
PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer


Abstract
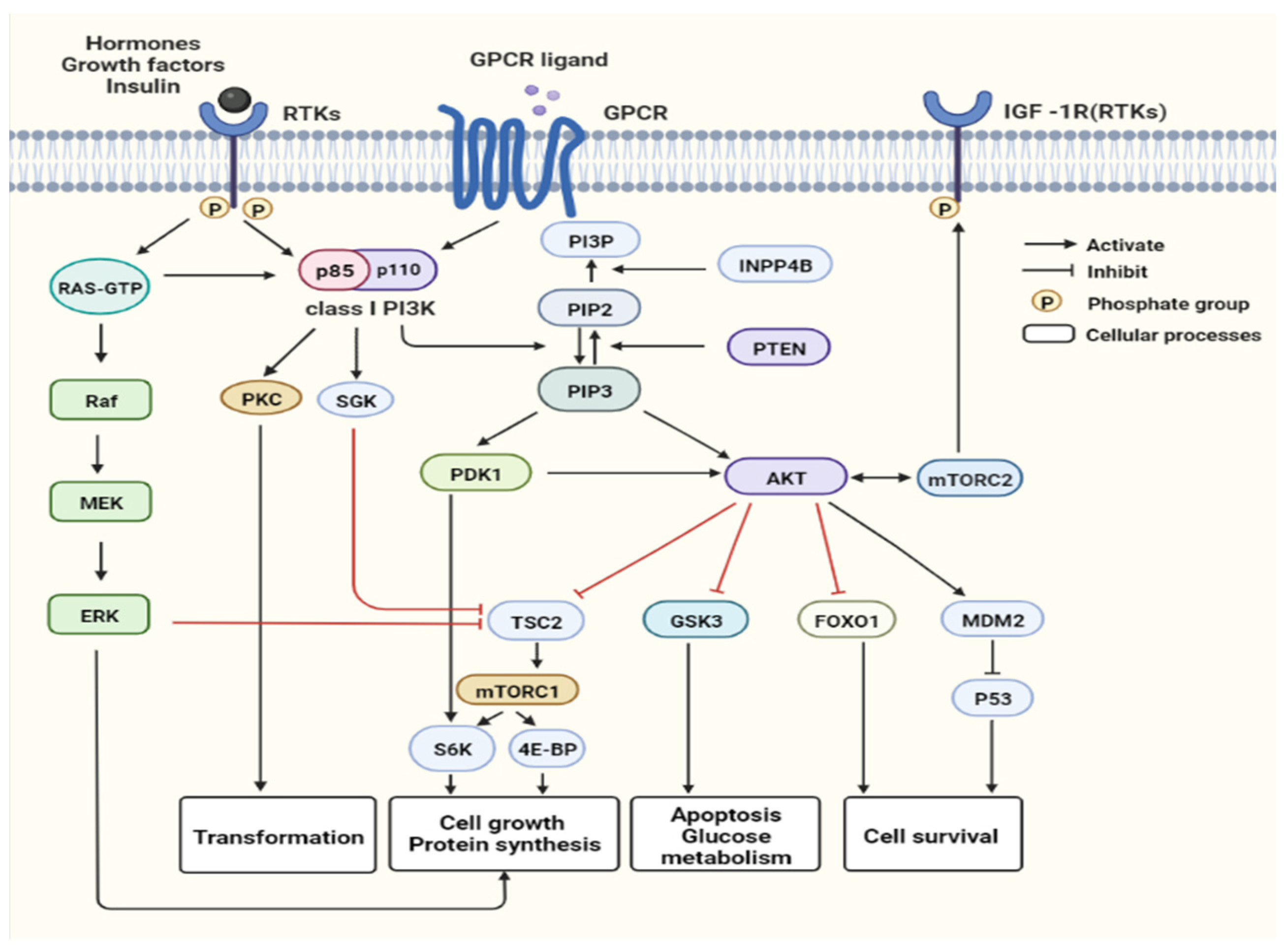
:1. Introduction
2. Changes of the PAM Pathway in Different Breast Cancer Subtypes
3. Preclinical and Clinical Development of PAM Pathway Inhibitors in Different Subtypes of Breast Cancer
3.1. PAM Inhibitors for Treating ER+/HER2− Breast Cancer
3.1.1. PI3K Inhibitors for Treating ER+/HER2− Breast Cancer
3.1.2. AKT Inhibitors for Treating ER+/HER2− Breast Cancer
3.1.3. mTOR Inhibitors for Treating ER+/HER2− Breast Cancer
3.2. PAM Inhibitors for Treating HER2+ Breast Cancer
3.3. PAM Inhibitors for Treating Triple-Negative Breast Cancer (TNBC)
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Samuels, Y. PIK3CA in cancer: The past 30 years. Semin. Cancer Biol. 2019, 59, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Coyaud, E.; Desmetz, C.; Laurent, E.; Béganton, B.; Coopman, P.; Raught, B.; Solassol, J. FKBP4 connects mTORC2 and PI3K to activate the PDK1/Akt-dependent cell proliferation signaling in breast cancer. Theranostics 2019, 9, 7003–7015. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, S.B.; Pedersen, N.M.; Liestøl, K.; Stenmark, H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp. Cell Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol -3, 4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic spindle assembly and genomic stability in breast cancer require PI3K-C2α scaffolding function. Cancer Cell. 2017, 32, 444–459.e7. [Google Scholar] [CrossRef] [PubMed]
- Kroesbergen, J.; Roozen, A.M.; Gelsema, W.J.; de Ligny, C.L. 99mTc bone scanning agents--IV. Chemical characterization of 99mTc(Sn)-pyrophosphate complexes. Int. J. Rad. Appl. Instrum. B 1988, 15, 209–214. [Google Scholar] [CrossRef]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K functions in cell biology and disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef] [PubMed]
- Young, C.D.; Arteaga, C.L.; Cook, R.S. Dual inhibition of type I and type III PI3 kinases increases tumor cell apoptosis in HER2+ breast cancers. Breast Cancer Res. 2015, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.A.; Wang, Y.W.; Chen, Y.L.; Chen, H.W.; Chuang, J.J.; Chang, H.Y.; Ho, C.L.; Chang, C.; Chow, N.H.; Lee, C.T. The role of phosphatidylinositol 3-kinase catalytic subunit type 3 in the pathogenesis of human cancer. Int. J. Mol. Sci. 2021, 22, 10964. [Google Scholar] [CrossRef]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019, 30 (Suppl. 10), x12–x20. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Zhang, H.; Wang, J.; Hua, H.; Jiang, Y. The role of network-forming collagens in cancer progression. Int. J. Cancer 2022, 151, 833–842. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Walter, K.; Spoerke, J.M.; O’Brien, C.; Huw, L.Y.; Hampton, G.M.; Lackner, M.R. Activating mutations in PIK3CB confer resistance to PI3K inhibition and define a novel oncogenic role for p110β. Cancer Res. 2016, 76, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, H.; Wang, J.; Liu, Y.; Luo, T.; Hua, H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J. Hematol. Oncol. 2022, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. Breast Cancer, Version 4.2021 Featured Updates to the NCCN Guidelines. JNCCN J. Natl. Compr. Cancer Netw. 2021, 19, 485–494. [Google Scholar] [CrossRef]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Dziubinski, M.; Yang, Z.; Ethier, S.P.; Wu, G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res. Treat. 2008, 112, 217–227. [Google Scholar] [CrossRef]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Functional specificity of Akt isoforms in cancer progression. Biomol. Concepts 2011, 2, 1–11. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tenorio, G.; Alkhori, L.; Olsson, B.; Waltersson, M.A.; Nordenskjöld, B.; Rutqvist, L.E.; Skoog, L.; Stål, O. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin. Cancer Res. 2007, 13, 3577–3584. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, J.; Zhong, X.R.; Luo, T.; Wang, Y.P.; Huang, H.F.; Yin, L.J.; Qiu, Y.; Bu, H.; Lv, Q.; et al. Correlation between activation of PI3K/AKT/mTOR pathway and prognosis of breast cancer in Chinese women. PLoS ONE 2015, 10, e0120511. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]
- Coussy, F.; El Botty, R.; Lavigne, M.; Gu, C.; Fuhrmann, L.; Briaux, A.; de Koning, L.; Dahmani, A.; Montaudon, E.; Morisset, L.; et al. Combination of PI3K and MEK inhibitors yields durable remission in PDX models of PIK3CA-mutated metaplastic breast cancers. J. Hematol. Oncol. 2020, 13, 13. [Google Scholar] [CrossRef]
- Crowder, R.J.; Phommaly, C.; Tao, Y.; Hoog, J.; Luo, J.; Perou, C.M.; Parker, J.S.; Miller, M.A.; Huntsman, D.G.; Lin, L.; et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009, 69, 3955–3962. [Google Scholar] [CrossRef]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.Y.; Zuo, W.J.; Li, S.; Qiao, F.; Liu, Y.R.; Cao, Z.G.; Zhou, S.L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef]
- Ellis, M.J.; Lin, L.; Crowder, R.; Tao, Y.; Hoog, J.; Snider, J.; Davies, S.; DeSchryver, K.; Evans, D.B.; Steinseifer, J.; et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2010, 119, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.M.; Tamura, K.; Oliveira, M.; Ciruelos, E.M.; Mayer, I.A.; Sablin, M.P.; Biganzoli, L.; Ambrose, H.J.; Ashton, J.; Barnicle, A.; et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Haibe-Kains, B.; Majjaj, S.; Lallemand, F.; Durbecq, V.; Larsimont, D.; Gonzalez-Angulo, A.M.; Pusztai, L.; Symmans, W.F.; Bardelli, A.; et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 10208–10213. [Google Scholar] [CrossRef]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Fu, J.; Li, Z.; Jiang, N.; Chen, Y.; Peng, J. The landscape of PDK1 in breast cancer. Cancers 2022, 14, 811. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Yan, G.; Wang, N.; Daliah, G.; Edick, A.M.; Poulet, S.; Boudreault, J.; Ali, S.; Burgos, S.A.; Lebrun, J.J. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021, 12, 3055. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR pathway causes drug resistance in breast cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.M.; Chen, H.; Hortobagyi, G.N.; et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol. Cancer Ther. 2011, 10, 1093–1101. [Google Scholar] [CrossRef]
- Graudenzi, A.; Cava, C.; Bertoli, G.; Fromm, B.; Flatmark, K.; Mauri, G.; Castiglioni, I. Pathway-based classification of breast cancer subtypes. Front. Biosci. (Landmark Ed.) 2017, 22, 1697–1712. [Google Scholar] [CrossRef]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Morita, T.Y.; Ohashi, A.; Haeno, H.; Hakozaki, Y.; Fujii, M.; Kashima, Y.; Kobayashi, S.S.; Mukohara, T. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci. Rep. 2020, 10, 21762. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Piscuoglio, S.; Geyer, F.C.; Burke, K.A.; Pareja, F.; Eberle, C.A.; Lim, R.S.; Natrajan, R.; Riaz, N.; Mariani, O.; et al. The Landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin. Cancer Res. 2017, 23, 3859–3870. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.S.; Jin, X.Y.; Wang, X.M.; Tou, L.Z.; Huang, J. Advances in endocrine and targeted therapy for hormone-receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer. Chin. Med. J. 2020, 133, 1099–1108. [Google Scholar] [CrossRef]
- Toomey, S.; Eustace, A.J.; Fay, J.; Sheehan, K.M.; Carr, A.; Milewska, M.; Madden, S.F.; Teiserskiene, A.; Kay, E.W.; O’Donovan, N.; et al. Impact of somatic PI3K pathway and ERBB family mutations on pathological complete response (pCR) in HER2-positive breast cancer patients who received neoadjuvant HER2-targeted therapies. Breast Cancer Res. 2017, 19, 87. [Google Scholar] [CrossRef]
- Ertay, A.; Liu, H.; Liu, D.; Peng, P.; Hill, C.; Xiong, H.; Hancock, D.; Yuan, X.; Przewloka, M.R.; Coldwell, M.; et al. WDHD1 is essential for the survival of PTEN-inactive triple-negative breast cancer. Cell Death Dis. 2020, 11, 1001. [Google Scholar] [CrossRef]
- Shoman, N.; Klassen, S.; McFadden, A.; Bickis, M.G.; Torlakovic, E.; Chibbar, R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod. Pathol. 2005, 18, 250–259. [Google Scholar] [CrossRef]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming endocrine resistance in breast cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020, 111, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef] [PubMed]
- García-García, C.; Ibrahim, Y.H.; Serra, V.; Calvo, M.T.; Guzmán, M.; Grueso, J.; Aura, C.; Pérez, J.; Jessen, K.; Liu, Y.; et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin. Cancer Res. 2012, 18, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Elkabets, M.; Vora, S.; Juric, D.; Morse, N.; Mino-Kenudson, M.; Muranen, T.; Tao, J.; Campos, A.B.; Rodon, J.; Ibrahim, Y.H.; et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. [published correction appears in Sci Transl Med. 2018 Nov 14;10,]. Sci. Transl. Med. 2013, 5, 196ra99. [Google Scholar] [CrossRef]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.S.; Chen, S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Endicott, S.J.; Ziemba, Z.J.; Beckmann, L.J.; Boynton, D.N.; Miller, R.A. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J. Cell Biol. 2020, 219, e202001031. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, X.; Meng, X.; Leslie, K.K. A mechanism for synergy with combined mTOR and PI3 kinase inhibitors. PLoS ONE 2011, 6, e26343. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.D.; Ling, M.T. Targeting drug-resistant prostate cancer with dual PI3K/mTOR inhibition. Curr. Med. Chem. 2014, 21, 3048–3056. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef]
- Boulay, A.; Rudloff, J.; Ye, J.; Zumstein-Mecker, S.; O’Reilly, T.; Evans, D.B.; Chen, S.; Lane, H.A. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin. Cancer Res. 2005, 11, 5319–5328. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. [published correction appears in Lancet Oncol. 2019 Feb;20, e71–e72]. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone- receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef]
- Vuylsteke, P.; Huizing, M.; Petrakova, K.; Roylance, R.; Laing, R.; Chan, S.; Abell, F.; Gendreau, S.; Rooney, I.; Apt, D.; et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann. Oncol. 2016, 27, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Mayer, I.A.; Prat, A.; Egle, D.; Blau, S.; Fidalgo, J.; Gnant, M.; Fasching, P.A.; Colleoni, M.; Wolff, A.C.; Winer, E.P.; et al. A Phase II Randomized study of neoadjuvant letrozole plus alpelisib for hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer (NEO-ORB). Clin. Cancer Res. 2019, 25, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.; Cortés, J.; Im, Y.H.; Diéras, V.; Harbeck, N.; Krop, I.E.; Wilson, T.R.; Cui, N.; Schimmoller, F.; Hsu, J.Y.; et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: The SANDPIPER trial. Ann. Oncol. 2021, 32, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Shor, R.E.; Dai, J.; Lee, S.Y.; Pisarsky, L.; Matei, I.; Lucotti, S.; Lyden, D.; Bissell, M.J.; Ghajar, C.M. The PI3K/mTOR inhibitor Gedatolisib eliminates dormant breast cancer cells in organotypic culture, but fails to prevent metastasis in preclinical settings. Mol. Oncol. 2022, 16, 130–147. [Google Scholar] [CrossRef]
- Clinical Trails.gov. Study of the Safety and Pharmacology of GDC-0980 in Combination with Paclitaxel with or without Bevacizumab in Patients with Locally Recurrent or Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01254526 (accessed on 26 March 2021).
- Clinical Trails.gov. A Study of LY2835219 (Abemaciclib) in Combination with Therapies for Breast Cancer That Has Spread. Available online: https://clinicaltrials.gov/ct2/show/NCT02057133 (accessed on 26 March 2021).
- Qin, H.; Liu, L.; Sun, S.; Zhang, D.; Sheng, J.; Li, B.; Yang, W. The impact of PI3K inhibitors on breast cancer cell and its tumor microenvironment. PeerJ 2018, 6, e5092. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First global approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Henry, N.L.; Somerfield, M.R.; Dayao, Z.; Elias, A.; Kalinsky, K.; McShane, L.M.; Moy, B.; Park, B.H.; Shanahan, K.M.; Sharma, P.; et al. Biomarkers for systemic therapy in metastatic breast cancer: ASCO guideline update. J. Clin. Oncol. 2022, JCO-22. [Google Scholar] [CrossRef]
- Song, K.W.; Edgar, K.A.; Hanan, E.J.; Hafner, M.; Oeh, J.; Merchant, M.; Sampath, D.; Nannini, M.A.; Hong, R.; Phu, L.; et al. RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077 (Inavolisib) efficacy. Cancer Discov. 2022, 12, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bhattacharyya, S.; Das, A.; Singh, G.; Bal, A. In vitro effect of PIK3CA/mTOR inhibition in triple-negative breast cancer subtype cell lines. Breast Dis. 2022, 41, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Mohammad, A.S.; Saralkar, P.; Sprowls, S.A.; Vickers, S.D.; John, D.; Tallman, R.M.; Lucke-Wold, B.P.; Jarrell, K.E.; Pinti, M.; et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol. Res. 2018, 132, 47–68. [Google Scholar] [CrossRef]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Cordeiro, M.N.D.S. AKT Inhibitors: The road ahead to computational modeling-guided discovery. Int. J. Mol. Sci. 2021, 22, 3944. [Google Scholar] [CrossRef]
- Xiang, R.F.; Wang, Y.; Zhang, N.; Xu, W.B.; Cao, Y.; Tong, J.; Li, J.M.; Wu, Y.L.; Yan, H. MK2206 enhances the cytocidal effects of bufalin in multiple myeloma by inhibiting the AKT/mTOR pathway. Cell Death Dis. 2017, 8, e2776. [Google Scholar] [CrossRef]
- Akcakanat, A.; Meric-Bernstam, F. MK-2206 window of opportunity study in breast cancer. Ann. Transl. Med. 2018, 6 (Suppl. S1), S57. [Google Scholar] [CrossRef]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II trial of neoadjuvant MK-2206, an AKT inhibitor, with Anastrozole in clinical stage II or III PIK3CA-mutant ER-positive and HER2-negative breast cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef]
- Turner, N.C.; Alarcón, E.; Armstrong, A.C.; Philco, M.; López Chuken, Y.A.; Sablin, M.P.; Tamura, K.; Gómez Villanueva, A.; Pérez-Fidalgo, J.A.; Cheung, S.; et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 774–780. [Google Scholar] [CrossRef]
- Jones, R.H.; Casbard, A.; Carucci, M.; Cox, C.; Butler, R.; Alchami, F.; Madden, T.A.; Bale, C.; Bezecny, P.; Joffe, J.; et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): A multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020, 21, 345–357. [Google Scholar] [CrossRef]
- Howell, S.J.; Casbard, A.; Carucci, M.; Ingarfield, K.; Butler, R.; Morgan, S.; Meissner, M.; Bale, C.; Bezecny, P.; Moon, S.; et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): Overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 2022, 23, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Brase, J.C.; Cheng, Y.; Nuciforo, P.; Paré, L.; Pascual, T.; Martínez, D.; Galván, P.; Vidal, M.; Adamo, B.; et al. Everolimus plus exemestane for hormone receptor-positive advanced breast cancer: A PAM50 intrinsic subtype analysis of BOLERO-2. Oncologist 2019, 24, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant plus vistusertib vs fulvestrant plus everolimus vs fulvestrant alone for women with hormone receptor-positive metastatic breast cancer: The MANTA phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, N.; Zhao, F.; Manola, J.; Klein, P.; Ramaswamy, B.; Brufsky, A.; Stella, P.J.; Burnette, B.; Telli, M.; Makower, D.F.; et al. Randomized phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer resistant to aromatase inhibitor therapy: Results of PrE0102. J. Clin. Oncol. 2018, 36, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; de Boer, R.H.; Hurvitz, S.; Yardley, D.A.; Kovalenko, E.; Ejlertsen, B.; Blau, S.; Özgüroğlu, M.; Landherr, L.; Ewertz, M.; et al. Everolimus plus exemestane vs everolimus or capecitabine monotherapy for estrogen receptor-positive, HER2-negative advanced breast cancer: The BOLERO-6 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1367–1374. [Google Scholar] [CrossRef]
- Schmid, P.; Sablin, M.P.; Bergh, J.; Im, S.A.; Lu, Y.S.; Martínez, N.; Neven, P.; Lee, K.S.; Morales, S.; Pérez-Fidalgo, J.A.; et al. A phase Ib/II study of xentuzumab, an IGF-neutralising antibody, combined with exemestane and everolimus in hormone receptor-positive, HER2-negative locally advanced/metastatic breast cancer. Breast Cancer Res. 2021, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Potter, D.A.; Salkeni, M.A.; Silverman, P.; Haddad, T.C.; Forget, F.; Awada, A.; Canon, J.L.; Danso, M.; Lortholary, A.; et al. Sapanisertib plus exemestane or fulvestrant in women with hormone receptor-positive/HER2-negative advanced or metastatic breast cancer. Clin. Cancer Res. 2021, 27, 3329–3338. [Google Scholar] [CrossRef]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef]
- Kalinsky, K.; Hong, F.; McCourt, C.K.; Sachdev, J.C.; Mitchell, E.P.; Zwiebel, J.A.; Doyle, L.A.; McShane, L.M.; Li, S.; Gray, R.J.; et al. Effect of capivasertib in patients with an AKT1 E17K-mutated tumor: NCI-MATCH subprotocol EAY131-Y nonrandomized trial. JAMA Oncol. 2021, 7, 271–278. [Google Scholar] [CrossRef]
- Hosford, S.R.; Miller, T.W. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharmgenom. Pers. Med. 2014, 7, 203–215. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I trial of the PARP inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2- and non-BRCA1/2-mutant cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Yoshino, K.I.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ballhausen, A.; Wheler, J.J.; Karp, D.D.; Piha-Paul, S.A.; Fu, S.; Pant, S.; Tsimberidou, A.M.; Hong, D.S.; Subbiah, V.; Holley, V.R.; et al. Phase I study of everolimus, letrozole, and trastuzumab in patients with hormone receptor-positive metastatic breast cancer or other solid tumors. Clin. Cancer Res. 2021, 27, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C.; et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef]
- Pistilli, B.; Pluard, T.; Urruticoechea, A.; Farci, D.; Kong, A.; Bachelot, T.; Chan, S.; Han, H.S.; Jerusalem, G.; Urban, P.; et al. Phase II study of buparlisib (BKM120) and trastuzumab in patients with HER2+ locally advanced or metastatic breast cancer resistant to trastuzumab-based therapy. Breast Cancer Res. Treat. 2018, 168, 357–364. [Google Scholar] [CrossRef]
- Guerin, M.; Rezai, K.; Isambert, N.; Campone, M.; Autret, A.; Pakradouni, J.; Provansal, M.; Camerlo, J.; Sabatier, R.; Bertucci, F.; et al. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur. J. Cancer 2017, 86, 28–36. [Google Scholar] [CrossRef]
- Jain, S.; Shah, A.N.; Santa-Maria, C.A.; Siziopikou, K.; Rademaker, A.; Helenowski, I.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2018, 171, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Tripathy, D.; Albain, K.S.; Symmans, W.F.; Rugo, H.S.; Melisko, M.E.; Wallace, A.M.; Schwab, R.; Helsten, T.; Forero-Torres, A.; et al. MK-2206 and standard neoadjuvant chemotherapy improves response in patients with human epidermal growth factor receptor 2-positive and/or hormone receptor-negative breast bancers in the I-SPY 2 trial. J. Clin. Oncol. 2020, 38, 1059–1069. [Google Scholar] [CrossRef]
- André, F.; Hurvitz, S.; Fasolo, A.; Tseng, L.M.; Jerusalem, G.; Wilks, S.; O’Regan, R.; Isaacs, C.; Toi, M.; Burris, H.A.; et al. Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2-overexpressing metastatic breast cancers: Combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J. Clin. Oncol. 2016, 34, 2115–2124. [Google Scholar] [CrossRef]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Acevedo-Gadea, C.; Hatzis, C.; Chung, G.; Fishbach, N.; Lezon-Geyda, K.; Zelterman, D.; DiGiovanna, M.P.; Harris, L.; Abu-Khalaf, M.M. Sirolimus and trastuzumab combination therapy for HER2-positive metastatic breast cancer after progression on prior trastuzumab therapy. Breast Cancer Res. Treat. 2015, 150, 157–167. [Google Scholar] [CrossRef]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef]
- Keegan, N.M.; Furney, S.J.; Walshe, J.M.; Gullo, G.; Kennedy, M.J.; Smith, D.; McCaffrey, J.; Kelly, C.M.; Egan, K.; Kerr, J.; et al. Phase Ib trial of copanlisib, a phosphoinositide-3 kinase (PI3K) inhibitor, with trastuzumab in advanced pre-treated HER2-positive breast cancer “PantHER”. Cancers 2021, 13, 1225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Chen, J.J.; Gao, X.; Huang, Z.; Shen, Q. Multifunctional nanoparticles loading with docetaxel and GDC0941 for reversing multidrug resistance mediated by PI3K/Akt signal pathway. Mol. Pharm. 2017, 14, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Fourneaux, B.; Chaire, V.; Lucchesi, C.; Karanian, M.; Pineau, R.; Laroche-Clary, A.; Italiano, A. Dual inhibition of the PI3K/AKT/mTOR pathway suppresses the growth of leiomyosarcomas but leads to ERK activation through mTORC2: Biological and clinical implications. Oncotarget 2017, 8, 7878–7890. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef]
- Gayle, S.S.; Arnold, S.L.; O’Regan, R.M.; Nahta, R. Pharmacologic inhibition of mTOR improves lapatinib sensitivity in HER2-overexpressing breast cancer cells with primary trastuzumab resistance. Anti-Cancer Agents Med. Chem. 2012, 12, 151–162. [Google Scholar] [CrossRef]
- Casadevall, D.; Hernández-Prat, A.; García-Alonso, S.; Arpí-Llucià, O.; Menéndez, S.; Qin, M.; Guardia, C.; Morancho, B.; Sánchez-Martín, F.J.; Zazo, S.; et al. mTOR inhibition and trastuzumab-emtansine (T-DM1) in HER2-positive breast cancer. Mol. Cancer Res. 2022, 6, 1108–1121. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: The PAKT trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- Ippen, F.M.; Grosch, J.K.; Subramanian, M.; Kuter, B.M.; Liederer, B.M.; Plise, E.G.; Mora, J.L.; Nayyar, N.; Schmidt, S.P.; Giobbie-Hurder, A.; et al. Targeting the PI3K/Akt/mTOR pathway with the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain metastases. Neuro. Oncol. 2019, 21, 1401–1411. [Google Scholar] [CrossRef]
- Oliveira, M.; Saura, C.; Nuciforo, P.; Calvo, I.; Andersen, J.; Passos-Coelho, J.L.; Gil, M.G.; Bermejo, B.; Patt, D.A.; Ciruelos, E.; et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann. Oncol. 2019, 30, 1289–1297. [Google Scholar] [CrossRef]
- Vicier, C.; Sfumato, P.; Isambert, N.; Dalenc, F.; Robert, M.; Levy, C.; Rezai, K.; Provansal, M.; Adélaïde, J.; Garnier, S.; et al. TAKTIC: A prospective, multicentre, uncontrolled, phase IB/II study of LY2780301, a p70S6K/AKT inhibitor, in combination with weekly paclitaxel in HER2-negative advanced breast cancer patients. Eur. J. Cancer 2021, 159, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC subtype, AR status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Basho, R.K.; Gilcrease, M.; Murthy, R.K.; Helgason, T.; Karp, D.D.; Meric-Bernstam, F.; Hess, K.R.; Herbrich, S.M.; Valero, V.; Albarracin, C.; et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: Evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017, 3, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, X.; Cao, S.; Sun, Y.; He, X.; Jiang, B.; Yu, Y.; Duan, J.; Qiu, F.; Kang, N. Berberine represses human gastric cancer cell growth in vitro and in vivo by inducing cytostatic autophagy via inhibition of MAPK/mTOR/p70S6K and Akt signaling pathways. Biomed. Pharmacother. 2020, 128, 110245. [Google Scholar] [CrossRef]
- Zecchin, D.; Moore, C.; Michailidis, F.; Horswell, S.; Rana, S.; Howell, M.; Downward, J. Combined targeting of G protein-coupled receptor and EGF receptor signaling overcomes resistance to PI3K pathway inhibitors in PTEN-null triple negative breast cancer. EMBO Mol. Med. 2020, 12, e11987. [Google Scholar] [CrossRef]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 2020, 27, 66–73.e7. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Xu, J.; Shen, Y.; Cahuzac, K.M.; Park, K.S.; Dale, B.; Liu, J.; Parsons, R.E.; Jin, J. Discovery of Potent, Selective, and In Vivo Efficacious AKT Kinase Protein Degraders via Structure-Activity Relationship Studies. J. Med. Chem. 2022, 65, 3644–3666. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Zhang, W.; Wang, X.; Chen, L.; Wang, S. BKM120 sensitizes BRCA-proficient triple negative breast cancer cells to olaparib through regulating FOXM1 and Exo1 expression. Sci. Rep. 2021, 11, 4774. [Google Scholar] [CrossRef] [PubMed]
- Gallyas FJr Sumegi, B.; Szabo, C. Role of Akt Activation in PARP Inhibitor Resistance in Cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Co-targeting EGFR and mTOR with gefitinib and everolimus in triple-negative breast cancer cells. Sci. Rep. 2020, 10, 6367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
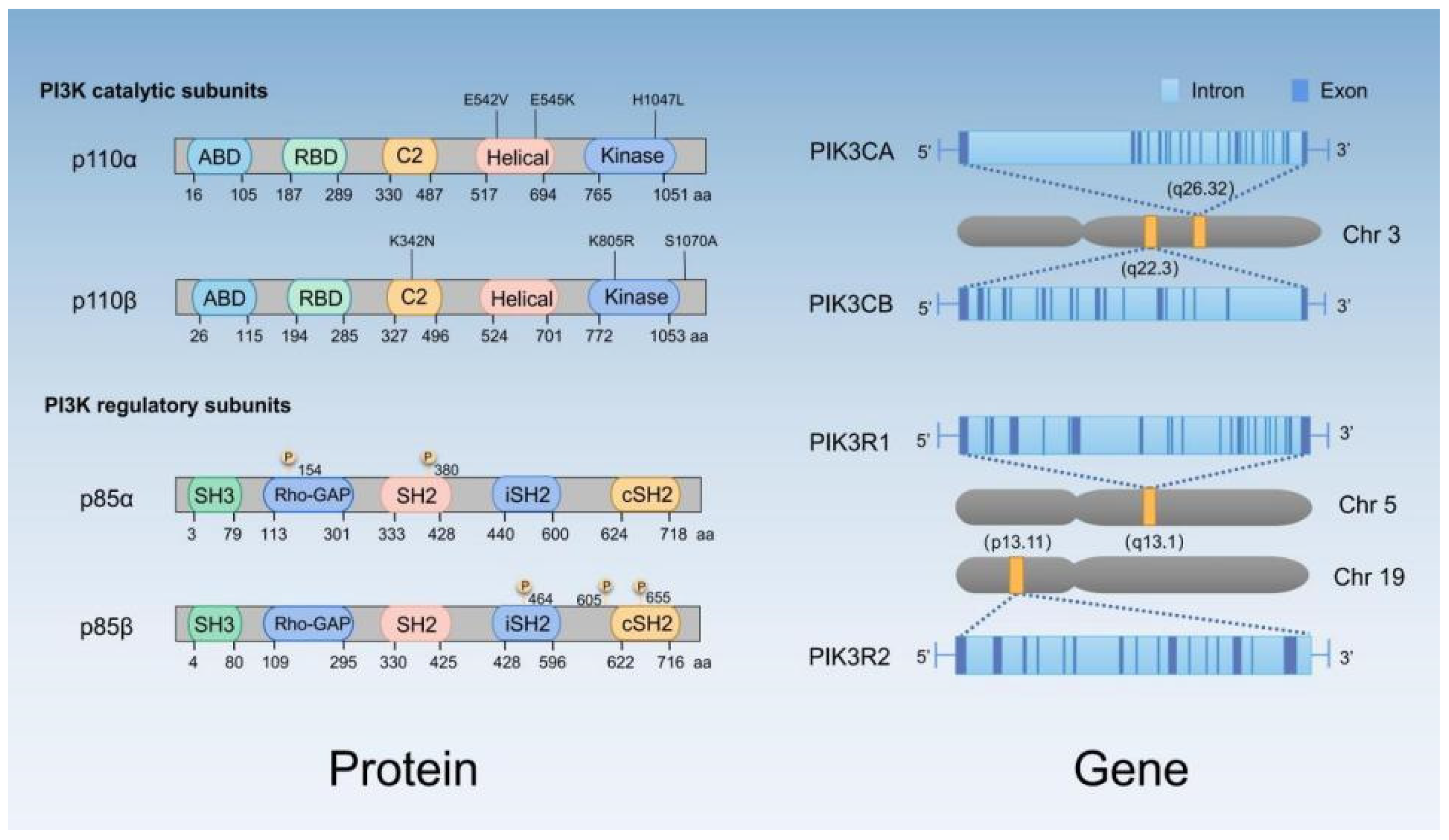
| Gene (Protein) | Alteration | Effect on Signaling | Correlation with Prognosis | Frequency | Reference | |||
|---|---|---|---|---|---|---|---|---|
| Luminal (ER+) | HER2+ | TNBC (ER−, PR−, HER2−) | ||||||
| A | B | |||||||
| PTEN | Inactivation and mutation/reduced expression | over activation of PI3K signaling | Negative in TNBC | 29–44% | 22% | 67% | [33,34,35] | |
| PIK3CA (p110α/PI3K) | Activating mutation | Hyperactivation of PI3K signaling | * Positive in luminal, negative in metastatic/HER2+ breast cancer | 47% | 33% | 23–39% | 8–25% | [33,36,37,38] |
| PIK3CB (p110β/PI3K) | Amplification /Mutation | PIP3 accumulates and activates AKT | Irrelevant | 5% | [29,39] | |||
| PIK3R1 (p85α/PI3K) | Inactivating mutation | Derepression of catalytic activity of p110α | - | 2% of Early breast cancer 11% of Metastatic breast cancer | [40,41] | |||
| AKT1 | Activating mutation | Hyperactivation of AKT | Irrelevant | 2.6–7.4% | [33,42,43,44] | |||
| AKT2 | Amplification | Irrelevant | 2.8–4% | [18] | ||||
| AKT3 | Amplification | Positive in luminal A breast cancer | 15% | [40] | ||||
| PDK1 | Amplification | Hyperactivation of AKT | - | 20–38% | [45] | |||
| (mTOR) | p-mTOR expression | Hyperactivation of mTOR | Negative in TNBC | 39% | 37.5–72.1% | [46] | ||
| Target | Drug | Study (Phase) | Patient Population | Regimen and Outcome | FDA/EMA Approval | Reference |
|---|---|---|---|---|---|---|
| Pan-PI3K | Buparisib (BKMI20) | BELLE-2 (III) | HR (+), HER2 (−), ABC/MBC (second line) | buparlisib + fulvestrant vs. placebo + fulvestrant (mPFS: 6.9 vs. 5.0 months; HR: 0.78; p = 0.00021) | N | [78] |
| BELLE-3 (II) | HR (+), HER2 (−), ABC/MBC relapsed on or after endocrine therapy and mTOR inhibitors | buparlisib vs. Placebo (mPFS: 3.9 vs. 1.8 months; HR: 0.67; p = 0.0003) | [79] | |||
| Pictilisib (GDC-0941) | FERGI (II) | HR (+), HER2 (−), ABC/MBC Al-resistant | pictilisib + fulvestrant vs. placebo + fulvestrant (mPFS:6.6 vs. 5.1 months; HR: 0.74; p = 0.096) | N | [80] | |
| PEGGY (II) | HR (+), HER2 (−) metastatic breast cancer | Pictilisib + paclitaxel vs. placebo + paclitaxel (mPFS:8.2 vs. 7.8 months; HR: 0.95) | [81] | |||
| PI3K (p110α) | Alpelisib (BYL719) | SOLAR-1 (III) | HR (+), HER2 (−), ABC Received endocrine therapy previously | PIK3CA-mutated: alpelisib vs. placebo (mPFS 11.0 vs. 5.7 months; HR: 0.65; p < 0.001); (mOS: 39.3 vs. 31.4 months; HR: 0.86; p = 0.15) | Y | [82] |
| BYLieve (II) | HR (+), HER2 (−), PIK3CA-mutant ABC progressed on/after prior therapy, including CDK inhibitors | proportion of without disease progression at 6 month was 50.4% (95% CI: 41.2–59.6). | [83] | |||
| NEO-ORB (II) | HR (+), HER2 (−) Postmenopausal women Tlc-T3 breast cancer | Alpelisib + letrozole vs. placebo + letrozde, ORR: 43% vs. 45%, PIK3CA-wild-type vs. mutant ORR: 63% vs. 61% | [84] | |||
| Taselisib (GDC0032) | SANDPIPER (III) | Postmenopausal women, disease recurrence/progression during/after AI | Taselisib vs. placebo (PFS: 7.4 vs. 5.4 months; HR: 0.70: p = 0.0037) | N | [85] | |
| PI3K-mTOR | Gedatolisib | NCT02684032 (I) | metastatic breast cancer | NA | N | [86] |
| Apitolisib | NCT01254526 (Ib) | locally recurrent breast cancer or metastatic breast cancer | NA | N | [87] | |
| Samotolisib | NCT02057133 (I) | In combination with: letrozole, anastrozole, tamoxifen, exemestane | NA | N | [88] |
| Target | Drug | Study (Phase) | Patient Population | Regimen and Outcome | FDA/EMA Approved | Reference |
|---|---|---|---|---|---|---|
| AKT1-3 | Capivasertib (AZD5363) | BEECH (II) | ER (+), HER2 (−) ABC/MBC (first-line) | capivasertib + paclitaxel vs. placebo + paclitaxel (mPFS: 10.9 vs. 8.4 months; HR: 0.80; p = 0.308) PIK3CA+ sub-population (mPFS: 10.9 vs. 10.8 months; HR: 1.11; p = 0.760) | N | [101] |
| FAKTION (II) | ER (+)/HER (−) ABC/MBC; Postmenopausal relapsed or progressed on AI | capivasertib + fulvestrant vs. placebo + fulvestrant (mPFS: 10.3 vs. 4.8 months; HR: 0.58; p = 0.0044; mOS: 29.3 vs. 23.4 months; HR: 0.66; p = 0.035) | [102,103] | |||
| Pan-AKT | MK-2206 | NCT01776008 (II) | Endocrine resistant, ER+ breast cancer | 0% pCR | N | - |
| mTOR (mTORC1) | Everolimus (RAD001) | BOLERO-2 (III) | ER (+)/HER2−, AI-resistant and postmenopausal ABC | Everolimus + exemestrane vs. placebo + exemestrane (final PFS:11.0 vs. 4.1 months; HR: 0.38; p < 0.0001) | [104] | |
| MANTA (II) | HR+, postmenopausal and AI-resistant locally ABC or MBC | Everolimus +fulvestrant vs. fulvestrant (mPFS: 12.3 vs. 5.4 months; HR: 0.63; p = 0.01) | A | [105] | ||
| PrE0102 (II) | ER (+)/HER2−, AI-resistant and postmenopausal MBC | Everolimus + fulvestrant vs. placebo + fulvestrant (mPFS:10.3 vs.5.1 months; HR: 0.61; p = 0.02) ORR: 18.2 vs. 12.3%; p = 0.47 | [106] | |||
| BOLERO-6 (III) | ER (+)/HER2−ABC | Everolimus + exemestrane vs. Exemestrane vs. capecitabine mOS: 23.1 vs. 29.3 months vs. 25.6 months | [107] | |||
| NCT02123823 (I-II) | ER (+)/HER2− ABC and MBC | xentuzumab + everolimus + exemestane, vs. exemestane + everolimus (mPFS: 7.3 vs. 5.6 months; p = 0.9057) | [108] | |||
| mTOR (mTORC1/2) | MLN0128 | NCT02049957 (I-II) | HR+/HER2− and AI-resistant MBC | everolimus-sensitive vs. everolimus-resistant cohorts, 1 CBR-16: 45% vs. 23%, 2 ORR: 8% vs. 2% | N | [109] |
| Apanisertib | NCT02756364 (II) | HR+/HER2− and AI-resistant MBC | NA | N | - | |
| Pan-mTOR | Temsirolimus | HORIZON (III) | HR+, postmenopausal and AI-naïve ABC | Temsirolimus vs. placebo + letrozole (mPFS: 9.0 vs. 5.6 months; HR: 0.7, p < 0.009) | N | [110] |
| Target | Drug | Study (Phase) | Patient Population | Regimen and Outcome | Reference |
|---|---|---|---|---|---|
| Pan-PI3K | Buparlisib (BKM120) | BKM120 (II) | Trastuzumab-resistant HER2+ breast cancer | buparlisib + trastuzumab: ORR:10% (ORR ≥ 25%) | [118] |
| PIKHER (II) | Trastuzumab-resistant HER2+ ABC | DCR: 79%; 95% CI: 57–92%, CBR: 29%; 95% CI: 12–51%. | [119] | ||
| Alpelisib | BYL-719 (I) | Trastuzumab- and taxane-resistant HER2+ MBC | Alpelisib + T-DM1: ORR: 43%. T-DM1-resistant (n = 10): ORR 30%. mPFS 8.1 months | [120] | |
| Pan-Akt | MK-2206 | SPY2 (II) | High-risk, early-stage Breast cancer with neoadjuvant therapy | MK-2206 vs. control: pCR 61.8% vs. 35% (control: standard taxane- and anthracycline-based neoadjuvant therapy) | [121] |
| AKT-1 | Ipatasertib (IPAT) | SOLTI-1507 (Ib) | HER2+ ABC or MBC with PIK3CA mutation | NA | - |
| mTOR | Everolimus | BOLERO-1 (III) | HER2+, HR- primary ABC | Everolimus vs. placebo + trastuzumab mPFS: 20.3 vs.13.1 months; HR: 0.66; p = 0.0049 | [122] |
| BOLERO-3 (III) | Taxane-pretreated and trastuzumab-resistant HER2+ ABC | Everolimus vs. Placebo + trastuzumab, vinorelbine mPFS: 7.0 vs.5.8 months; HR: 0.78; p = 0.0067 | [123] | ||
| Sirolimus | M124188 (II) | HER2(+) MBC | Trastuzumab + sirolimus ORR: 1/9 (11%) CBR: 4/9 (44%) | [124] |
| Target | Drug | Study (Phase) | Patient Population | Regimen and Outcome | Reference |
|---|---|---|---|---|---|
| Pan-PI3K | Buparlisib (BKM120) | NCT01790932 (II) | Metastatic TNBC | CBR:12% (6 patients, all SD ≥ 4 months) mPFS: 1.8 months (95% CI: 1.6–2.3) mOS: 11.2 months (95% CI: 6.2–25) | [134] |
| AKT1-3 | Capivasertib | (II) | Metastatic TNBC | Capivasertib vs. Placebo + paclitaxel (mPFS: 5.9 vs. 4.2 months; p = 0.06) (mOS 19.1 vs. 12.6 months; p = 0.04) | [135] |
| Pan-AKT | GDC-0068 (Ipatasertib) | LOTU (II) | Metastatic TNBC | Ipatasertib vs. placebo + paclitaxel (mPFS 6.2 vs. 4.9 months; HR: 0.60; p = 0.037) | [136] |
| FAIRLANE (II) | Early TNBC | Ipatasertib + paclitaxel vs. placebo + paclitaxel pCR rates: 17% vs. 13% | [137] | ||
| LY2780301 | TAKTIC (Ib/II) | HER2-ABC | 6-month ORR:63.9% [48.8–76.8] | [138] | |
| mTOR | Everolimus (DAE) | NCT00930930 (II) | II/III TNBC (Neoadjuvant therapy) | Everolimus vs. placebo (5pCR: 36% vs. 49%) | [139] |
| Temsirolimus (DAT) | NCT00761644 (II) | Metaplastic TNBC | Doxorubicin + bevacizumab + DAT or DAE ORR: 21%; CBR: 40% PI3K pathway alteration: ORR: (31% vs. 0%; p = 0.04); CBR (44% vs. 45%; p > 0.99). | [140] | |
| PI3K-mTOR | Eganelisib | NCT03719326 (I/Ib) | Advanced or metastatic TNBC | In combination with pegylated liposomal doxorubicin (PLD) or A2aR/A2bR antagonist-1(AB928) NA | - |
| Target | Drug | HR+, HER2− | HER2+ | Triple Negative Breast Cancer |
|---|---|---|---|---|
| Class I PI3K | Buparisib | NCT01633060 | - | NCT01790932 |
| NCT01339442 | ||||
| NCT01610284 | ||||
| Pictilisib | NCT01437566 | NCT00928330 | NCT01918306 | |
| NCT01740336 | ||||
| Alpelisib | NCT02379247 | NCT05230810 | NCT02038010 | |
| NCT03386162 | ||||
| NCT04208178 | ||||
| Taselisib | NCT02340221 | NCT02390427 | NCT02457910 | |
| NCT02273973 | ||||
| PI3K-mTOR | Gedatolisib | NCT02684032 | NCT03698383 | NCT03243331 |
| NCT01920061 | ||||
| Apitolisib | NCT01254526 | - | - | |
| Samotolisib | NCT02057133 | - | - | |
| Eganelisib | - | - | NCT03961698 | |
| AKT | Capivasertib | NCT01277757 | - | NCT03997123 |
| NCT01992952 | NCT03742102 | |||
| MK-2206 | NCT01776008 | - | - | |
| Ipatasertib | - | NCT03840200 | NCT02301988 | |
| NCT03800836 | ||||
| LY2780301 | - | - | NCT01980277 | |
| mTOR | Everolimus | NCT02216786 | NCT00912340 | NCT01931163 |
| NCT01797120 | NCT00876395 | |||
| NCT01783444 | ||||
| NCT02123823 | ||||
| MLN0128 | NCT02049957 | - | NCT02719691 | |
| NCT02988986 | ||||
| Apanisertib | NCT02756364 | - | - | |
| Temsirolimus | NCT02152943 | NCT00411788 | NCT02723877 | |
| NCT01248494 | ||||
| Sirolimus | NCT00411788 | NCT01783444 | - | |
| Ridaforolimus | - | NCT00736970 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, K.; Wu, Y.; He, P.; Fan, Y.; Zhong, X.; Zheng, H.; Luo, T. PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer. Cells 2022, 11, 2508. https://doi.org/10.3390/cells11162508
Zhu K, Wu Y, He P, Fan Y, Zhong X, Zheng H, Luo T. PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer. Cells. 2022; 11(16):2508. https://doi.org/10.3390/cells11162508
Chicago/Turabian StyleZhu, Kunrui, Yanqi Wu, Ping He, Yu Fan, Xiaorong Zhong, Hong Zheng, and Ting Luo. 2022. "PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer" Cells 11, no. 16: 2508. https://doi.org/10.3390/cells11162508