
Growth State-Dependent Expression of Arachidonate Lipoxygenases in the Human Endothelial Cell Line EA.hy926


Abstract
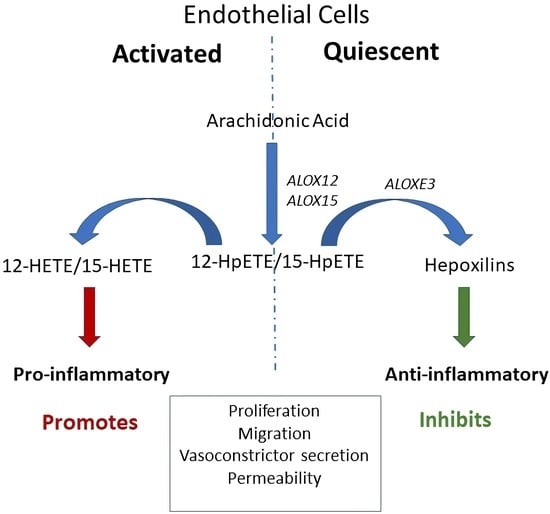
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction, cDNA Synthesis, and Reverse-Transcription PCR (RT-PCR)
2.3. Plasmids and Transfection
2.4. Dicer-Substrate siRNA (DsiRNA) Mediated Knockdown
2.5. Western Blotting
2.6. Endothelial Cell Perfusion for Simulation of Laminar Flow
2.7. Statistical Analysis
3. Results
3.1. RT-PCR Detects ALOXE3, ALOX15, and ALOX15B mRNAs in EA.hy926 Cells
3.2. Differential Cadherin (CDH5) Expression Distinguishes the Activated (Non-Confluent) and Quiescent (Confluent) EA.hy926 Cell Growth States
3.3. ALOX15B mRNA, but Not ALOX15 mRNA, Is Translated in EA.hy926 Cells
3.4. Identification of ALOXE3 Protein in Endothelial Cells
3.5. Shear Force Influences the Levels of ALOXE3 Protein in Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Chia, P.Y.; Teo, A.; Yeo, T.W. Overview of the Assessment of Endothelial Function in Humans. Front. Med. 2020, 7, 542567. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdev, U.; Lotze, M.T. Perpetual change: Autophagy, the endothelium, and response to vascular injury. J. Leukoc. Biol. 2017, 102, 221–235. [Google Scholar] [CrossRef]
- Ricard, N.; Bailly, S.; Guignabert, C.; Simons, M. The quiescent endothelium: Signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021, 18, 565–580. [Google Scholar] [CrossRef]
- Greenspan, L.J.; Weinstein, B.M. To be or not to be: Endothelial cell plasticity in development, repair, and disease. Angiogenesis 2021, 24, 251–269. [Google Scholar] [CrossRef]
- Islam, S.; Boström, K.I.; Di Carlo, D.; Simmons, C.A.; Tintut, Y.; Yao, Y.; Hsu, J.J. The Mechanobiology of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Front. Physiol. 2021, 12, 734215. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. 2020, 134, 2399–2418. [Google Scholar] [CrossRef]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Wu, B.; Mottola, G.; Schaller, M.; Upchurch, G.R., Jr.; Conte, M.S. Resolution of vascular injury: Specialized lipid mediators and their evolving therapeutic implications. Mol. Asp. Med. 2017, 58, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Wang, X.; Du, J.; Cui, Q.; Huang, Y.; Jin, H. Metabolic Reprogramming of Vascular Endothelial Cells: Basic Research and Clinical Applications. Front. Cell Dev. Biol. 2021, 9, 626047. [Google Scholar] [CrossRef]
- Desjardins, F.; Balligand, J.L. Nitric oxide-dependent endothelial function and cardiovascular disease. Acta Clin. Belg. 2006, 61, 326–334. [Google Scholar] [CrossRef]
- Cuneo, A.A.; Autieri, M.V. Expression and function of anti-inflammatory interleukins: The other side of the vascular response to injury. Curr. Vasc. Pharmacol. 2009, 7, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Esser-von Bieren, J. Eicosanoids in tissue repair. Immunol. Cell Biol. 2019, 97, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, M.S.; Desai, T.A.; Wu, B.; Schaller, M.; Werlin, E. Pro-resolving lipid mediators in vascular disease. J. Clin. Investig. 2018, 128, 3727–3735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogatcheva, N.V.; Sergeeva, M.G.; Dudek, S.M.; Verin, A.D. Arachidonic acid cascade in endothelial pathobiology. Microvasc. Res. 2005, 69, 107–127. [Google Scholar] [CrossRef]
- Chatterjee, A.; Komshian, S.; Sansbury, B.E.; Wu, B.; Mottola, G.; Chen, M.; Spite, M.; Conte, M.S. Biosynthesis of proresolving lipid mediators by vascular cells and tissues. FASEB J. 2017, 31, 3393–3402. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D. Eicosanoid blood vessel regulation in physiological and pathological states. Clin. Sci. 2020, 134, 2707–2727. [Google Scholar] [CrossRef]
- Alfranca, A.; Iñiguez, M.A.; Fresno, M.; Redondo, J.M. Prostanoid signal transduction and gene expression in the endothelium: Role in cardiovascular diseases. Cardiovasc. Res. 2006, 70, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.F.; Steele, C.; Belton, O.; Fitzgerald, D.J. Induction of cyclooxygenase-1 and -2 modulates angiogenic responses to engagement of alphavbeta3. Br. J. Haematol. 2003, 121, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.J.; Fleming, W.E.; McGuire, J.C.; Ohlmann, G.M.; Gray, G.D. The role of cyclooxygenase and lipoxygenase pathways in the adhesive interaction between bovine polymorphonuclear leukocytes and bovine endothelial cells. Prostaglandins Leukot. Med. 1986, 21, 221–230. [Google Scholar] [CrossRef]
- Setty, B.N.; Dampier, C.D.; Stuart, M.J. Arachidonic acid metabolites are involved in mediating red blood cell adherence to endothelium. J. Lab. Clin. Med. 1995, 125, 608–617. [Google Scholar] [PubMed]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Thiele, B.J. The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett. 1999, 449, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Porter, K.M.; Kang, B.Y.; Adesina, S.E.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Chronic hypoxia promotes pulmonary artery endothelial cell proliferation through H2O2-induced 5-lipoxygenase. PLoS ONE 2014, 9, e98532. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, J.W.; Liu, Z.H.; Shen, Y.; Ding, F.H.; Gu, G.; Liu, J.; Qiu, J.P.; Gao, J.; Zhang, R.Y.; et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis 2018, 278, 197–209. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Saleh, H.; El-Shafey, M.; Hussein, K.A.; El-Masry, K.; Baban, B.; Sheibani, N.; Wang, M.H.; Tawfik, A.; Al-Shabrawey, M. Targeting of 12/15-Lipoxygenase in retinal endothelial cells, but not in monocytes/macrophages, attenuates high glucose-induced retinal leukostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 636–645. [Google Scholar] [CrossRef]
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Taylor, C.G.; Aukema, H.M.; Zahradka, P. Importance of extracellular matrix and growth state for the EA.hy926 endothelial cell response to polyunsaturated fatty acids. PLoS ONE 2018, 13, e0197613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauls, S.D.; Rodway, L.A.; Winter, T.; Taylor, C.G.; Zahradka, P.; Aukema, H.M. Alpha-linolenic acid enhances the phagocytic and secretory functions of alternatively activated macrophages in part via changes to the oxylipin profile. Int. J. Biochem. Cell Biol. 2020, 119, 105662. [Google Scholar] [CrossRef] [PubMed]
- Sabbir, M.G.; Taylor, C.G.; Zahradka, P. Antisense overlapping long non-coding RNA regulates coding arachidonate 12-lipoxygenase gene by translational interference. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158987. [Google Scholar] [CrossRef]
- Hartig, S.M. Basic image analysis and manipulation in ImageJ. Curr. Protoc. Mol. Biol. 2013, 14, 14–15. [Google Scholar] [CrossRef]
- Sabbir, M.G.; Taylor, C.G.; Zahradka, P. Hypomorphic CAMKK2 in EA.hy926 endothelial cells causes abnormal transferrin trafficking, iron homeostasis and glucose metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118763. [Google Scholar] [CrossRef]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Gorman, R.R.; Oglesby, T.D.; Bundy, G.L.; Hopkins, N.K. Evidence for 15-HETE synthesis by human umbilical vein endothelial cells. Circulation 1985, 72, 708–712. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.W.; Kühn, H.; Kaiser, S.; Hennig, B.; Daugherty, A.; Toborek, M. Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J. Lipid Res. 2001, 42, 783–791. [Google Scholar] [CrossRef]
- Bacus, S.S.; Kiguchi, K.; Chin, D.; King, C.R.; Huberman, E. Differentiation of cultured human breast cancer cells (AU-565 and MCF-7) associated with loss of cell surface HER-2/neu antigen. Mol. Carcinog. 1990, 3, 350–362. [Google Scholar] [CrossRef]
- Cailleau, R.; Young, R.; Olivé, M.; Reeves, W.J., Jr. Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Brüne, B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front. Pharmacol. 2019, 10, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef]
- Pauls, S.D.; Du, Y.; Clair, L.; Winter, T.; Aukema, H.M.; Taylor, C.G.; Zahradka, P. Impact of Age, Menopause, and Obesity on Oxylipins Linked to Vascular Health. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 883–897. [Google Scholar] [CrossRef]
- Le, D.E.; García-Jaramillo, M.; Bobe, G.; Magana, A.A.; Vaswani, A.; Minnier, J.; Jump, D.B.; Rinkevich, D.; Alkayed, N.J.; Maier, C.S.; et al. Plasma Oxylipins: A Potential Risk Assessment Tool in Atherosclerotic Coronary Artery Disease. Front. Cardiovasc. Med. 2021, 8, 645786. [Google Scholar] [CrossRef]
- Kundumani-Sridharan, V.; Dyukova, E.; Hansen, D.E., 3rd; Rao, G.N. 12/15-Lipoxygenase mediates high-fat diet-induced endothelial tight junction disruption and monocyte transmigration: A new role for 15(S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J. Biol. Chem. 2013, 288, 15830–15842. [Google Scholar] [CrossRef] [Green Version]
- Chawengsub, Y.; Gauthier, K.M.; Campbell, W.B. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H495–H507. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Zhu, P.; Shah, S.J.; Blair, I.A. 15-oxo-Eicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol. Pharmacol. 2009, 76, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, H.; Gimbrone, M.A., Jr.; Schafer, A.I. Vascular lipoxygenase activity: Synthesis of 15-hydroxyeicosatetraenoic acid from arachidonic acid by blood vessels and cultured vascular endothelial cells. Thromb. Res. 1987, 45, 803–816. [Google Scholar] [CrossRef]
- Kakularam, K.R.; Karst, F.; Polamarasetty, A.; Ivanov, I.; Heydeck, D.; Kuhn, H. Paralog- and ortholog-specificity of inhibitors of human and mouse lipoxygenase-isoforms. Biomed. Pharmacother. 2022, 145, 112434. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Gu, J.L.; Natarajan, R.; Berliner, J.A.; Nadler, J.L. A leukocyte type of 12-lipoxygenase is expressed in human vascular and mononuclear cells. Evidence for upregulation by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 942–948. [Google Scholar] [CrossRef]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef]
- Du, Y.; Taylor, C.G.; Aukema, H.M.; Zahradka, P. PD146176 affects human EA.hy926 endothelial cell function by differentially modulating oxylipin production of LOX, COX and CYP epoxygenase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159156. [Google Scholar] [CrossRef]
- Singh, N.K.; Rao, G.N. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog. Lipid Res. 2019, 73, 28–45. [Google Scholar] [CrossRef]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, D.; Fürstenberger, G.; Krieg, P. Inducible expression of 15-lipoxygenase-2 and 8-lipoxygenase inhibits cell growth via common signaling pathways. J. Lipid Res. 2007, 48, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.G.; Bhatia, B.; Tang, S.; Schneider-Broussard, R. 15-lipoxygenase 2 (15-LOX2) is a functional tumor suppressor that regulates human prostate epithelial cell differentiation, senescence, and growth (size). Prostaglandins Other Lipid Mediat. 2007, 82, 135–146. [Google Scholar] [CrossRef]
- Guo, Y.; Nie, D. Tumor-suppressing 15-lipoxygenase-2: Time for prime time? Cell Cycle 2014, 13, 1836–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, R.; Cumbana, R.; Lehmann, C.; Kutzner, L.; Toewe, A.; Ferreirós, N.; Parnham, M.J.; Schebb, N.H.; Steinhilber, D.; Kahnt, A.S. Long-term stimulation of toll-like receptor-2 and -4 upregulates 5-LO and 15-LO-2 expression thereby inducing a lipid mediator shift in human monocyte-derived macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158702. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Nobili, E.; Folkersen, L.; Newman, J.W.; Pedersen, T.L.; Ekstrand, J.; Swedenborg, J.; Kühn, H.; Wheelock, C.E.; Hansson, G.K.; et al. 12- and 15-lipoxygenases in human carotid atherosclerotic lesions: Associations with cerebrovascular symptoms. Atherosclerosis 2011, 215, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Vijil, C.; Hermansson, C.; Jeppsson, A.; Bergström, G.; Hultén, L.M. Arachidonate 15-lipoxygenase enzyme products increase platelet aggregation and thrombin generation. PLoS ONE 2014, 9, e88546. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Garcia, A.; Thomas, C.P.; Keeney, D.S.; Zheng, Y.; Brash, A.R. The importance of the lipoxygenase-hepoxilin pathway in the mammalian epidermal barrier. Biochim. Biophys. Acta 2014, 1841, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Krieg, P.; Rosenberger, S.; de Juanes, S.; Latzko, S.; Hou, J.; Dick, A.; Kloz, U.; van der Hoeven, F.; Hausser, I.; Esposito, I.; et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J. Investig. Dermatol. 2013, 133, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Biringer, R.G. The enzymology of human eicosanoid pathways: The lipoxygenase branches. Mol. Biol. Rep. 2020, 47, 7189–7207. [Google Scholar] [CrossRef]
- Pace-Asciak, C.R. Pathophysiology of the hepoxilins. Biochim. Biophys. Acta 2015, 1851, 383–396. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Dyukova, E.; Singh, N.K.; Ohba, M.; Mobley, J.A.; Rao, G.N. Vascular endothelial tight junctions and barrier function are disrupted by 15(S)-hydroxyeicosatetraenoic acid partly via protein kinase C ε-mediated zona occludens-1 phosphorylation at threonine 770/772. J. Biol. Chem. 2014, 289, 3148–3163. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.M.; Reynaud, D.; Pace-Asciak, C.R. In vivo stimulation of 12(S)-lipoxygenase in the rat skin by bradykinin and platelet activating factor: Formation of 12(S)-HETE and hepoxilins, and actions on vascular permeability. Biochim. Biophys. Acta 1999, 1436, 354–362. [Google Scholar] [CrossRef]
- Yu, Z.; Schneider, C.; Boeglin, W.E.; Brash, A.R. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids 2007, 42, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.L.; Spitzbarth, N.; Nithipatikom, K.; Falck, J.R.; Campbell, W.B. Metabolism of 12-hydroperoxyeicosatetraenoic acid to vasodilatory trioxilin C3 by rabbit aorta. Biochim. Biophys. Acta 2003, 1622, 6–13. [Google Scholar] [CrossRef]
- Margalit, A.; Granot, Y. Endogenous hepoxilin A3, produced under short duration of high shear-stress, inhibits thrombin-induced aggregation in human platelets. Biochim. Biophys. Acta 1994, 1190, 173–176. [Google Scholar] [CrossRef]
- Nigam, S.; Zafiriou, M.P.; Deva, R.; Ciccoli, R.; Roux-Van der Merwe, R. Structure, biochemistry and biology of hepoxilins: An update. Febs. J. 2007, 274, 3503–3512. [Google Scholar] [CrossRef] [Green Version]
- Siangjong, L.; Goldman, D.H.; Kriska, T.; Gauthier, K.M.; Smyth, E.M.; Puli, N.; Kumar, G.; Falck, J.R.; Campbell, W.B. Vascular hepoxilin and trioxilins mediate vasorelaxation through TP receptor inhibition in mouse arteries. Acta Physiol. 2017, 219, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Hatley, M.E.; Srinivasan, S.; Reilly, K.B.; Bolick, D.T.; Hedrick, C.C. Increased production of 12/15 lipoxygenase eicosanoids accelerates monocyte/endothelial interactions in diabetic db/db mice. J. Biol. Chem. 2003, 278, 25369–25375. [Google Scholar] [CrossRef] [Green Version]
- Patricia, M.K.; Kim, J.A.; Harper, C.M.; Shih, P.T.; Berliner, J.A.; Natarajan, R.; Nadler, J.L.; Hedrick, C.C. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2615–2622. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Khan, H.; Xiao, J.; Cheang, W.S. Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 12029. [Google Scholar] [CrossRef]
- Sasson, S.; Eckel, J. Disparate effects of 12-lipoxygenase and 12-hydroxyeicosatetraenoic acid in vascular endothelial and smooth muscle cells and in cardiomyocytes. Arch. Physiol. Biochem. 2006, 112, 119–129. [Google Scholar] [CrossRef]
- Zheng, Q.; Zou, Y.; Teng, P.; Chen, Z.; Wu, Y.; Dai, X.; Li, X.; Hu, Z.; Wu, S.; Xu, Y.; et al. Mechanosensitive channel PIEZO1 senses shear force to induce KLF2/4 expression via CaMKII/MEKK3/ERK5 axis in endothelial cells. Cells 2022, 11, 2191. [Google Scholar] [CrossRef]
- Lu, Y.W.; Martino, N.; Gerlach, B.D.; Lamar, J.M.; Vincent, P.A.; Adam, A.P.; Schwarz, J.J. MEF2 is essential for endothelial homeostasis and the atheroprotective gene expression program. Artierioscler. Thromb. Vasc. Biol. 2021, 41, 1105–1123. [Google Scholar] [CrossRef] [PubMed]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional vascular endothelium as a driver of atherosclerosis: Emerging insights into pathogenesis and treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.-M.; Yuan, H.-Q.; Ren, Z.; Qu, S.-L.; Liu, L.-S.; Wei, D.-H.; Yin, K.; Fu, M.; Jiang, Z.-S. Endothelial to mesenchymal transition in atherosclerotic remodeling. Clin. Chim. Acta 2019, 490, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Moro, K.; Nagahashi, M.; Ramanathan, R.; Takabe, K.; Wakai, T. Resolvins and omega three polyunsaturated fatty acids: Clinical implications in inflammatory diseases and cancer. World J. Clin. Cases 2016, 4, 155–164. [Google Scholar] [CrossRef]








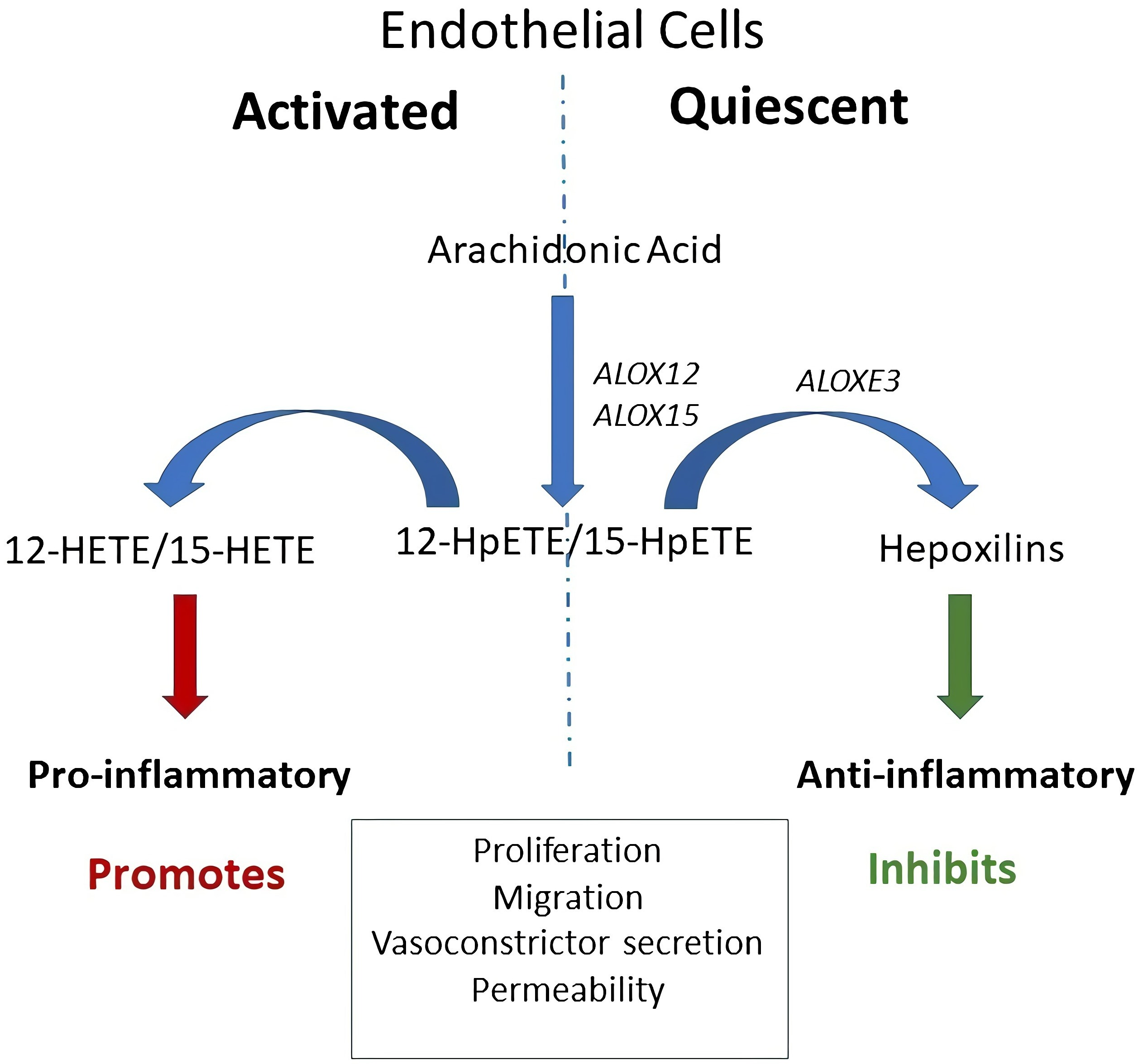{kind=link}
{kind=link}
{kind=link}
{kind=link}
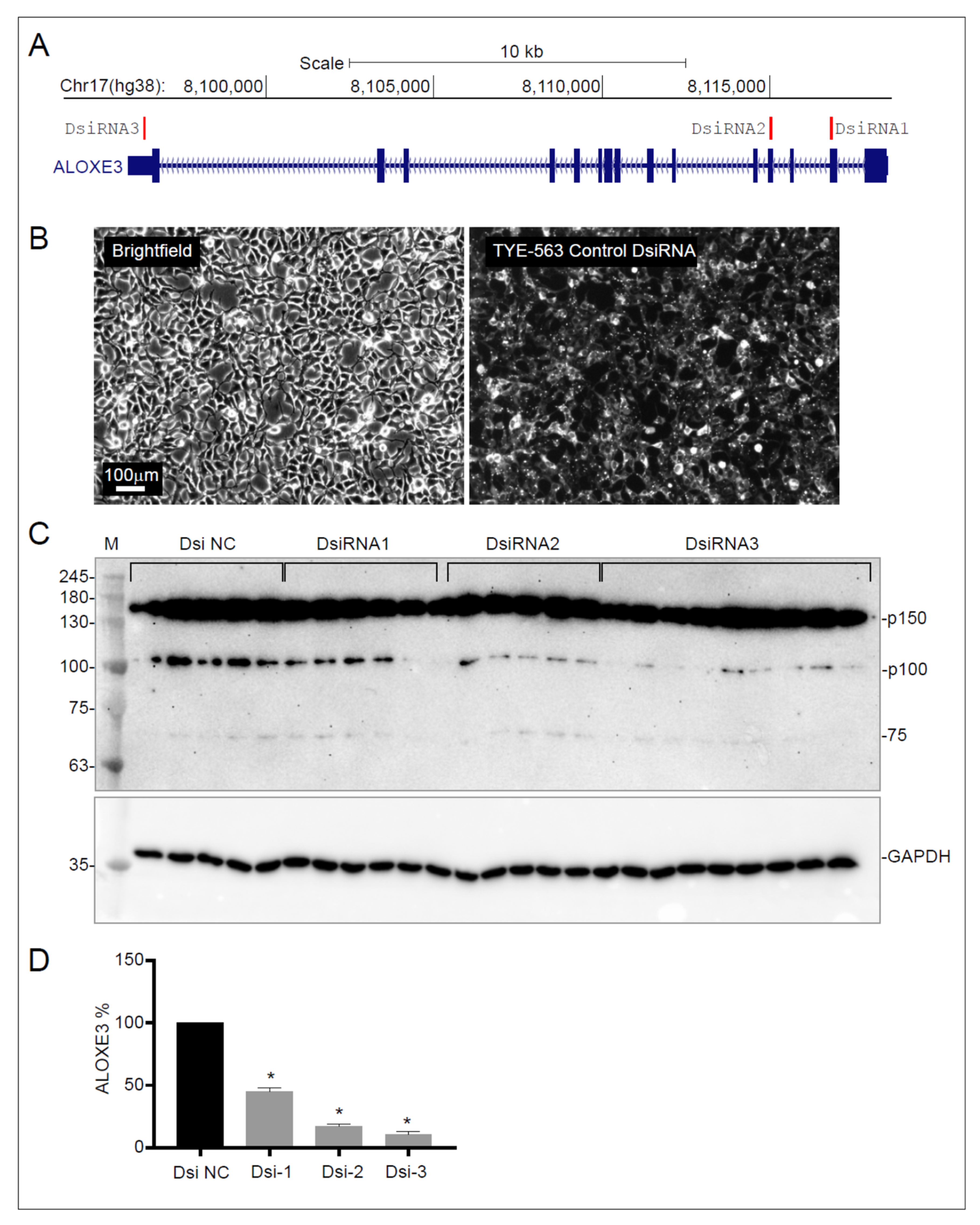{kind=link}
{kind=link}
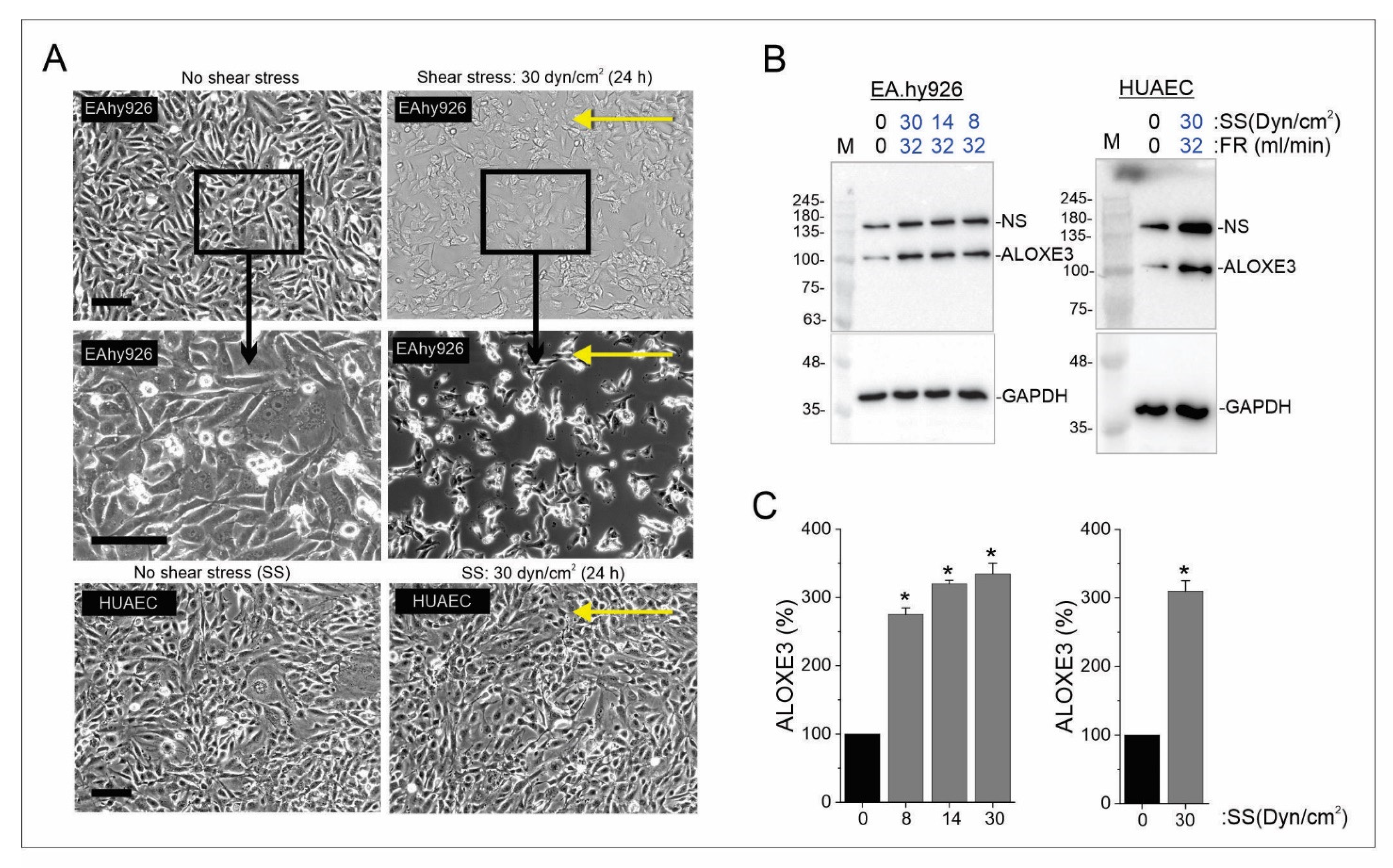{kind=link}
{kind=link}
| Primer Sequences for PCR | ||||
|---|---|---|---|---|
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) | Amplicon length (base pairs) | |
| ALOXE3 | GACGTCAACAGCTTTCAGGAGATG | AGATCGTCTTGGTGGCTGAGTAT | 184 | |
| ALOX5 | GGCAGGAAGACCTGATGTTTGG | GCAGCTCAAAGTCCACGATGAA | 189 | |
| ALOX12 | GAGAAGAGGCTGGACTTTGAATGG | CCATTGAGGAACTGGTAGCTGAAC | 200 | |
| ALOX12B | GGAAGAGGCTGAAGGACATTAGGA | TGAGGTACTGGTACCCAAAGAAGG | 111 | |
| ALOX15 | CCTTCCTAACCTACAGCTCCTTCT | TCACAGCCACGTCTGTCTTATAGT | 168 | |
| ALOX15B | GATACACCCTGCACATCAACACA | TCAGGCAGACACAGGAGAGAATAG | 154 | |
| GAPDH | ACGGGAAGCTTGTCATCAATGG | CCAGTAGAGGCAGGGATGATGT | 445 | |
| Sequences for DsiRNA | ||||
| DsiRNA1 | UUCCCCUGCUAUCAGUGGAUUGAAGUGAAGGGGACGAUAGUCACCUAACUUC | |||
| DsiRNA2 | CUGCAUGGUAGACGUCAACAGCUTTGGGACGUACCAUCUGCAGUUGUCGAAA | |||
| DsiRNA3 | AAGGAAGGAACCGCUUCACUUCUTGCUUUCCUUCCUUGGCGAAGUGAAGAAC | |||
| Antibodies | ||||
| Name | Source 1 | Species | Type | Catalog number |
| ALOXE3 | ThermoFisher | Rabbit | Polyclonal | PA5-21833 |
| ALOX15 | ThermoFisher | Mouse | Monoclonal | MA5-25853 |
| ALOX15B | SantaCruz Biotechnology | Mouse | Monoclonal (Clone-D9) | sc-271290 |
| Flag | Sigma-Aldrich | Mouse | monoclonal | F3165 |
| NPM1 | SantaCruz Biotechnology | Mouse | Monoclonal (Clone-NA24) | sc-53175 |
| GAPDH | SantaCruz Biotechnology | Mouse | Monoclonal (Clone-D9) | sc-25778 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabbir, M.G.; Wigle, J.T.; Taylor, C.G.; Zahradka, P. Growth State-Dependent Expression of Arachidonate Lipoxygenases in the Human Endothelial Cell Line EA.hy926. Cells 2022, 11, 2478. https://doi.org/10.3390/cells11162478
Sabbir MG, Wigle JT, Taylor CG, Zahradka P. Growth State-Dependent Expression of Arachidonate Lipoxygenases in the Human Endothelial Cell Line EA.hy926. Cells. 2022; 11(16):2478. https://doi.org/10.3390/cells11162478
Chicago/Turabian StyleSabbir, Mohammad G., Jeffrey T. Wigle, Carla G. Taylor, and Peter Zahradka. 2022. "Growth State-Dependent Expression of Arachidonate Lipoxygenases in the Human Endothelial Cell Line EA.hy926" Cells 11, no. 16: 2478. https://doi.org/10.3390/cells11162478