
HSF1 Can Prevent Inflammation following Heat Shock by Inhibiting the Excessive Activation of the ATF3 and JUN&FOS Genes

 , , , ,
, , , ,
Abstract
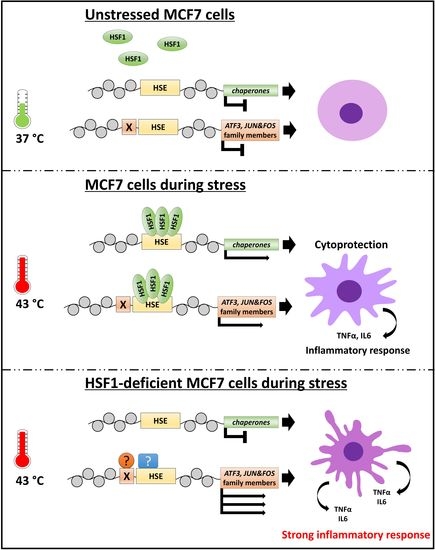
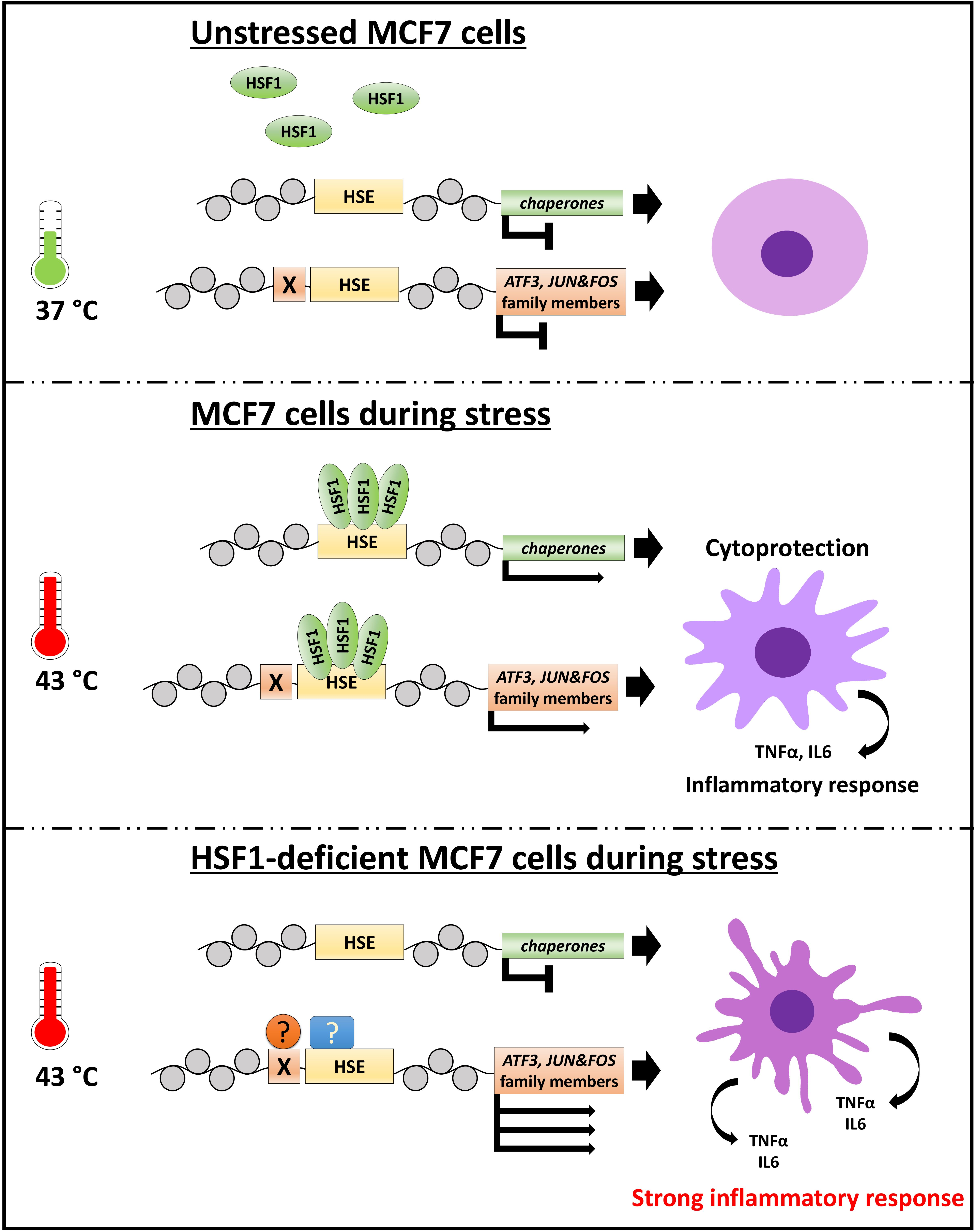
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Treatments
2.2. Global Gene Expression Profiling and Analysis
2.3. RNA Isolation, cDNA Synthesis, and RT-qPCR
2.4. Protein Extraction and Western Blotting
2.5. Chromatin Immunoprecipitation (ChIP), Global Profiling of Chromatin Binding Sites, and ChIP-qPCR
2.6. ChIP-Seq and RNA-Seq Data Integration
2.7. Proximity Ligation Assay
2.8. Measurement of Secreted Cytokines by ELISA
2.9. TCGA (The Cancer Genome Atlas) Data Analysis: Data Retrieval, Selection of Cases, and Differential Expression Analysis
3. Results
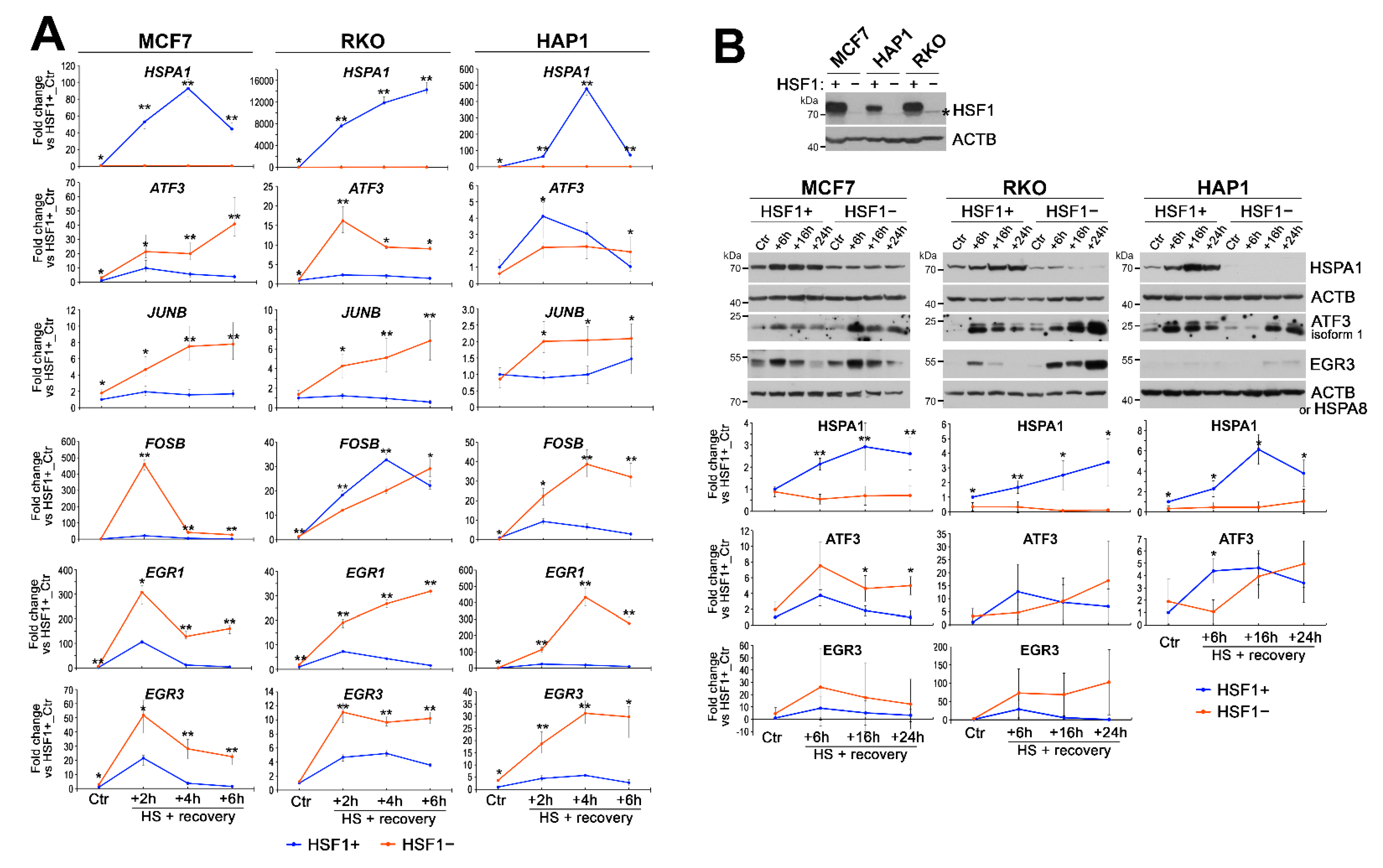
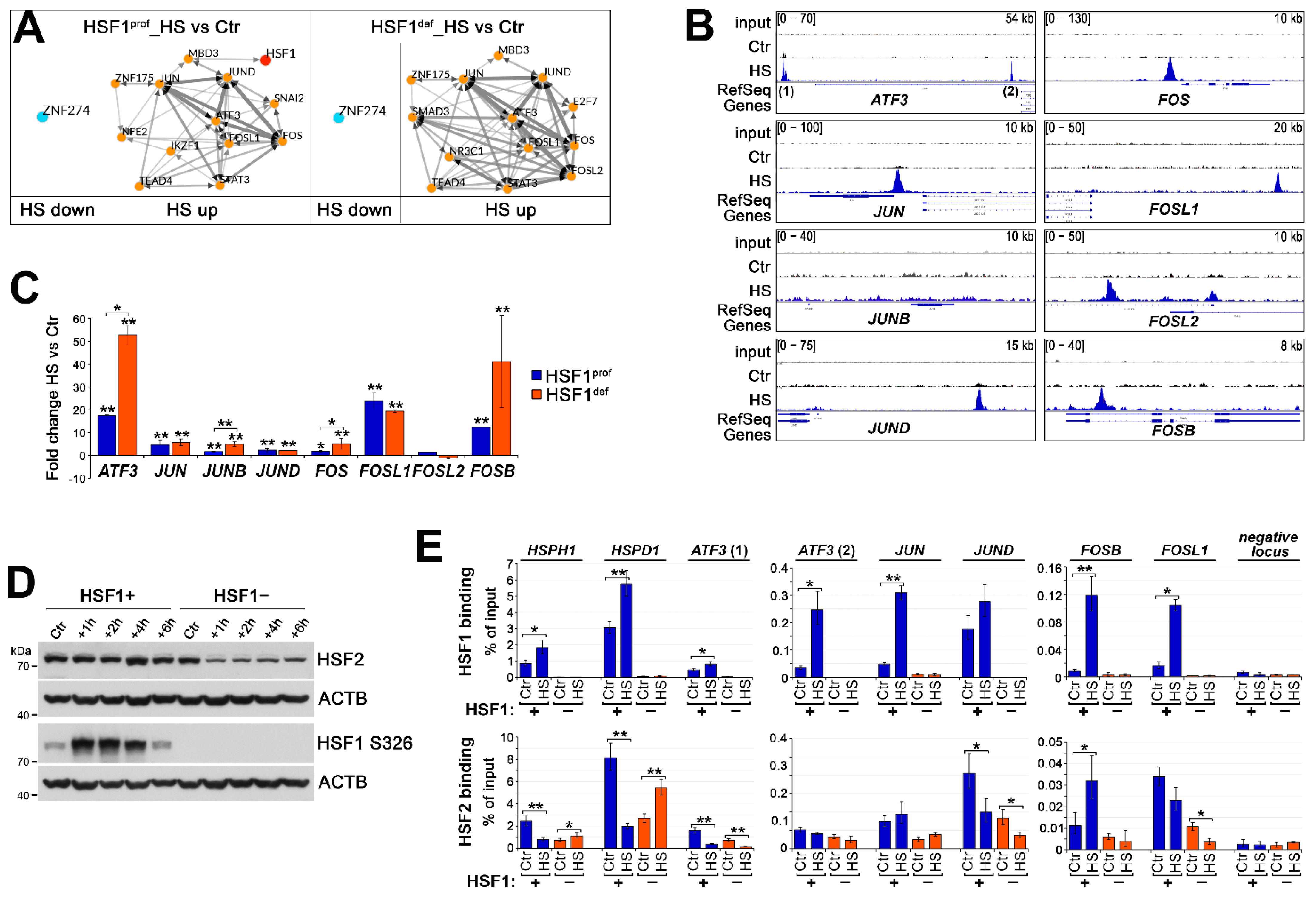
3.1. HSF1 Deficiency Generally Impairs Transcriptional Response to Heat Shock but Results in Enhanced Induction of a Subset of Genes
3.2. HSF1 Has an Activating Function in the Heat-Induced Transcription but May Inhibit the Overactivation of Certain Genes including Those Coding for Transcription Regulators
3.3. Heat Shock-Induced Inflammatory Response Could Be Stronger in HSF1-Deficient Cells
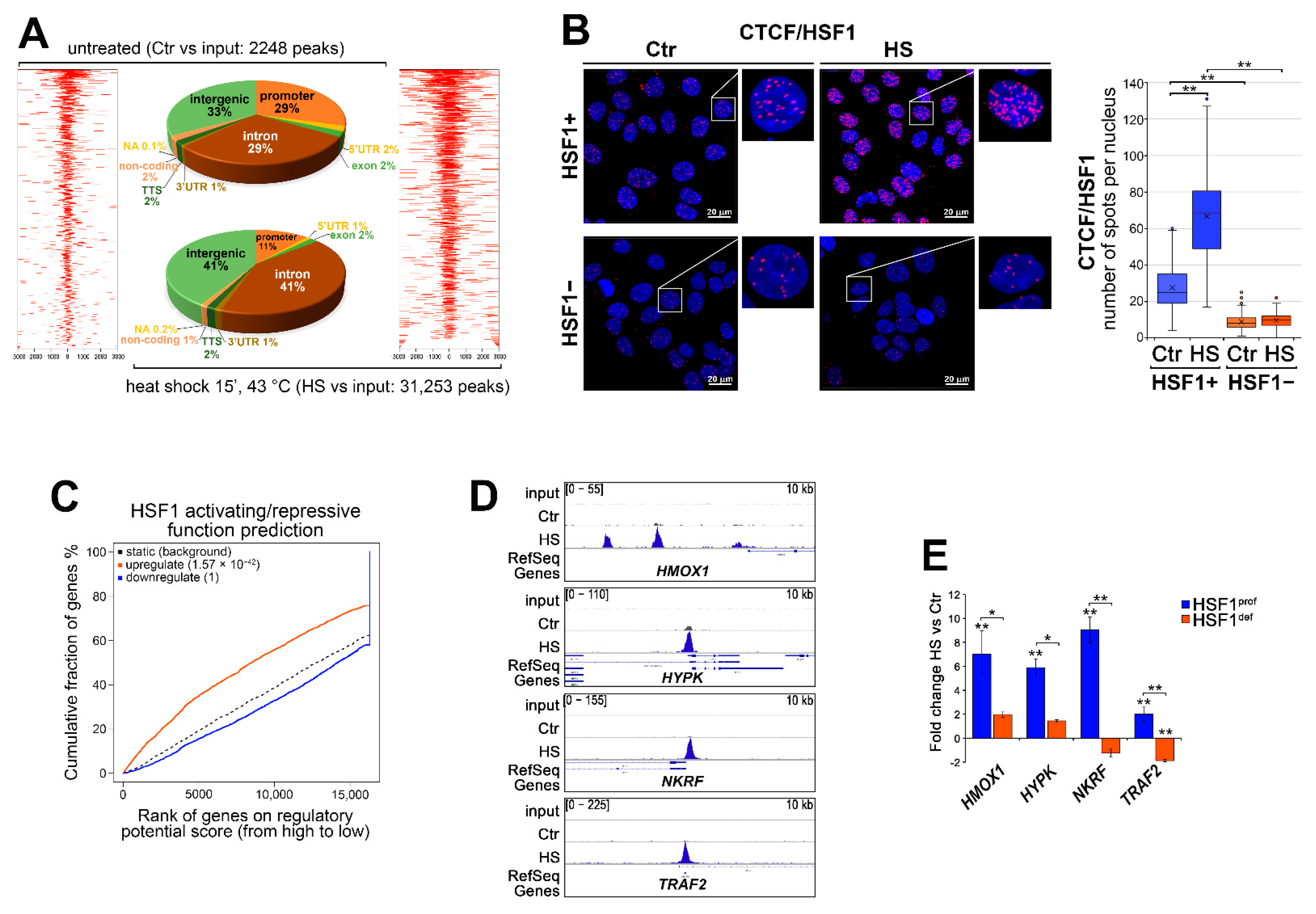
3.4. Transcriptional Regulatory Network Created during Heat Shock May Be Repressed by HSF1
3.5. Subsets of Heat Shock-Regulated Genes Are Expressed Differently in Human Cancers with Different Levels of HSF1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rupik, W.; Jasik, K.; Bembenek, J.; Widłak, W. The Expression Patterns of Heat Shock Genes and Proteins and Their Role during Vertebrate’s Development. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 159, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Jäger, R.; Mosser, D.D.; Samali, A. Regulation of Apoptosis by Heat Shock Proteins. IUBMB Life 2014, 66, 327–338. [Google Scholar] [CrossRef]
- Pincus, D. Regulation of Hsf1 and the Heat Shock Response. Adv. Exp. Med. Biol. 2020, 1243, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, L.; Zhang, J.; Moskophidis, D.; Mivechi, N.F. Targeted Disruption of Hsf1 Leads to Lack of Thermotolerance and Defines Tissue-Specific Regulation for Stress-Inducible Hsp Molecular Chaperones. J. Cell. Biochem. 2002, 86, 376–393. [Google Scholar] [CrossRef]
- Barna, J.; Csermely, P.; Vellai, T. Roles of Heat Shock Factor 1 beyond the Heat Shock Response. Cell. Mol. Life Sci. CMLS 2018, 75, 2897–2916. [Google Scholar] [CrossRef]
- Vydra, N.; Toma, A.; Widlak, W. Pleiotropic Role of HSF1 in Neoplastic Transformation. Curr. Cancer Drug Targets 2014, 14, 144–155. [Google Scholar] [CrossRef]
- Puustinen, M.C.; Sistonen, L. Molecular Mechanisms of Heat Shock Factors in Cancer. Cells 2020, 9, E1202. [Google Scholar] [CrossRef]
- Cyran, A.M.; Zhitkovich, A. Heat Shock Proteins and HSF1 in Cancer. Front. Oncol. 2022, 12, 860320. [Google Scholar] [CrossRef]
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat Shock Transcription Factor 1 as a Therapeutic Target in Neurodegenerative Diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944. [Google Scholar] [CrossRef]
- Janus, P.; Toma-Jonik, A.; Vydra, N.; Mrowiec, K.; Korfanty, J.; Chadalski, M.; Widłak, P.; Dudek, K.; Paszek, A.; Rusin, M.; et al. Pro-Death Signaling of Cytoprotective Heat Shock Factor 1: Upregulation of NOXA Leading to Apoptosis in Heat-Sensitive Cells. Cell Death Differ. 2020, 27, 2280–2292. [Google Scholar] [CrossRef]
- Widlak, W.; Vydra, N.; Malusecka, E.; Dudaladava, V.; Winiarski, B.; Scieglińska, D.; Widlak, P. Heat Shock Transcription Factor 1 Down-Regulates Spermatocyte-Specific 70 KDa Heat Shock Protein Expression Prior to the Induction of Apoptosis in Mouse Testes. Genes Cells 2007, 12, 487–499. [Google Scholar] [CrossRef]
- Kmiecik, S.W.; Mayer, M.P. Molecular Mechanisms of Heat Shock Factor 1 Regulation. Trends Biochem. Sci. 2022, 47, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Trinklein, N.D.; Murray, J.I.; Hartman, S.J.; Botstein, D.; Myers, R.M. The Role of Heat Shock Transcription Factor 1 in the Genome-Wide Regulation of the Mammalian Heat Shock Response. Mol. Biol. Cell 2004, 15, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Page, T.J.; Sikder, D.; Yang, L.; Pluta, L.; Wolfinger, R.D.; Kodadek, T.; Thomas, R.S. Genome-Wide Analysis of Human HSF1 Signaling Reveals a Transcriptional Program Linked to Cellular Adaptation and Survival. Mol. Biosyst. 2006, 2, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Salamanca, H.H.; Duarte, F.M.; Danko, C.G.; Lis, J.T. Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-Wide Transcriptional Regulation. Mol. Cell 2016, 62, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Vilaboa, N.; Boré, A.; Martin-Saavedra, F.; Bayford, M.; Winfield, N.; Firth-Clark, S.; Kirton, S.B.; Voellmy, R. New Inhibitor Targeting Human Transcription Factor HSF1: Effects on the Heat Shock Response and Tumor Cell Survival. Nucleic Acids Res. 2017, 45, 5797–5817. [Google Scholar] [CrossRef]
- Vydra, N.; Janus, P.; Kus, P.; Stokowy, T.; Mrowiec, K.; Toma-Jonik, A.; Krzywon, A.; Cortez, A.J.; Wojtas, B.; Gielniewski, B.; et al. Heat Shock Factor 1 (HSF1) Cooperates with Estrogen Receptor α (ERα) in the Regulation of Estrogen Action in Breast Cancer Cells. eLife 2021, 10, e69843. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinforma. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Zyla, J.; Marczyk, M.; Domaszewska, T.; Kaufmann, S.H.E.; Polanska, J.; Weiner, J. Gene Set Enrichment for Reproducible Science: Comparison of CERNO and Eight Other Algorithms. Bioinforma. Oxf. Engl. 2019, 35, 5146–5154. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J., 3rd; Domaszewska, T. Tmod: An R Package for General and Multivariate Enrichment Analysis. PeerJ Preprints 2016, 4, e2420v1. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. In Use R! 2nd ed.; Springer International Publishing: New York, NY, USA, 2016; ISBN 978-3-319-24275-0. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription Factor Enrichment Analysis by Orthogonal Omics Integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef] [PubMed]
- Kus-Liśkiewicz, M.; Polańska, J.; Korfanty, J.; Olbryt, M.; Vydra, N.; Toma, A.; Widłak, W. Impact of Heat Shock Transcription Factor 1 on Global Gene Expression Profiles in Cells Which Induce Either Cytoprotective or Pro-Apoptotic Response Following Hyperthermia. BMC Genom. 2013, 14, 456. [Google Scholar] [CrossRef]
- Vydra, N.; Janus, P.; Toma-Jonik, A.; Stokowy, T.; Mrowiec, K.; Korfanty, J.; Długajczyk, A.; Wojtaś, B.; Gielniewski, B.; Widłak, W. 17β-Estradiol Activates HSF1 via MAPK Signaling in ERα-Positive Breast Cancer Cells. Cancers 2019, 11, 1533. [Google Scholar] [CrossRef]
- Wang, S.; Sun, H.; Ma, J.; Zang, C.; Wang, C.; Wang, J.; Tang, Q.; Meyer, C.A.; Zhang, Y.; Liu, X.S. Target Analysis by Integration of Transcriptome and ChIP-Seq Data with BETA. Nat. Protoc. 2013, 8, 2502–2515. [Google Scholar] [CrossRef]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An Integrative Platform for Transcriptional Regulation Studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Ostman, A.; Landegren, U. Protein Detection Using Proximity-Dependent DNA Ligation Assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR Is a Web App for Creating, Exploring, Labeling and Sharing Volcano Plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Vihervaara, A.; Mahat, D.B.; Guertin, M.J.; Chu, T.; Danko, C.G.; Lis, J.T.; Sistonen, L. Transcriptional Response to Stress Is Pre-Wired by Promoter and Enhancer Architecture. Nat. Commun. 2017, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Burchfiel, E.T.; Vihervaara, A.; Guertin, M.J.; Gomez-Pastor, R.; Thiele, D.J. Comparative Interactomes of HSF1 in Stress and Disease Reveal a Role for CTCF in HSF1-Mediated Gene Regulation. J. Biol. Chem. 2021, 296, 100097. [Google Scholar] [CrossRef] [PubMed]
- Klenova, E.M.; Nicolas, R.H.; Sally, U.; Carne, A.F.; Lee, R.E.; Lobanenkov, V.V.; Goodwin, G.H. Molecular Weight Abnormalities of the CTCF Transcription Factor: CTCF Migrates Aberrantly in SDS-PAGE and the Size of the Expressed Protein Is Affected by the UTRs and Sequences within the Coding Region of the CTCF Gene. Nucleic Acids Res. 1997, 25, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Roos-Mattjus, P.; Sistonen, L. Interplay between Mammalian Heat Shock Factors 1 and 2 in Physiology and Pathology. FEBS J. 2021. [Google Scholar] [CrossRef]
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, E2385. [Google Scholar] [CrossRef]
- Chen, F.; Fan, Y.; Cao, P.; Liu, B.; Hou, J.; Zhang, B.; Tan, K. Pan-Cancer Analysis of the Prognostic and Immunological Role of HSF1: A Potential Target for Survival and Immunotherapy. Oxid. Med. Cell. Longev. 2021, 2021, 5551036. [Google Scholar] [CrossRef]
- Hentze, N.; Le Breton, L.; Wiesner, J.; Kempf, G.; Mayer, M.P. Molecular Mechanism of Thermosensory Function of Human Heat Shock Transcription Factor Hsf1. eLife 2016, 5, e11576. [Google Scholar] [CrossRef]
- Sawai, M.; Ishikawa, Y.; Ota, A.; Sakurai, H. The Proto-Oncogene JUN Is a Target of the Heat Shock Transcription Factor HSF1. FEBS J. 2013, 280, 6672–6680. [Google Scholar] [CrossRef]
- Takii, R.; Inouye, S.; Fujimoto, M.; Nakamura, T.; Shinkawa, T.; Prakasam, R.; Tan, K.; Hayashida, N.; Ichikawa, H.; Hai, T.; et al. Heat Shock Transcription Factor 1 Inhibits Expression of IL-6 through Activating Transcription Factor 3. J. Immunol. 2010, 184, 1041–1048. [Google Scholar] [CrossRef]
- Chen, C.; Xie, Y.; Stevenson, M.A.; Auron, P.E.; Calderwood, S.K. Heat Shock Factor 1 Represses Ras-Induced Transcriptional Activation of the c-Fos Gene. J. Biol. Chem. 1997, 272, 26803–26806. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Perie, L.; Mueller, E. The MRNA Levels of Heat Shock Factor 1 Are Regulated by Thermogenic Signals via the CAMP-Dependent Transcription Factor ATF3. J. Biol. Chem. 2020, 295, 5984–5994. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 Drives a Transcriptional Program Distinct from Heat Shock to Support Highly Malignant Human Cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Korfanty, J.; Stokowy, T.; Widlak, P.; Gogler-Piglowska, A.; Handschuh, L.; Podkowiński, J.; Vydra, N.; Naumowicz, A.; Toma-Jonik, A.; Widlak, W. Crosstalk between HSF1 and HSF2 during the Heat Shock Response in Mouse Testes. Int. J. Biochem. Cell Biol. 2014, 57C, 76–83. [Google Scholar] [CrossRef]
- Stephanou, A.; Latchman, D.S. Transcriptional Regulation of the Heat Shock Protein Genes by STAT Family Transcription Factors. Gene Expr. 1999, 7, 311–319. [Google Scholar] [PubMed]
- Tacchini, L.; Fusar-Poli, D.; Sironi, M.; Mantovani, A.; Bernelli-Zazzera, A. Activation of Signal Transducer and Activator of Transcription 3 in Rat Liver after Heat Shock and Reperfusion Stress. Int. J. Biochem. Cell Biol. 2003, 35, 316–323. [Google Scholar] [CrossRef]
- Matozaki, M.; Saito, Y.; Yasutake, R.; Munira, S.; Kaibori, Y.; Yukawa, A.; Tada, M.; Nakayama, Y. Involvement of Stat3 Phosphorylation in Mild Heat Shock-Induced Thermotolerance. Exp. Cell Res. 2019, 377, 67–74. [Google Scholar] [CrossRef]
- Lyu, X.; Rowley, M.J.; Corces, V.G. Architectural Proteins and Pluripotency Factors Cooperate to Orchestrate the Transcriptional Response of HESCs to Temperature Stress. Mol. Cell 2018, 71, 940–955.e7. [Google Scholar] [CrossRef]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 Is Required for Extra-Embryonic Development, Postnatal Growth and Protection during Inflammatory Responses in Mice. EMBO J. 1999, 18, 5943–5952. [Google Scholar] [CrossRef]
- Murapa, P.; Ward, M.R.; Gandhapudi, S.K.; Woodward, J.G.; D’Orazio, S.E.F. Heat Shock Factor 1 Protects Mice from Rapid Death during Listeria Monocytogenes Infection by Regulating Expression of Tumor Necrosis Factor Alpha during Fever. Infect. Immun. 2011, 79, 177–184. [Google Scholar] [CrossRef]
- Chu, W.-M. Tumor Necrosis Factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.-C. ATF3 Mediates the Inhibitory Action of TNF-α on Osteoblast Differentiation through the JNK Signaling Pathway. Biochem. Biophys. Res. Commun. 2018, 499, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory Feedback Control of NF-ΚB Signalling in Health and Disease. Biochem. J. 2021, 478, 2619–2664. [Google Scholar] [CrossRef] [PubMed]
- Kardyńska, M.; Paszek, A.; Śmieja, J.; Spiller, D.; Widłak, W.; White, M.R.H.; Paszek, P.; Kimmel, M. Quantitative Analysis Reveals Crosstalk Mechanisms of Heat Shock-Induced Attenuation of NF-ΚB Signaling at the Single Cell Level. PLoS Comput. Biol. 2018, 14, e1006130. [Google Scholar] [CrossRef]
- Paszek, A.; Kardyńska, M.; Bagnall, J.; Śmieja, J.; Spiller, D.G.; Widłak, P.; Kimmel, M.; Widlak, W.; Paszek, P. Heat Shock Response Regulates Stimulus-Specificity and Sensitivity of the pro-Inflammatory NF-ΚB Signalling. Cell Commun. Signal. CCS 2020, 18, 77. [Google Scholar] [CrossRef]
- Turner, D.A.; Paszek, P.; Woodcock, D.J.; Nelson, D.E.; Horton, C.A.; Wang, Y.; Spiller, D.G.; Rand, D.A.; White, M.R.H.; Harper, C.V. Physiological Levels of TNFalpha Stimulation Induce Stochastic Dynamics of NF-KappaB Responses in Single Living Cells. J. Cell Sci. 2010, 123, 2834–2843. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank Product | Gene Symbol | Fold Change (FC) 1 | Difference (FC HSF1def/HSF1 prof) | Difference (padj) | Difference Confirmed by RT-qPCR | ||
|---|---|---|---|---|---|---|---|
| HSF1prof | HSF1def | ||||||
| 1. | 0.00818 | LINC00324 | 2.87 | 10.98 | 3.82 | 0.030 | + |
| 2. | 0.00749 | TRIB1 2 | 2.30 | 8.54 | 3.71 | 0.020 | + |
| 3. | 0.00060 | FOSB 2 | 12.47 | 41.14 | 3.30 | 0.173 | + |
| 4. | 0.00410 | EGR2 2 | 13.37 | 41.43 | 3.10 | 0.567 | na |
| 5. | 0.00005 | ATF3 2 | 17.40 | 52.79 | 3.03 | 0.003 | + |
| 6. | 0.01065 | DUSP10 | 1.70 | 5.12 | 3.02 | 0.0056 | + |
| 7. | 0.00229 | RRAD | 480.7 | 1413.7 | 2.94 | 0.845 | na |
| 8. | 0.00134 | GEM | 24.72 | 72.56 | 2.94 | 0.362 | na |
| 9. | 0.004546 | FOS 2 | 1.82 | 5.12 | 2.82 | 0.023 | na |
| 10. | 0.003438 | ERRFI1 2 | 1.97 | 5.01 | 2.54 | 3.03 × 10−7 | na |
| 11. | 0.01135 | CCDC89 | 4.54 | 10.97 | 2.41 | 0.603 | na |
| 12. | 0.00363 | CSRNP1 2 | 4.25 | 9.35 | 2.20 | 0.153 | na |
| 13. | 0.00023 | NR4A1 2 | 17.61 | 38.63 | 2.19 | 0.375 | na |
| 14. | 0.01285 | ID2 2 | 2.44 | 5.35 | 2.19 | 0.348 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janus, P.; Kuś, P.; Vydra, N.; Toma-Jonik, A.; Stokowy, T.; Mrowiec, K.; Wojtaś, B.; Gielniewski, B.; Widłak, W. HSF1 Can Prevent Inflammation following Heat Shock by Inhibiting the Excessive Activation of the ATF3 and JUN&FOS Genes. Cells 2022, 11, 2510. https://doi.org/10.3390/cells11162510
Janus P, Kuś P, Vydra N, Toma-Jonik A, Stokowy T, Mrowiec K, Wojtaś B, Gielniewski B, Widłak W. HSF1 Can Prevent Inflammation following Heat Shock by Inhibiting the Excessive Activation of the ATF3 and JUN&FOS Genes. Cells. 2022; 11(16):2510. https://doi.org/10.3390/cells11162510
Chicago/Turabian StyleJanus, Patryk, Paweł Kuś, Natalia Vydra, Agnieszka Toma-Jonik, Tomasz Stokowy, Katarzyna Mrowiec, Bartosz Wojtaś, Bartłomiej Gielniewski, and Wiesława Widłak. 2022. "HSF1 Can Prevent Inflammation following Heat Shock by Inhibiting the Excessive Activation of the ATF3 and JUN&FOS Genes" Cells 11, no. 16: 2510. https://doi.org/10.3390/cells11162510