
Hyperoxia Reprogrammes Microvascular Endothelial Cell Response to Hypoxia in an Organ-Specific Manner


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Brain MVECs
2.2. Isolation of Lung MVEC
2.3. MVEC Culture and Hypoxia Treatment
2.4. Viability Time Course Using Propidium Iodide
2.5. Real Time Viability
2.6. qPCR
2.7. Western Blot Analysis
2.8. Western Blot for Mitochondrial Electron Transfer Chain Complexes
2.9. Glucose Uptake Assays
2.10. Glycolytic and Mitochondrial Stress Tests
2.11. Real Time Hypoxia Response Curves
2.12. Mitochondrial Staining
2.13. Migration Assays
2.14. Succinate Dehydrogenase Assay
2.15. Succinate Assay
2.16. Statistical Analysis
3. Results
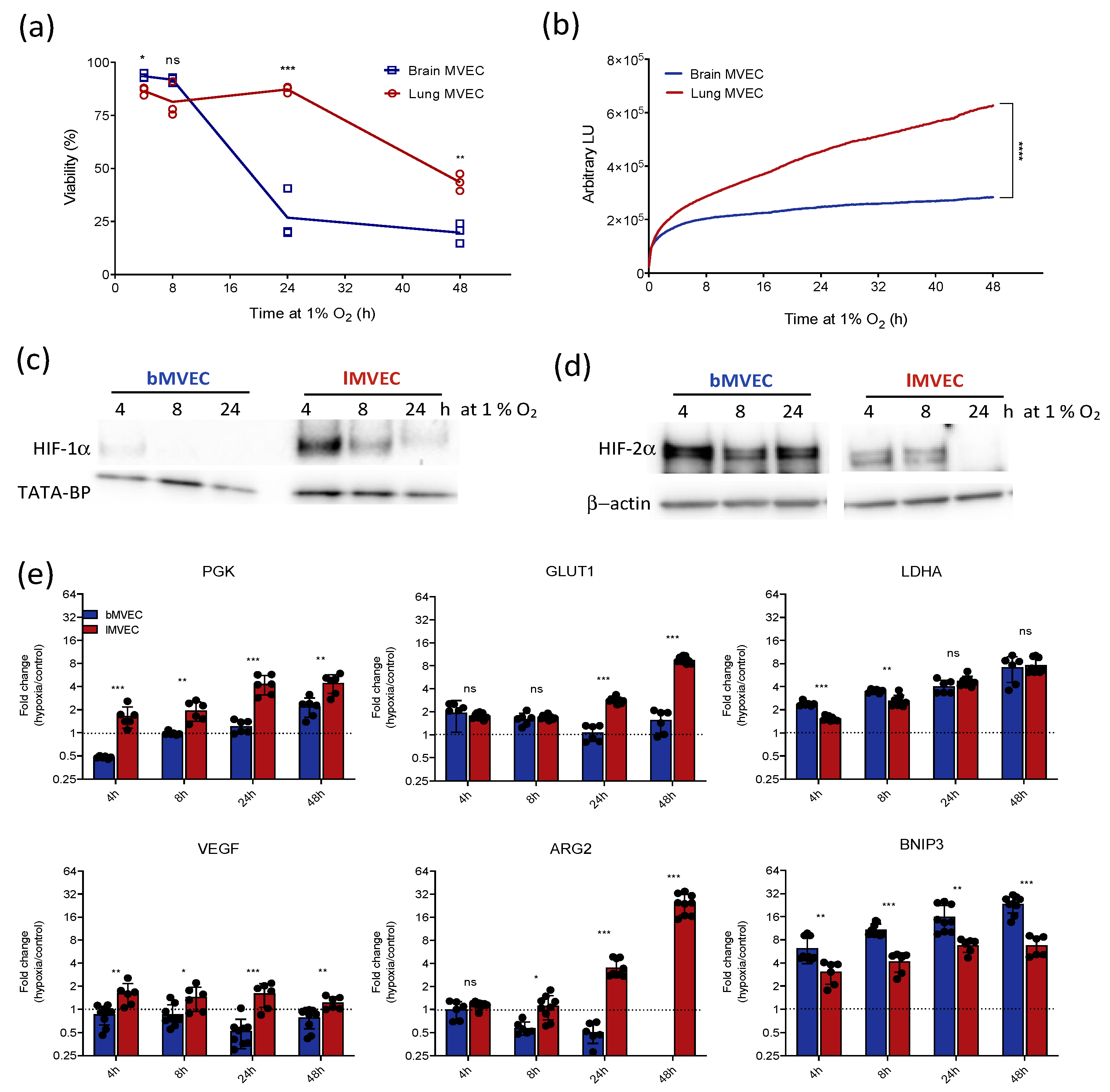
3.1. Brain and Lung Microvascular Endothelial Cells Respond Differently to Hypoxic Stress
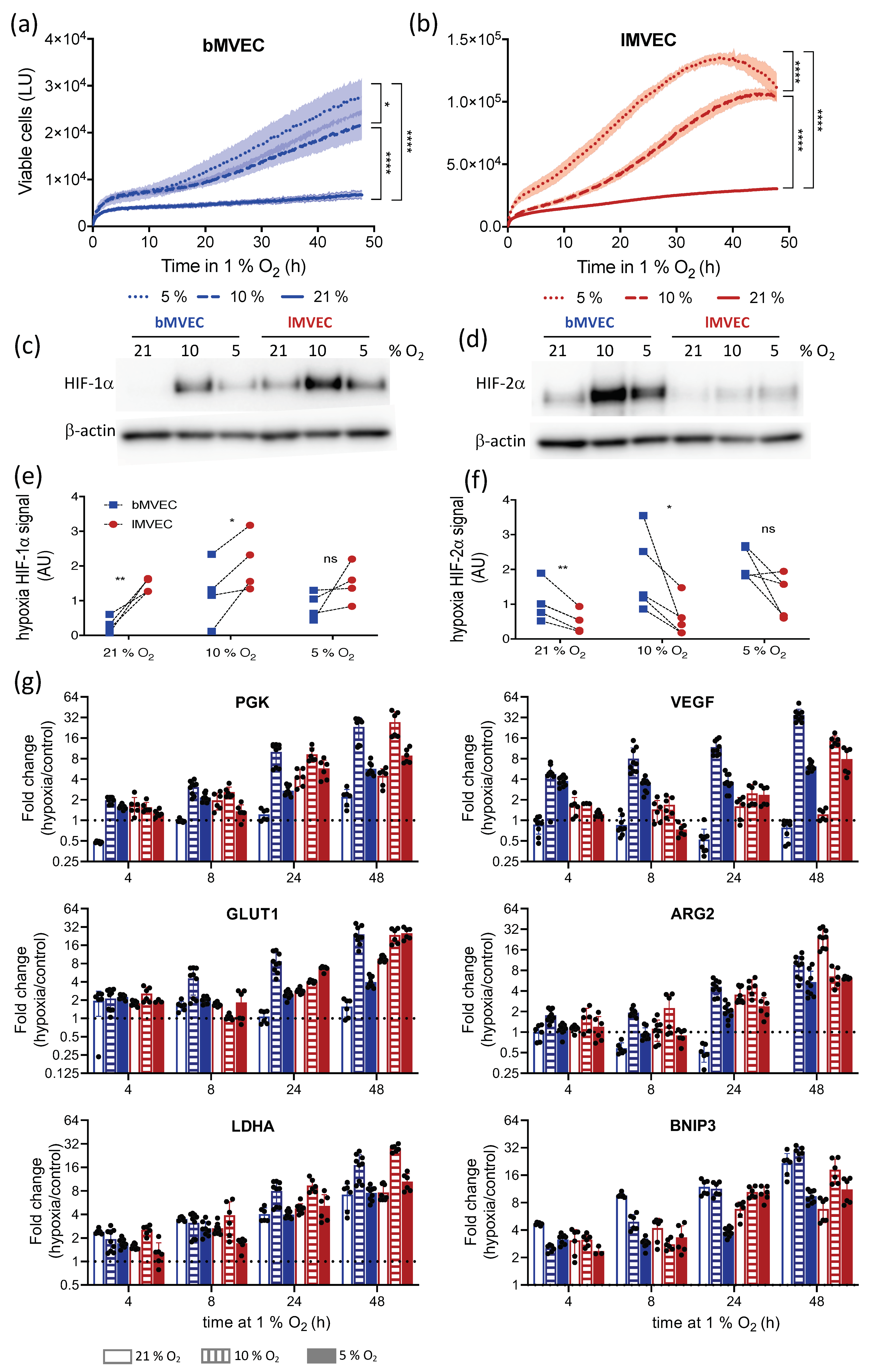
3.2. Oxygen Priming Differentially Conditions the Hypoxia Response of Brain and Lung MVEC
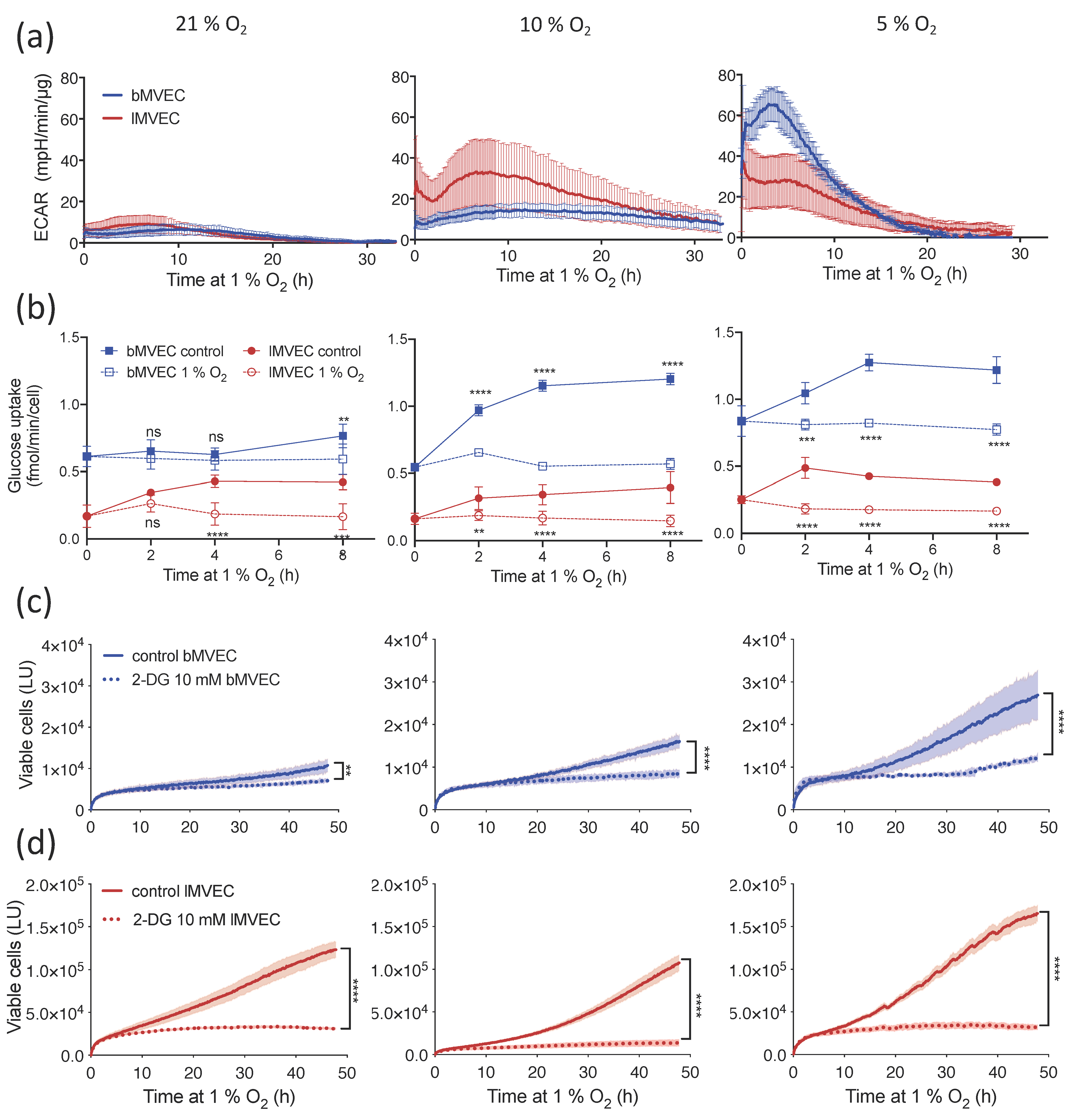
3.3. Glycolytic Activity of MVEC Is Altered as a Result of Oxygen Priming
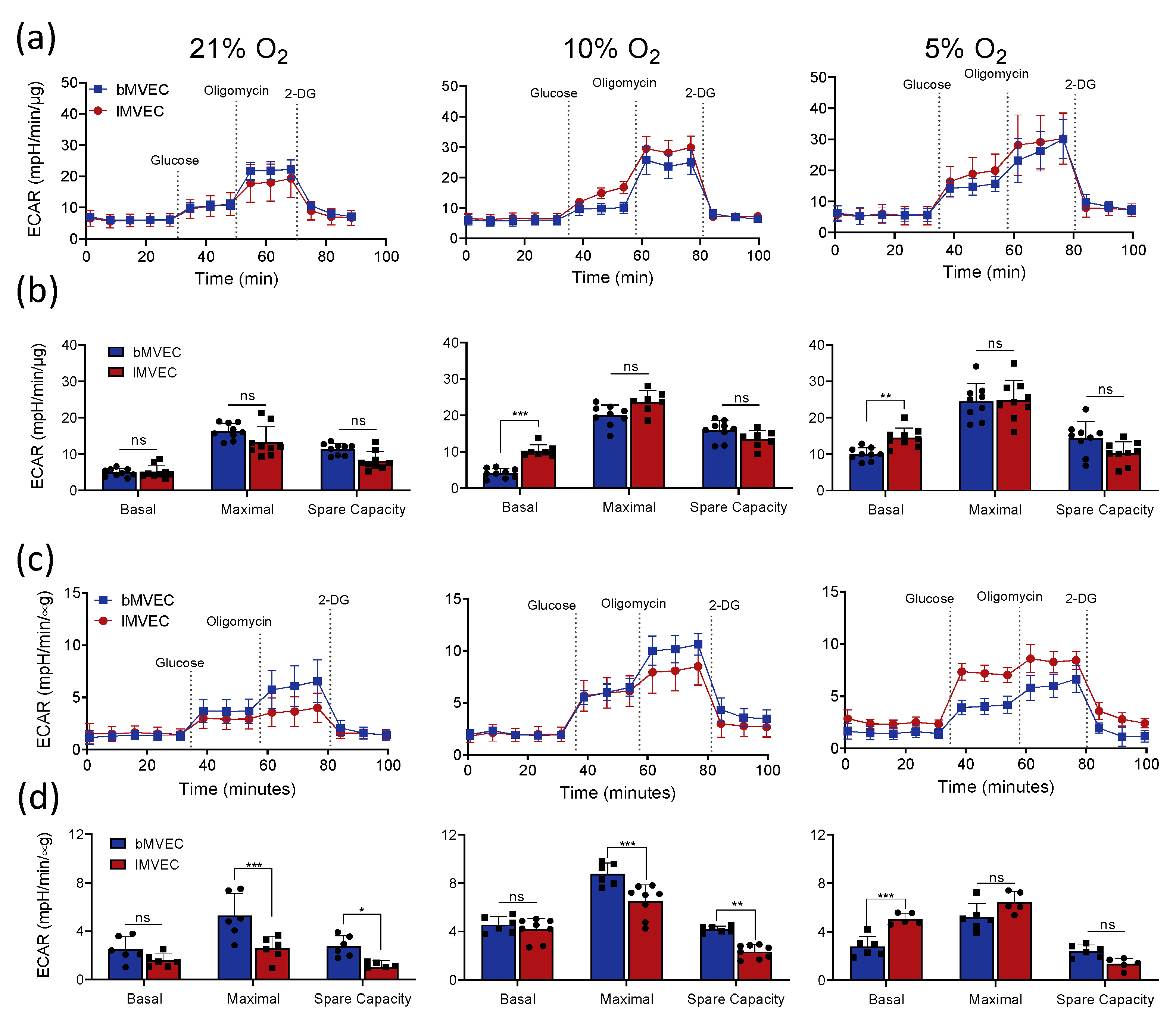
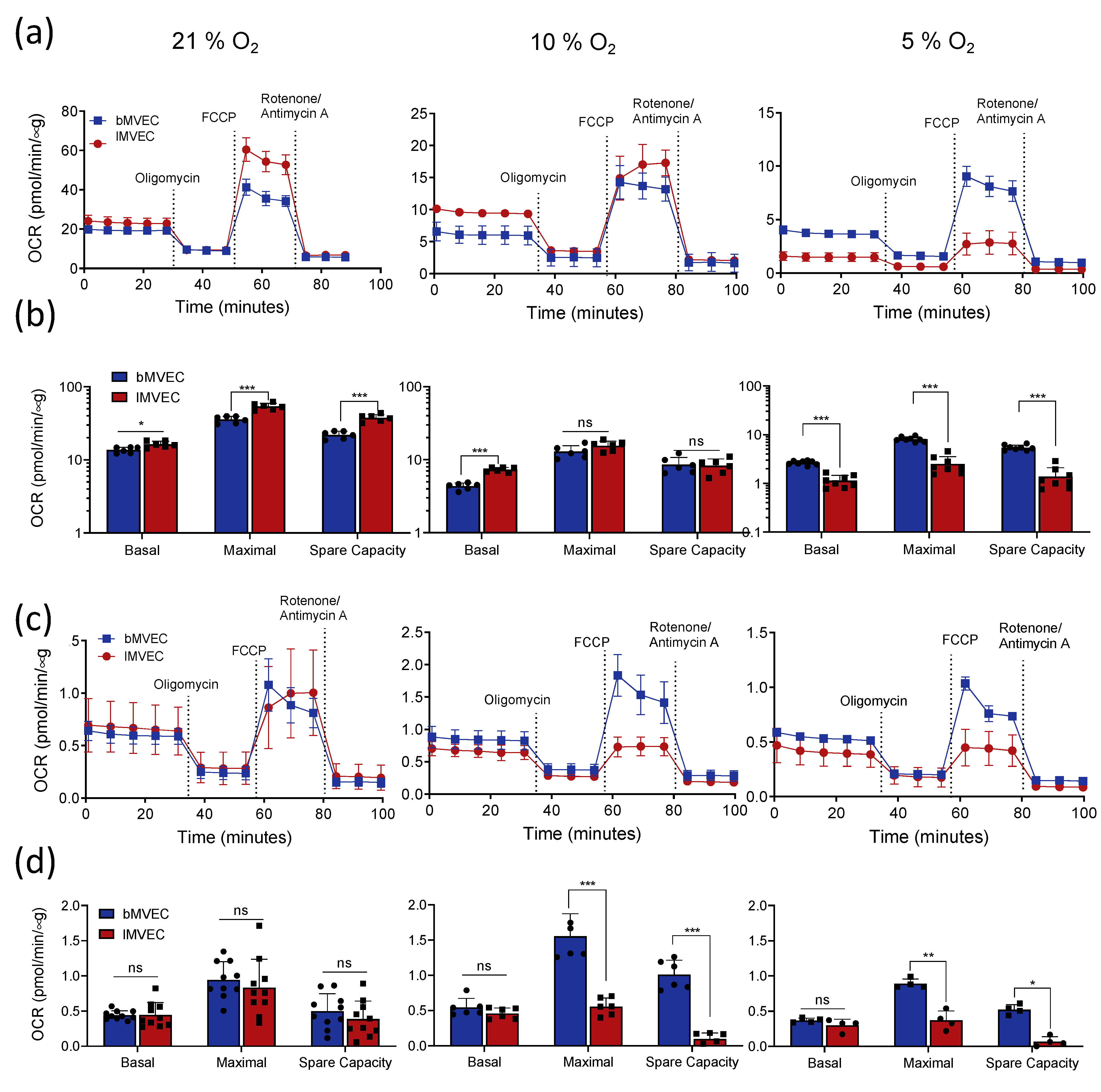
3.4. Effects of Oxygen Priming on Mitochondrial Respiration
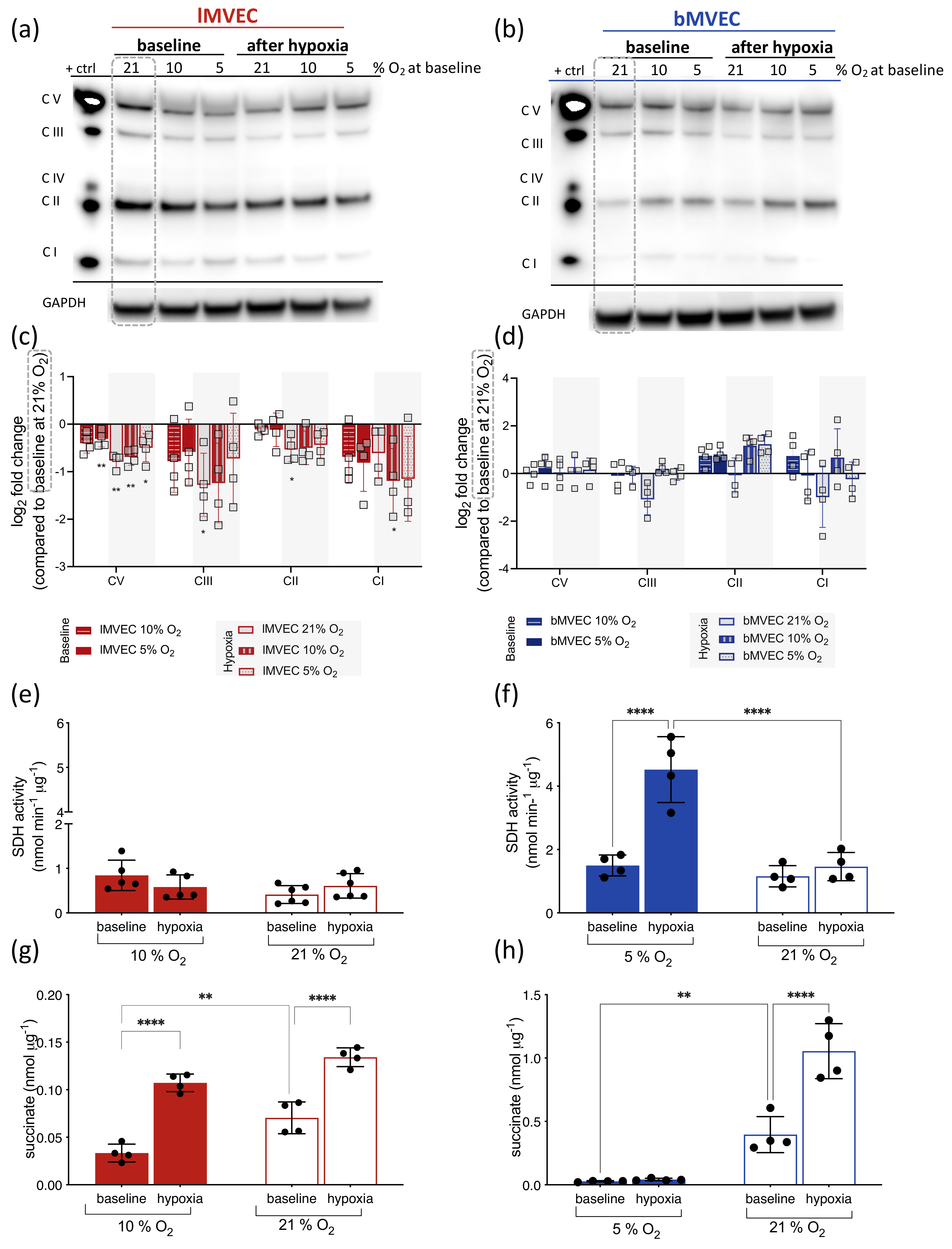
3.5. Effects of O2 on Mitochondrial ETC Complexes Is Organ-Specific
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Reiterer, M.; Branco, C.M. Endothelial cells and organ function: Applications and implications of understanding unique and reciprocal remodelling. FEBS J. 2020, 287, 1088–1100. [Google Scholar] [CrossRef]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’Hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab. 2020, 31, 1136–1153.e1137. [Google Scholar] [CrossRef]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.-S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular Signatures of Tissue-Specific Microvascular Endothelial Cell Heterogeneity in Organ Maintenance and Regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef]
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef]
- Ding, B.-S.; Nolan, D.J.; Guo, P.; Babazadeh, A.O.; Cao, Z.; Rosenwaks, Z.; Crystal, R.G.; Simons, M.; Sato, T.N.; Worgall, S.; et al. Endothelial-Derived Angiocrine Signals Induce and Sustain Regenerative Lung Alveolarization. Cell 2011, 147, 539–553. [Google Scholar] [CrossRef]
- Rojas-Camayo, J.; Mejia, C.R.; Callacondo, D.; Dawson, J.A.; Posso, M.; Galvan, C.A.; Davila-Arango, N.; Bravo, E.A.; Loescher, V.Y.; Padilla-Deza, M.M.; et al. Reference values for oxygen saturation from sea level to the highest human habitation in the Andes in acclimatised persons. Thorax 2018, 73, 776–778. [Google Scholar] [CrossRef]
- Prefaut, C.; Durand, F.; Mucci, P.; Caillaud, C. Exercise-Induced Arterial Hypoxaemia in Athletes. Sports Med. 2000, 30, 47–61. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Mistry, N.; Mazer, C.D.; Sled, J.G.; Lazarus, A.H.; Cahill, L.S.; Solish, M.; Zhou, Y.Q.; Romanova, N.; Hare, A.G.M.; Doctor, A.; et al. Red blood cell antibody-induced anemia causes differential degrees of tissue hypoxia in kidney and brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R611–R622. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. (Berl.) 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Kent, B.D.; Mitchell, P.D.; McNicholas, W.T. Hypoxemia in patients with COPD: Cause, effects, and disease progression. Int J. Chron. Obs. Pulmon Dis. 2011, 6, 199–208. [Google Scholar] [CrossRef]
- Ding, B.S.; Gomi, K.; Rafii, S.; Crystal, R.G.; Walters, M.S. Endothelial MMP14 is required for endothelial-dependent growth support of human airway basal cells. J. Cell Sci. 2015, 128, 2983–2988. [Google Scholar] [CrossRef]
- Cai, J.; Boulton, M. The pathogenesis of diabetic retinopathy: Old concepts and new questions. Eye (Lond.) 2002, 16, 242–260. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Mach, W.J.; Thimmesch, A.R.; Pierce, J.T.; Pierce, J.D. Consequences of Hyperoxia and the Toxicity of Oxygen in the Lung. Nurs. Res. Pract. 2011, 2011, 260482. [Google Scholar] [CrossRef]
- Brugniaux, J.V.; Coombs, G.B.; Barak, O.F.; Dujic, Z.; Sekhon, M.S.; Ainslie, P.N. Highs and lows of hyperoxia: Physiological, performance, and clinical aspects. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2018, 315, R1–R27. [Google Scholar] [CrossRef]
- Amarelle, L.; Quintela, L.; Hurtado, J.; Malacrida, L. Hyperoxia and Lungs: What We Have Learned From Animal Models. Front. Med. 2021, 8, 606678. [Google Scholar] [CrossRef]
- Dings, J.; Meixensberger, J.; Jäger, A.; Roosen, K. Clinical Experience with 118 Brain Tissue Oxygen Partial Pressure Catheter Probes. Neurosurgery 1998, 43, 1082–1094. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Dunn, J.F.; Vasconez, J.; Castillo, D.; Viscor, G. Partial pressure of oxygen in the human body: A general review. Am. J. Blood Res. 2019, 9, 1–14. [Google Scholar]
- Ast, T.; Mootha, V.K. Oxygen and mammalian cell culture: Are we repeating the experiment of Dr. Ox? Nat. Metab. 2019, 1, 858–860. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Allegranzi, B.; Zayed, B.; Bischoff, P.; Kubilay, N.Z.; de Jonge, S.; de Vries, F.; Gomes, S.M.; Gans, S.; Wallert, E.D.; Wu, X.; et al. New WHO recommendations on intraoperative and postoperative measures for surgical site infection prevention: An evidence-based global perspective. Lancet Infect. Dis. 2016, 16, e288–e303. [Google Scholar] [CrossRef]
- Hopf, H.W. Wound Tissue Oxygen Tension Predicts the Risk of Wound Infection in Surgical Patients. Arch. Surg. 1997, 132, 997. [Google Scholar] [CrossRef]
- Thom, S.R. Analytic Reviews: Hyperbaric Oxygen Therapy. J. Intensive Care Med. 1989, 4, 58–74. [Google Scholar] [CrossRef]
- Goldman, R.J. Hyperbaric Oxygen Therapy for Wound Healing and Limb Salvage: A Systematic Review. PMR 2009, 1, 471–489. [Google Scholar] [CrossRef]
- Chen, J.-R.; Xu, H.-Z.; Ding, J.-B.; Qin, Z.-Y. Radiotherapy after hyperbaric oxygenation in malignant gliomas. Curr. Med. Res. Opin. 2015, 31, 1977–1984. [Google Scholar] [CrossRef]
- Thom, S.R.; Milovanova, T.N.; Yang, M.; Bhopale, V.M.; Sorokina, E.M.; Uzun, G.; Malay, D.S.; Troiano, M.A.; Hardy, K.R.; Lambert, D.S.; et al. Vasculogenic stem cell mobilization and wound recruitment in diabetic patients: Increased cell number and intracellular regulatory protein content associated with hyperbaric oxygen therapy. Wound Repair Regen. 2011, 19, 149–161. [Google Scholar] [CrossRef]
- Banham, N.D. Oxygen toxicity seizures: 20 years’ experience from a single hyperbaric unit. Diving Hyperb. Med. 2011, 41, 202–210. [Google Scholar]
- Kistler, G.S.; Caldwell, P.R.B.; Weibel, E.R. Development of Fine Structural Damage to Alveolar and Capillary Lining Cells in Oxygen-Poisoned Rat Lungs. J. Cell Biol. 1967, 32, 605–628. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmad, A.; Ghosh, M.; Leslie, C.C.; White, C.W. Extracellular ATP-mediated signaling for survival in hyperoxia-induced oxidative stress. J. Biol. Chem. 2004, 279, 16317–16325. [Google Scholar] [CrossRef]
- Narula, P.; Xu, J.; Kazzaz, J.A.; Robbins, C.G.; Davis, J.M.; Horowitz, S. Synergistic cytotoxicity from nitric oxide and hyperoxia in cultured lung cells. Am. J. Physiol. 1998, 274, L411–L416. [Google Scholar] [CrossRef]
- Coisne, C.; Dehouck, L.; Faveeuw, C.; Delplace, Y.; Miller, F.; Landry, C.; Morissette, C.; Fenart, L.; Cecchelli, R.; Tremblay, P.; et al. Mouse syngenic in vitro blood–brain barrier model: A new tool to examine inflammatory events in cerebral endothelium. Lab. Investig. 2005, 85, 734–746. [Google Scholar] [CrossRef]
- Watson, P.M.D.; Paterson, J.C.; Thom, G.; Ginman, U.; Lundquist, S.; Webster, C.I. Modelling the endothelial blood-CNS barriers: A method for the production of robust in vitromodels of the rat blood-brain barrier and blood-spinal cord barrier. BMC Neurosci. 2013, 14, 59. [Google Scholar] [CrossRef]
- Ruck, T.; Bittner, S.; Epping, L.; Herrmann, A.M.; Meuth, S.G. Isolation of Primary Murine Brain Microvascular Endothelial Cells. J. Vis. Exp. 2014, e52204. [Google Scholar] [CrossRef]
- Welser-Alves, J.V.; Boroujerdi, A.; Milner, R. Isolation and Culture of Primary Mouse Brain Endothelial Cells. Methods Mol. Biol. 2014, 345–356. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: Evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J. Histochem. Cytochem. 1989, 37, 159–164. [Google Scholar] [CrossRef]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial Cell HIF-1α and HIF-2α Differentially Regulate Metastatic Success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef]
- Wild, J.M.; Fichele, S.; Woodhouse, N.; Paley, M.N.J.; Kasuboski, L.; van Beek, E.J.R. 3D volume-localizedpO2 measurement in the human lung with3He MRI. Magn. Reson. Med. 2005, 53, 1055–1064. [Google Scholar] [CrossRef]
- Newby, D.; Marks, L.; Lyall, F. Dissolved oxygen concentration in culture medium: Assumptions and pitfalls. Placenta 2005, 26, 353–357. [Google Scholar] [CrossRef]
- Duellman, S.J.; Zhou, W.; Meisenheimer, P.; Vidugiris, G.; Cali, J.J.; Gautam, P.; Wennerberg, K.; Vidugiriene, J. Bioluminescent, Nonlytic, Real-Time Cell Viability Assay and Use in Inhibitor Screening. Assay Drug Dev. Technol. 2015, 13, 456–465. [Google Scholar] [CrossRef]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF)-1α and HIF-2α Expression in Lung Epithelial Cells. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial cell–specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Reiterer, M.; Colaço, R.; Emrouznejad, P.; Jensen, A.; Rundqvist, H.; Johnson, R.S.; Branco, C. Author Correction: Acute and chronic hypoxia differentially predispose lungs for metastases. Sci. Rep. 2020, 10, 1627. [Google Scholar] [CrossRef]
- Krotova, K.; Patel, J.M.; Block, E.R.; Zharikov, S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am. J. Physiol.-Cell Physiol. 2010, 299, C1541–C1548. [Google Scholar] [CrossRef]
- Bellot, G.g.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.l.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Cater, D.; Garattini, S.; Marina, F.; Silver, I. Changes of oxygen tension in brain and somatic tissues induced by vasodilator and vasoconstrictor drugs. Proc. R. Soc. London. Ser. B. Biol. Sci. 1961, 155, 136–157. [Google Scholar]
- Smith, R.H.; Guilbeau, E.J.; Reneau, D.D. The oxygen tension field within a discrete volume of cerebral cortex. Microvasc. Res. 1977, 13, 233–240. [Google Scholar] [CrossRef]
- Wenger, R.; Kurtcuoglu, V.; Scholz, C.; Marti, H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of Metabolism by Hypoxia-Inducible Factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef]
- Jiang, Y.-Z.; Wang, K.; Li, Y.; Dai, C.-F.; Wang, P.; Kendziorski, C.; Chen, D.-B.; Zheng, J. Transcriptional and Functional Adaptations of Human Endothelial Cells to Physiological Chronic Low Oxygen1. Biol. Reprod. 2013, 88, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Gaber, T. Hypoxia/HIF Modulates Immune Responses. Biomedicines 2021, 9, 260. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzym. 2014, 542, 91–114. [Google Scholar] [CrossRef]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-like receptor 4 has an essential role in early skin wound healing. J. Investig. Derm. 2013, 133, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Cowburn, A.S.; Palazon, A.; Madhu, B.; Tyrakis, P.A.; Macías, D.; Bargiela, D.M.; Pietsch, S.; Gralla, M.; Evans, C.E.; et al. The Factor Inhibiting HIF Asparaginyl Hydroxylase Regulates Oxidative Metabolism and Accelerates Metabolic Adaptation to Hypoxia. Cell Metab. 2018, 27, 898–913.e897. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar Shear Stress Inhibits Endothelial Cell Metabolism via KLF2-Mediated Repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of Endothelial Cell Metabolism in Vessel Sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouché, A.; García-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Kuschinsky, W. Brain glucose transporters: Relationship to local energy demand. News Physiol. Sci. 2001, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lei, L.; Liu, D.; Jovin, I.; Russell, R.; Johnson, R.S.; Di Lorenzo, A.; Giordano, F.J. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1 -dependent function. Proc. Natl. Acad. Sci. USA 2012, 109, 17478–17483. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F.; Matschinsky, F.M. Hyperbaric oxygen toxicity in brain: A case of hyperoxia induced hypoglycemic brain syndrome. Med. Hypotheses 2019, 132, 109375. [Google Scholar] [CrossRef]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Nanadikar, M.S.; Vergel Leon, A.M.; Borowik, S.; Hillemann, A.; Zieseniss, A.; Belousov, V.V.; Bogeski, I.; Rehling, P.; Dudek, J.; Katschinski, D.M. O(2) affects mitochondrial functionality ex vivo. Redox Biol. 2019, 22, 101152. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Smith, K.A.; Schumacker, P.T. Sensors and signals: The role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J. Physiol. 2019, 597, 1033–1043. [Google Scholar] [CrossRef]
- Dröse, S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 578–587. [Google Scholar] [CrossRef]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guerrero, F.; Mazur, A.; Lambrechts, K.; Buzzacott, P.; Belhomme, M.; Theron, M. Reactive Oxygen Species, Mitochondria, and Endothelial Cell Death during In Vitro Simulated Dives. Med. Sci. Sports Exerc. 2015, 47, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef]
- Bukeirat, M.; Sarkar, S.N.; Hu, H.; Quintana, D.D.; Simpkins, J.W.; Ren, X. MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. J. Cereb. Blood Flow Metab. 2016, 36, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Rellick, S.L.; Hu, H.; Simpkins, J.W.; Ren, X. Evaluation of Bioenergetic Function in Cerebral Vascular Endothelial Cells. J. Vis. Exp. 2016, 54847. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef]
- Sharma, S.; Wang, J.; Cortes Gomez, E.; Taggart, R.T.; Baysal, B.E. Mitochondrial complex II regulates a distinct oxygen sensing mechanism in monocytes. Hum. Mol. Genet. 2017, 26, 1328–1339. [Google Scholar] [CrossRef]
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The genetic basis of isolated mitochondrial complex II deficiency. Mol. Genet. Metab 2020, 131, 53–65. [Google Scholar] [CrossRef]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef]
- Ma, C.; Beyer, A.M.; Durand, M.; Clough, A.V.; Zhu, D.; Norwood Toro, L.; Terashvili, M.; Ebben, J.D.; Hill, R.B.; Audi, S.H.; et al. Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Adrario, E.; Girardis, M.; Romano, R.; Pelaia, P.; Singer, M.; Donati, A. Arterial hyperoxia and mortality in critically ill patients: A systematic review and meta-analysis. Crit. Care 2014, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Helmerhorst, H.J.F.; Roos-Blom, M.-J.; van Westerloo, D.J.; de Jonge, E. Association Between Arterial Hyperoxia and Outcome in Subsets of Critical Illness. Crit. Care Med. 2015, 43, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Luchtel, R.; Gulani, P. Considerations for target oxygen saturation in COVID-19 patients: Are we under-shooting? BMC Med. 2020, 18, 260. [Google Scholar] [CrossRef]
- Ni, Y.-N.; Wang, Y.-M.; Liang, B.-M.; Liang, Z.-A. The effect of hyperoxia on mortality in critically ill patients: A systematic review and meta analysis. BMC Pulm. Med. 2019, 19, 53. [Google Scholar] [CrossRef]
- Rasmussen, B.S.; Perner, A.; Wetterslev, J.; Meyhoff, C.S.; Schjørring, O.L. Oxygenation targets in acutely ill patients: Still a matter of debate. Lancet 2018, 392, 2436–2437. [Google Scholar] [CrossRef]
- Wijesinghe, M.; Perrin, K.; Ranchord, A.; Simmonds, M.; Weatherall, M.; Beasley, R. Routine use of oxygen in the treatment of myocardial infarction: Systematic review. Heart 2008, 95, 198–202. [Google Scholar] [CrossRef]
- Volk, T.; Peters, J.; Sessler, D.I. The WHO recommendation for 80% perioperative oxygen is poorly justified. Der Anaesthesist 2017, 66, 227–229. [Google Scholar] [CrossRef]
- Mattishent, K.; Thavarajah, M.; Sinha, A.; Peel, A.; Egger, M.; Solomkin, J.; de Jonge, S.; Latif, A.; Berenholtz, S.; Allegranzi, B.; et al. Safety of 80% vs 30–35% fraction of inspired oxygen in patients undergoing surgery: A systematic review and meta-analysis. Br. J. Anaesth. 2019, 122, 311–324. [Google Scholar] [CrossRef]
- Choudry, M.; Tang, X.; Santorian, T.; Wasnik, S.; Xiao, J.; Xing, W.; Lau, K.W.; Mohan, S.; Baylink, D.J.; Qin, X. Deficient arginase II expression without alteration in arginase I expression attenuated experimental autoimmune encephalomyelitis in mice. Immunology 2018, 155, 85–98. [Google Scholar] [CrossRef]
- Gupta, K.K.; Donahue, D.L.; Sandoval-Cooper, M.J.; Castellino, F.J.; Ploplis, V.A. Abrogation of plasminogen activator inhibitor-1-vitronectin interaction ameliorates acute kidney injury in murine endotoxemia. PLoS ONE 2015, 10, e0120728. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Cowburn, A.S.; Torres-Torrelo, H.; Ortega-Sáenz, P.; López-Barneo, J.; Johnson, R.S. HIF-2α is essential for carotid body development and function. Elife 2018, 7, e34681. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiterer, M.; Eakin, A.; Johnson, R.S.; Branco, C.M. Hyperoxia Reprogrammes Microvascular Endothelial Cell Response to Hypoxia in an Organ-Specific Manner. Cells 2022, 11, 2469. https://doi.org/10.3390/cells11162469
Reiterer M, Eakin A, Johnson RS, Branco CM. Hyperoxia Reprogrammes Microvascular Endothelial Cell Response to Hypoxia in an Organ-Specific Manner. Cells. 2022; 11(16):2469. https://doi.org/10.3390/cells11162469
Chicago/Turabian StyleReiterer, Moritz, Amanda Eakin, Randall S. Johnson, and Cristina M. Branco. 2022. "Hyperoxia Reprogrammes Microvascular Endothelial Cell Response to Hypoxia in an Organ-Specific Manner" Cells 11, no. 16: 2469. https://doi.org/10.3390/cells11162469