. 2007, 23, 1147–1154. [Google Scholar] [CrossRef]
- Zakkar, M.; Chaudhury, H.; Sandvik, G.; Enesa, K.; Luong, L.A.; Cuhlmann, S.; Mason, J.C.; Krams, R.; Clark, A.R.; Haskard, D.O. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ. Res. 2008, 103, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Luong, L.A.; Chaudhury, H.; Ruud, O.; Punjabi, P.P.; Anderson, J.R.; Mullholand, J.W.; Clements, A.T.; Krams, R.; Foin, N. Dexamethasone arterializes venous endothelial cells by inducing mitogen-activated protein kinase phosphatase-1: A novel antiinflammatory treatment for vein grafts? Circulation 2011, 123, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Model, L.; Ziegler, K.; Eghbalieh, S.D.; Dardik, A. Mechanisms of Vein Graft Adaptation to the Arterial Circulation–Insights Into the Neointimal Algorithm and Management Strategies. Circ. J. 2010, 74, 1501–1512. [Google Scholar] [CrossRef]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St. Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef]
- Wolff, R.A.; Malinowski, R.L.; Heaton, N.S.; Hullett, D.A.; Hoch, J.R. Transforming growth factor-β1 antisense treatment of rat vein grafts reduces the accumulation of collagen and increases the accumulation of h-caldesmon. J. Vasc. Surg. 2006, 43, 1028–1036. [Google Scholar] [CrossRef]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Kuo, H.-M.; Lin, C.-Y.; Lam, H.-C.; Lin, P.-R.; Chan, H.-H.; Tseng, J.-C.; Sun, C.-K.; Hsu, T.-F.; Wu, C.-C.; Yang, C.-Y. PTEN overexpression attenuates angiogenic processes of endothelial cells by blockade of endothelin-1/endothelin B receptor signaling. Atherosclerosis 2012, 221, 341–349. [Google Scholar] [CrossRef]
- Girão-Silva, T.; Fonseca-Alaniz, M.; Ribeiro-Silva, J.; Lee, J.; Patil, N.; Dallan, L.; Baker, A.; Harmsen, M.; Krieger, J.; Miyakawa, A. High stretch induces endothelial dysfunction accompanied by oxidative stress and actin remodeling in human saphenous vein endothelial cells. Sci. Rep. 2021, 11, 13493. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Guida, G.; Suleiman, M.; Angelini, G.D. Cardiopulmonary bypass and oxidative stress. Oxid. Med. Cell. Longev. 2015, 2015, 189863. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.O.; Sala-Newby, G.B.; Ladak, S.; Angelini, G.D.; Caputo, M.; Suleiman, M.-S.; Evans, P.C.; George, S.J.; Zakkar, M. Nrf2-Keap-1 imbalance under acute shear stress induces inflammatory response in venous endothelial cells. Perfusion 2021, 37, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [PubMed]
- Tekin, I.; Demir, M.; Özdem, S. Effect of different storage solutions on oxidative stress in human saphenous vein grafts. J. Cardiothorac. Surg. 2022, 17, 7. [Google Scholar] [CrossRef]
- Favaloro, R.G. Saphenous vein graft in the surgical treatment of coronary artery disease. Operative technique. J. Thorac. Cardiovasc. Surg. 1969, 58, 178–185. [Google Scholar] [CrossRef]
- Dries, D.; Mohammad, S.F.; Woodward, S.C.; Nelson, R.M.; Johnston, P.S. The influence of harvesting technique on endothelial preservation in saphenous veins. J. Surg. Res. 1992, 52, 219–225. [Google Scholar] [CrossRef]
- Cambria, R.P.; Megerman, J.; Abbott, W.M. Endothelial preservation in reversed and in situ autogenous vein grafts. A quantitative experimental study. Ann. Surg. 1985, 202, 50–55. [Google Scholar] [CrossRef]
- Angelini, G.D.; Passani, S.L.; Breckenridge, I.M.; Newby, A.C. Nature and pressure dependence of damage induced by distension of human saphenous vein coronary artery bypass grafts. Cardiovasc. Res. 1987, 21, 902–907. [Google Scholar] [CrossRef]
- Topal, G.; Loesch, A.; Dashwood, M.R. COVID-19—Endothelial Axis and Coronary Artery Bypass Graft Patency: A Target for Therapeutic Intervention? Braz. J. Cardiovasc. Surg. 2020, 35, 757–763. [Google Scholar] [CrossRef]
- De Vries, M.R.; Simons, K.H.; Jukema, J.W.; Braun, J.; Quax, P.H. Vein graft failure: From pathophysiology to clinical outcomes. Nat. Rev. Cardiol. 2016, 13, 451–470. [Google Scholar] [CrossRef] [PubMed]
- Souza, D. A new no-touch preparation technique: Technical notes. Scand. J. Thorac. Cardiovasc. Surg. 1996, 30, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Dreifaldt, M.; Souza, D.S.; Loesch, A.; Muddle, J.R.; Karlsson, M.G.; Filbey, D.; Bodin, L.; Norgren, L.; Dashwood, M.R. The “no-touch” harvesting technique for vein grafts in coronary artery bypass surgery preserves an intact vasa vasorum. J. Thorac. Cardiovasc. Surg. 2011, 141, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Loesch, A.; Dashwood, M.R. Vasa vasorum inside out/outside in communication: A potential role in the patency of saphenous vein coronary artery bypass grafts. J. Cell Commun. Signal. 2018, 12, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Gollasch, M.; Dubrovska, G. Paracrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol. Sci. 2004, 25, 647–653. [Google Scholar] [CrossRef]
- Dashwood, M.R.; Loesch, A. Inducible nitric oxide synthase and vein graft performance in patients undergoing coronary artery bypass surgery: Physiological or pathophysiological role? Curr. Vasc. Pharmacol. 2014, 12, 144–151. [Google Scholar] [CrossRef]
- Dashwood, M.R.; Dooley, A.; Shi-Wen, X.; Abraham, D.J.; Souza, D.S. Does periadventitial fat-derived nitric oxide play a role in improved saphenous vein graft patency in patients undergoing coronary artery bypass surgery? J. Vasc. Res. 2007, 44, 175–181. [Google Scholar] [CrossRef]
- Angelini, G.; Breckenridge, I.M.; Butchart, E.G.; Armistead, S.H.; Middleton, K.M.; Henderson, A.H.; Newby, A.C. Metabolic damage to human saphenous vein during preparation for coronary artery bypass grafting. Cardiovasc. Res. 1985, 19, 326–334. [Google Scholar] [CrossRef]
- Tian, M.; Wang, X.; Sun, H.; Feng, W.; Song, Y.; Lu, F.; Wang, L.; Wang, Y.; Xu, B.; Wang, H.; et al. No-Touch Versus Conventional Vein Harvesting Techniques at 12 Months After Coronary Artery Bypass Grafting Surgery: Multicenter Randomized, Controlled Trial. Circulation 2021, 144, 1120–1129. [Google Scholar] [CrossRef]
- Allen, K.; Cheng, D.; Cohn, W.; Connolly, M.; Edgerton, J.; Falk, V.; Martin, J.; Ohtsuka, T.; Vitali, R. Endoscopic vascular harvest in coronary artery bypass grafting surgery: A consensus statement of the International Society of Minimally Invasive Cardiothoracic Surgery (ISMICS) 2005. Innovations 2005, 1, 51–60. [Google Scholar] [CrossRef]
- Hashmi, S.F.; Krishnamoorthy, B.; Critchley, W.R.; Walker, P.; Bishop, P.W.; Venkateswaran, R.V.; Fildes, J.E.; Yonan, N. Histological and immunohistochemical evaluation of human saphenous vein harvested by endoscopic and open conventional methods. Interact. Cardiovasc. Thorac. Surg. 2014, 20, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.R.; Kendrick, D.E.; Allemang, M.T.; Gosling, A.F.; Kim, A.H.; Hausladen, A.; Kashyap, V.S. Endothelial Function Is Preserved in Veins Harvested by Either Endoscopic or Surgical Techniques. Ann. Vasc. Surg. 2017, 44, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Rousou, L.J.; Taylor, K.B.; Lu, X.G.; Healey, N.; Crittenden, M.D.; Khuri, S.F.; Thatte, H.S. Saphenous vein conduits harvested by endoscopic technique exhibit structural and functional damage. Ann. Thorac. Surg. 2009, 87, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lamm, P.; Juchem, G.; Milz, S.; Reichart, B. Continuous graft perfusion: Optimizing the quality of saphenous vein grafts. Heart Surg. Forum 2002, 5 (Suppl. S4), S355–S361. [Google Scholar] [PubMed]
- Krishnamoorthy, B.; Critchley, W.R.; Thompson, A.J.; Payne, K.; Morris, J.; Venkateswaran, R.V.; Caress, A.L.; Fildes, J.E.; Yonan, N. Study comparing vein integrity and clinical outcomes in open vein harvesting and 2 types of endoscopic vein harvesting for coronary artery bypass grafting: The VICO randomized clinical trial (vein integrity and clinical outcomes). Circulation 2017, 136, 1688–1702. [Google Scholar] [CrossRef]
- Bouhout, I.; Ali, W.B.; Perrault, L.P. The effect of storage solutions on endothelial function and saphenous vein graft patency. Indian J. Thorac. Cardiovasc. Surg. 2018, 34, 258–265. [Google Scholar] [CrossRef]
- Carpentier, S.; Murawsky, M.; Carpentier, A. Cytotoxicity of cardioplegic solutions: Evaluation by tissue culture. Circulation 1981, 64, II90-5. [Google Scholar]
- Wilbring, M.; Ebner, A.; Schoenemann, K.; Knaut, M.; Tugtekin, S.M.; Zatschler, B.; Waldow, T.; Alexiou, K.; Matschke, K.; Deussen, A. Heparinized blood better preserves cellular energy charge and vascular functions of intraoperatively stored saphenous vein grafts in comparison to isotonic sodium-chloride-solution. Clin. Hemorheol. Microcirc. 2013, 55, 445–455. [Google Scholar] [CrossRef]
- Takeuchi, K.; Sakamoto, S.; Nagayoshi, Y.; Nishizawa, H.; Matsubara, J. Reactivity of the human internal thoracic artery to vasodilators in coronary artery bypass grafting. Eur. J. Cardio-Thorac. Surg. 2004, 26, 956–959. [Google Scholar] [CrossRef]
- Baumann, F.G.; Catinella, F.P.; Cunningham, J.N., Jr.; Spencer, F.C. Vein contraction and smooth muscle cell extensions as causes of endothelial damage during graft preparation. Ann. Surg. 1981, 194, 199–211. [Google Scholar] [CrossRef]
- NICE. DuraGraft for Preserving Vascular Grafts. Available online: https://www.nice.org.uk/advice/mib184 (accessed on 8 May 2019).
- Aschacher, T.; Baranyi, U.; Aschacher, O.; Eichmair, E.; Messner, B.; Zimpfer, D.; Moayedifar, R.; Laufer, G.; Emmert, M.Y.; Sandner, S.E. A Novel Endothelial Damage Inhibitor Reduces Oxidative Stress and Improves Cellular Integrity in Radial Artery Grafts for Coronary Artery Bypass. Front. Cardiovasc. Med. 2021, 8, 736503. [Google Scholar] [CrossRef] [PubMed]
- Toto, F.; Torre, T.; Turchetto, L.; Lo Cicero, V.; Soncin, S.; Klersy, C.; Demertzis, S.; Ferrari, E. Efficacy of Intraoperative Vein Graft Storage Solutions in Preserving Endothelial Cell Integrity during Coronary Artery Bypass Surgery. J. Clin. Med. 2022, 11, 1093. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Ruel, M.; Jneid, H.; Ferguson, T.; Hiratzka, L.; Ikonomidis, J.; Lopez-Jimenez, F.; McNallan, S.; Patel, M.; Roger, V. American Heart Association Council on Cardiovascular Surgery and Anesthesia. Secondary prevention after coronary artery bypass graft surgery: A scientific statement from the American Heart Association. Circulation 2015, 131, 927–964. [Google Scholar] [CrossRef] [Green Version]
- Fuster, V.; Dyken, M.L.; Vokonas, P.S.; Hennekens, C. Aspirin as a therapeutic agent in cardiovascular disease. Special Writing Group. Circulation 1993, 87, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Ellinsworth, D.C.; Shukla, N.; Fleming, I.; Jeremy, J.Y. Interactions between thromboxane A2, thromboxane/prostaglandin (TP) receptors, and endothelium-derived hyperpolarization. Cardiovasc. Res. 2014, 102, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hennekens, C.H.; Schneider, W.R.; Pokov, A.; Hetzel, S.; DeMets, D.; Serebruany, V.; Schröder, H. A randomized trial of aspirin at clinically relevant doses and nitric oxide formation in humans. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 344–348. [Google Scholar] [CrossRef]
- Hetzel, S.; DeMets, D.; Schneider, R.; Borzak, S.; Schneider, W.; Serebruany, V.; Schröder, H.; Hennekens, C.H. Aspirin increases nitric oxide formation in chronic stable coronary disease. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 217–221. [Google Scholar] [CrossRef]
- Yang, Z.; Kozai, T.; van der Loo, B.; Viswambharan, H.; Lachat, M.; Turina, M.I.; Malinski, T.; Luscher, T.F. HMG-CoA reductase inhibition improves endothelial cell function and inhibits smooth muscle cell proliferation in human saphenous veins. J. Am. Coll. Cardiol. 2000, 36, 1691–1697. [Google Scholar] [CrossRef]
- Anderson, T.J.; Meredith, I.T.; Yeung, A.C.; Frei, B.; Selwyn, A.P.; Ganz, P. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N. Engl. J. Med. 1995, 332, 488–493. [Google Scholar] [CrossRef]
- The Post Coronary Artery Bypass Graft Trial Investigators. The Effect of Aggressive Lowering of Low-Density Lipoprotein Cholesterol Levels and Low-Dose Anticoagulation on Obstructive Changes in Saphenous-Vein Coronary-Artery Bypass Grafts. N. Engl. J. Med. 1997, 336, 153–163. [Google Scholar] [CrossRef]
- Alderman, M.H.; Madhavan, S.; Ooi, W.L.; Cohen, H.; Sealey, J.E.; Laragh, J.H. Association of the Renin-Sodium Profile with the Risk of Myocardial Infarction in Patients with Hypertension. N. Engl. J. Med. 1991, 324, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- O’Donohoe, M.K.; Schwartz, L.B.; Radic, Z.S.; Mikat, E.M.; McCann, R.L.; Hagen, P.O. Chronic ACE inhibition reduces intimal hyperplasia in experimental vein grafts. Ann. Surg. 1991, 214, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell-Boswell, M.; Robertson, A.L., Jr. Effects of angiotensin II and vasopressin on human smooth muscle cells in vitro. Exp. Mol. Pathol. 1981, 35, 265–276. [Google Scholar] [CrossRef]
- Daemen, M.J.; Lombardi, D.M.; Bosman, F.T.; Schwartz, S.M. Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ. Res. 1991, 68, 450–456. [Google Scholar] [CrossRef]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of Angiotensin II and Interleukin 6 in Human Coronary Atherosclerotic Plaques. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef]
- Hitomi, H.; Fukui, T.; Moriwaki, K.; Matsubara, K.; Sun, G.P.; Rahman, M.; Nishiyama, A.; Kiyomoto, H.; Kimura, S.; Ohmori, K.; et al. Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J. Hypertens. 2006, 24, 1089–1095. [Google Scholar] [CrossRef]
- Sun, J.-Z.; Cao, L.-H.; Liu, H. ACE inhibitors in cardiac surgery: Current studies and controversies. Hypertens. Res. 2011, 34, 15–22. [Google Scholar] [CrossRef]
- Enseleit, F.; Lüscher, T.F.; Ruschitzka, F. Angiotensin-converting enzyme inhibition and endothelial dysfunction: Focus on ramipril. Eur. Heart J. Suppl. 2003, 5 (Suppl. SA), A31–A36. [Google Scholar] [CrossRef]
- O’Driscoll, G.; Green, D.; Maiorana, A.; Stanton, K.; Colreavy, F.; Taylor, R. Improvement in endothelial function by angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1999, 33, 1506–1511. [Google Scholar] [CrossRef]
- Lee, A.F.; Dick, J.B.; Bonnar, C.E.; Struthers, A.D. Lisinopril improves arterial function in hyperlipidaemia. Clin. Sci. 1999, 96, 441–448. [Google Scholar] [CrossRef]
- Oosterga, M.; Voors, A.A.; Pinto, Y.M.; Buikema, H.; Grandjean, J.G.; Kingma, J.H.; Crijns, H.J.; van Gilst, W.H. Effects of quinapril on clinical outcome after coronary artery bypass grafting (The QUO VADIS Study). QUinapril on Vascular Ace and Determinants of Ischemia. Am. J. Cardiol. 2001, 87, 542–546. [Google Scholar] [CrossRef]
- Rouleau, J.L.; Warnica, W.J.; Baillot, R.; Block, P.J.; Chocron, S.; Johnstone, D.; Myers, M.G.; Calciu, C.D.; Dalle-Ave, S.; Martineau, P.; et al. Effects of angiotensin-converting enzyme inhibition in low-risk patients early after coronary artery bypass surgery. Circulation 2008, 117, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Trevelyan, J.; Needham, E.; Morris, A.; Mattu, R. Comparison of the effect of enalapril and losartan in conjunction with surgical coronary revascularisation versus revascularisation alone on systemic endothelial function. Heart 2005, 91, 1053–1057. [Google Scholar] [CrossRef]
- Ge, J.; Huang, D.; Liang, C.; Luo, Y.; Jia, Q.; Wang, K. Upregulation of lectinlike oxidized low-density lipoprotein receptor-1 expression contributes to the vein graft atherosclerosis: Modulation by losartan. Atherosclerosis 2004, 177, 263–268. [Google Scholar] [CrossRef]
- Southerland, K.W.; Frazier, S.B.; Bowles, D.E.; Milano, C.A.; Kontos, C.D. Gene therapy for the prevention of vein graft disease. Transl. Res. 2013, 161, 321–338. [Google Scholar] [CrossRef]
- Ohta, S.; Komori, K.; Yonemitsu, Y.; Onohara, T.; Matsumoto, T.; Sugimachi, K. Intraluminal gene transfer of endothelial cell-nitric oxide synthase suppresses intimal hyperplasia of vein grafts in cholesterol-fed rabbit: A limited biological effect as a result of the loss of medial smooth muscle cells. Surgery 2002, 131, 644–653. [Google Scholar] [CrossRef]
- Eichstaedt, H.C.; Liu, Q.; Chen, Z.; Bobustuc, G.C.; Terry, T.; Willerson, J.T.; Zoldhelyi, P. Gene transfer of COX-1 improves lumen size and blood flow in carotid bypass grafts. J. Surg. Res. 2010, 161, 162–167. [Google Scholar] [CrossRef]
- Zoldhelyi, P.; McNatt, J.; Xu, X.-M.; Loose-Mitchell, D.; Meidell, R.S.; Clubb Jr, F.J.; Buja, L.M.; Willerson, J.T.; Wu, K.K. Prevention of arterial thrombosis by adenovirus-mediated transfer of cyclooxygenase gene. Circulation 1996, 93, 10–17. [Google Scholar] [CrossRef]
- Kim, A.Y.; Walinsky, P.L.; Kolodgie, F.D.; Bian, C.; Sperry, J.L.; Deming, C.B.; Peck, E.A.; Shake, J.G.; Ang, G.B.; Sohn, R.H. Early loss of thrombomodulin expression impairs vein graft thromboresistance: Implications for vein graft failure. Circ. Res. 2002, 90, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Wyatt, M.J.; Newby, A.C. Reduction of early vein graft thrombosis by tissue plasminogen activator gene transfer. Thromb. Haemost. 2009, 102, 145–152. [Google Scholar] [PubMed]
- Schepers, A.; Eefting, D.; Bonta, P.; Grimbergen, J.; De Vries, M.; Van Weel, V.; De Vries, C.; Egashira, K.; Van Bockel, J.; Quax, P. Anti–MCP-1 gene therapy inhibits vascular smooth muscle cells proliferation and attenuates vein graft thickening both in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Tatewaki, H.; Egashira, K.; Kimura, S.; Nishida, T.; Morita, S.; Tominaga, R. Blockade of monocyte chemoattractant protein-1 by adenoviral gene transfer inhibits experimental vein graft neointimal formation. J. Vasc. Surg. 2007, 45, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, A.; Mann, M.J.; Dell’Acqua, G.; Dzau, V.J. Long-term stabilization of vein graft wall architecture and prolonged resistance to experimental atherosclerosis after E2F decoy oligonucleotide gene therapy. J. Thorac. Cardiovasc. Surg. 2001, 121, 714–722. [Google Scholar] [CrossRef]
- Alexander, J.H.; Hafley, G.; Harrington, R.A.; Peterson, E.D.; Ferguson, T.B., Jr.; Lorenz, T.J.; Goyal, A.; Gibson, M.; Mack, M.J.; Gennevois, D. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: A randomized controlled trial. JAMA 2005, 294, 2446–2454. [Google Scholar]
- Paschalaki, K.E.; Randi, A.M. Recent advances in endothelial colony forming cells toward their use in clinical translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Z.; Davison, F.; Hu, Y. Circulating progenitor cells regenerate endothelium of vein graft atherosclerosis, which is diminished in ApoE-deficient mice. Circ. Res. 2003, 93, e76–e86. [Google Scholar] [CrossRef]
- Kraus, X.; van de Flierdt, E.; Renzelmann, J.; Thoms, S.; Witt, M.; Scheper, T.; Blume, C. Peripheral blood derived endothelial colony forming cells as suitable cell source for pre-endothelialization of arterial vascular grafts under dynamic flow conditions. Microvasc. Res. 2022, 143, 104402. [Google Scholar] [CrossRef]
- Feng, Y.; Gordts, S.C.; Chen, F.; Hu, Y.; Van Craeyveld, E.; Jacobs, F.; Carlier, V.; Feng, Y.; Zhang, Z.; Xu, Q. Topical HDL administration reduces vein graft atherosclerosis in apo E deficient mice. Atherosclerosis 2011, 214, 271–278. [Google Scholar] [CrossRef]
- Brown, M.A.; Zhang, L.; Levering, V.W.; Wu, J.-H.; Satterwhite, L.L.; Brian, L.; Freedman, N.J.; Truskey, G.A. Human umbilical cord blood–derived endothelial cells reendothelialize vein grafts and prevent thrombosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Fashina, O.; Abbasciano, R.G.; McQueen, L.W.; Ladak, S.; George, S.J.; Suleiman, S.; Punjabi, P.P.; Angelini, G.D.; Zakkar, M. Large animal model of vein grafts intimal hyperplasia: A systematic review. Perfusion 2022, 02676591221091200. [Google Scholar] [CrossRef] [PubMed]
- McQueen, L.W.; Ladak, S.S.; Abbasciano, R.; George, S.J.; Suleiman, M.-S.; Angelini, G.D.; Murphy, G.J.; Zakkar, M. Next-Generation and Single-Cell Sequencing Approaches to Study Atherosclerosis and Vascular Inflammation Pathophysiology: A Systematic Review. Front. Cardiovasc. Med. 2022, 9, 849675. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladak, S.S.; McQueen, L.W.; Layton, G.R.; Aujla, H.; Adebayo, A.; Zakkar, M. The Role of Endothelial Cells in the Onset, Development and Modulation of Vein Graft Disease. Cells 2022, 11, 3066. https://doi.org/10.3390/cells11193066
Ladak SS, McQueen LW, Layton GR, Aujla H, Adebayo A, Zakkar M. The Role of Endothelial Cells in the Onset, Development and Modulation of Vein Graft Disease. Cells. 2022; 11(19):3066. https://doi.org/10.3390/cells11193066
Chicago/Turabian StyleLadak, Shameem S., Liam W. McQueen, Georgia R. Layton, Hardeep Aujla, Adewale Adebayo, and Mustafa Zakkar. 2022. "The Role of Endothelial Cells in the Onset, Development and Modulation of Vein Graft Disease" Cells 11, no. 19: 3066. https://doi.org/10.3390/cells11193066