
Simple Detection of Unstained Live Senescent Cells with Imaging Flow Cytometry

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results
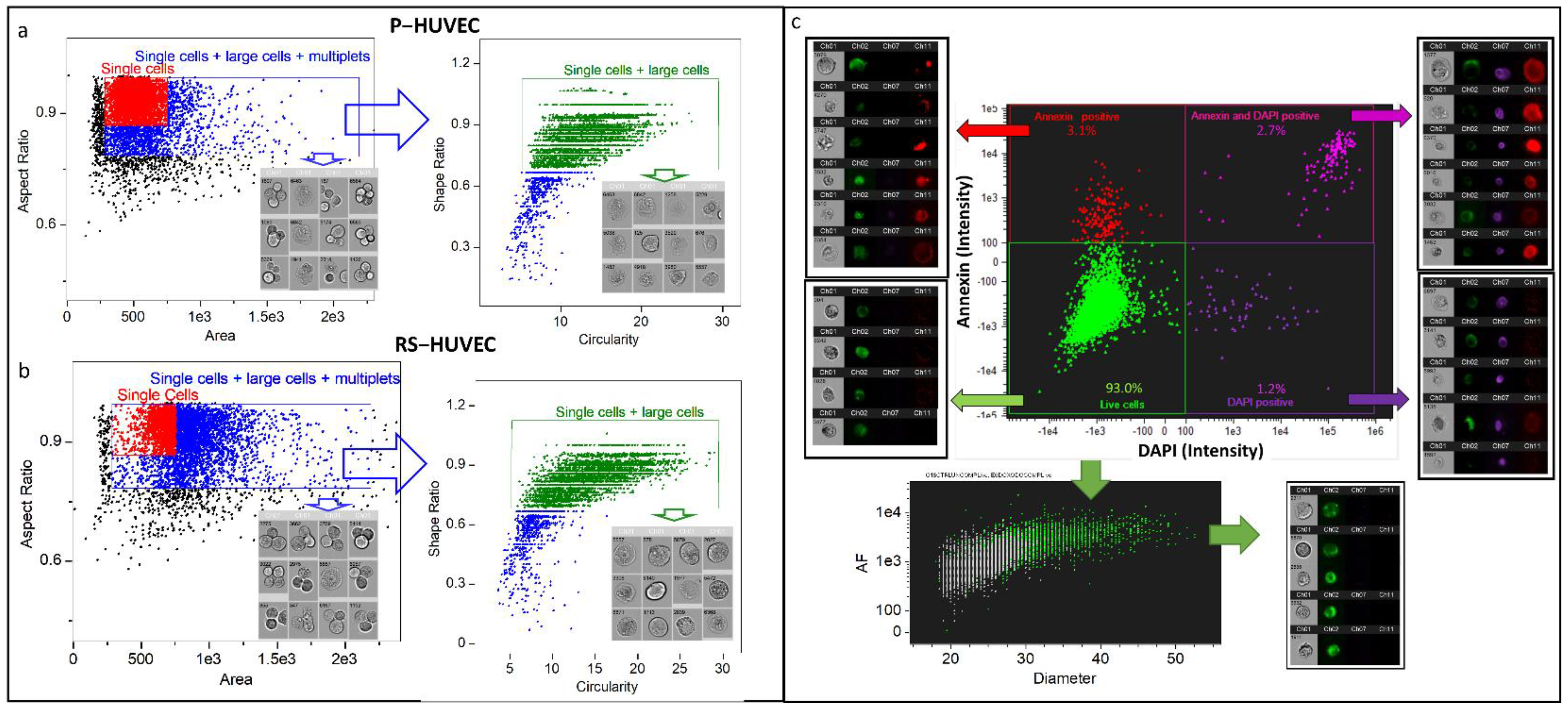
2.1. Analysis of Senescent Cells Is Biased by Multiplets That Can Be Removed with a Specific Gating Strategy
2.2. Quantification of Cellular Senescence in Various Human and Murine Senescence Models
2.3. Quantification of Cellular Senescence in Ex-Vivo Samples
2.4. Comparison of the Effect of Common Senolytics on Mouse Senescent Fibroblasts
3. Discussion
4. Methods
4.1. Cell Culture and Growth Conditions
4.1.1. Human Bone Marrow (BM)-Derived Mesenchymal Stem Cells (MSCs) and Human Umbilical Vein Endothelial Cells (HUVEC)
4.1.2. Human Dermal Fibroblasts (HuDe)
4.1.3. Murine Ear Fibroblasts (MearFs)
4.1.4. Ex Vivo Cells from Mouse Ear Biopsies
4.1.5. Ex Vivo Cardiac Pericytes
4.2. Senescence Biomarkers Assays
4.2.1. Senescence Associated β-Galactosidase Staining
4.2.2. p16ink4a and p21Cip1 qRT-PCR
4.2.3. KI-67 and γH2AX in Cardiac Pericytes
4.3. FlowSight Analysis
4.3.1. Senescence Associated β-Galactosidase Flow Cytometry Assays
4.3.2. Identification of Senescent Cells by Artificial Intelligence (AI) and Machine Learning (ML)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Robbins, E.; Levine, E.M.; Eagle, H. Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med. 1970, 131, 1211–1222. [Google Scholar] [CrossRef]
- Rattan, S.I.S.; Keeler, K.D.; Buchanan, J.H.; Holliday, R. Autofluorescence as an index of ageing in human fibroblasts in culture. Biosci. Rep. 1982, 2, 561–567. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Kohli, J.; Wang, B.; Brandenburg, S.M.; Basisty, N.; Evangelou, K.; Varela-Eirin, M.; Campisi, J.; Schilling, B.; Gorgoulis, V.; Demaria, M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 2021, 16, 2471–2498. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Noppe, G.; Dekker, P.; de Koning-Treurniet, C.; Blom, J.; van Heemst, D.; Dirks, R.W.; Tanke, H.J.; Westendorp, R.G.J.; Maier, A.B. Rapid flow cytometric method for measuring senescence associated beta-galactosidase activity in human fibroblasts. Cytom. A 2009, 75, 910–916. [Google Scholar] [CrossRef]
- Majore, I.; Moretti, P.; Hass, R.; Kasper, C. Identification of subpopulations in mesenchymal stem cell-like cultures from human umbilical cord. Cell Commun. Signal. 2009, 7, 6. [Google Scholar] [CrossRef]
- Malavolta, M.; Costarelli, L.; Giacconi, R.; Basso, A.; Piacenza, F.; Pierpaoli, E.; Provinciali, M.; Ogo, O.A.O.A.; Ford, D. Changes in Zn homeostasis during long term culture of primary endothelial cells and effects of Zn on endothelial cell senescence. Exp. Gerontol. 2017, 99, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Barteneva, N.S.; Fasler-Kan, E.; Vorobjev, I.A. Imaging Flow Cytometry: Coping with Heterogeneity in Biological Systems. J. Histochem. Cytochem. 2012, 60, 723. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Zhai, W.; Tan, J.; Russell, T.; Chen, S.; McGonagle, D.; Win Naing, M.; Yong, D.; Jones, E. Multi-pronged approach to human mesenchymal stromal cells senescence quantification with a focus on label-free methods. Sci. Rep. 2021, 11, 1054. [Google Scholar] [CrossRef]
- Bertolo, A.; Baur, M.; Guerrero, J.; Pötzel, T.; Stoyanov, J. Autofluorescence is a Reliable in vitro Marker of Cellular Senescence in Human Mesenchymal Stromal Cells. Sci. Rep. 2019, 9, 2074. [Google Scholar] [CrossRef]
- Bertolo, A.; Guerrero, J.; Stoyanov, J. Autofluorescence-based sorting removes senescent cells from mesenchymal stromal cell cultures. Sci. Rep. 2020, 10, 19084. [Google Scholar] [CrossRef]
- Kauffman, M.K.; Kauffman, M.E.; Zhu, H.; Jia, Z.; Li, Y.R. Fluorescence-Based Assays for Measuring Doxorubicin in Biological Systems. React. Oxyg. Species (Apex) 2016, 2, 432–439. [Google Scholar] [CrossRef]
- Xu, X.; Sun, X.; Wan, X.; Chen, X.; Jiang, X. Mitomycin induces alveolar epithelial cell senescence by down-regulating GSK3β signaling. Toxicol. Lett. 2021, 352, 61–69. [Google Scholar] [CrossRef]
- Rolle, I.G.; Crivellari, I.; Zanello, A.; Mazzega, E.; Dalla, E.; Bulfoni, M.; Avolio, E.; Battistella, A.; Lazzarino, M.; Cellot, A.; et al. Heart failure impairs the mechanotransduction properties of human cardiac pericytes. J. Mol. Cell. Cardiol. 2021, 151, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Caragnano, A.; Aleksova, A.; Bulfoni, M.; Cervellin, C.; Rolle, I.G.; Veneziano, C.; Barchiesi, A.; Mimmi, M.C.; Vascotto, C.; Finato, N.; et al. Autophagy and Inflammasome Activation in Dilated Cardiomyopathy. J. Clin. Med. 2019, 8, 1519. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Faragher, R.G.A. Cellular senescence: From growth arrest to immunogenic conversion. Age 2015, 37, 27. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.O.; Perdue, B.D.; Uruchurtu-Valdivia, R.A. Isolation by flow sorting of cytokinetic and morphological heterogeneity in late-passage cultures of human diploid fibroblasts (IMR-90). Anat. Rec. 1983, 206, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Nilsson, E.; Docke, W.D.; Brunk, U.T. Lipofuscin accumulation and ageing of fibroblasts. Gerontology 1995, 41, 95–108. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Gianfranceschi, G.; Caragnano, A.; Piazza, S.; Manini, I.; Ciani, Y.; Verardo, R.; Toffoletto, B.; Finato, N.; Livi, U.; Beltrami, C.A.; et al. Critical role of lysosomes in the dysfunction of human Cardiac Stem Cells obtained from failing hearts. Int. J. Cardiol. 2016, 216, 140–150. [Google Scholar] [CrossRef]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef]
- Saxena, S.; Vekaria, H.; Sullivan, P.G.; Seifert, A.W. Connective tissue fibroblasts from highly regenerative mammals are refractory to ROS-induced cellular senescence. Nat. Commun. 2019, 10, 4400. [Google Scholar] [CrossRef] [PubMed]
- Elbakrawy, E.; Bains, S.K.; Bright, S.; Al-Abedi, R.; Mayah, A.; Goodwin, E.; Kadhim, M. Radiation-Induced Senescence Bystander Effect: The Role of Exosomes. Biology 2020, 9, 191. [Google Scholar] [CrossRef]
- Rossi, O.; Barbieri, O.; Frosina, G. Time-course of spontaneous transformation of CD-1 mouse embryonic fibroblasts. Anticancer Res. 2003, 23, 1373–1377. [Google Scholar]
- Sun, H.; Taneja, R. Analysis of transformation and tumorigenicity using mouse embryonic fibroblast cells. Methods Mol. Biol. 2007, 383, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Von Zglinicki, T.; Passos, J.F. Cell sorting of young and senescent cells. Methods Mol. Biol. 2013, 1048, 31–47. [Google Scholar] [CrossRef]
- Wang, D.; Lu, P.; Liu, Y.; Chen, L.; Zhang, R.; Sui, W.; Dumitru, A.G.; Chen, X.; Wen, F.; Ouyang, H.W.; et al. Isolation of Live Premature Senescent Cells Using FUCCI Technology. Sci. Rep. 2016, 6, 30705. [Google Scholar] [CrossRef] [PubMed]
- Kovacovicova, K.; Vinciguerra, M. Isolation of senescent cells by iodixanol (OptiPrep) density gradient-based separation. Cell Prolif. 2019, 52, e12674. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.-H.; Que, F.-C.; Gu, C.-P.; Zhong, D.-S.; Zhou, D.; Kong, Y.; Yu, L.; Liu, S.-W. ABT-263 induces G1/G0-phase arrest, apoptosis and autophagy in human esophageal cancer cells in vitro. Acta Pharmacol. Sin. 2017, 38, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Grezella, C.; Fernandez-Rebollo, E.; Franzen, J.; Ventura Ferreira, M.S.; Beier, F.; Wagner, W. Effects of senolytic drugs on human mesenchymal stromal cells. Stem Cell Res. Ther. 2018, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Fixation and permeabilization of cells and tissues. Cold Spring Harb. Protoc. 2008, 2008, pdb-top36. [Google Scholar] [CrossRef]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef]
- Althubiti, M.; Macip, S. Detection of Senescent Cells by Extracellular Markers Using a Flow Cytometry-Based Approach. Methods Mol. Biol. 2017, 1534, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, A.; Mikami, H.; Hiramatsu, K.; Sakuma, S.; Kasai, Y.; Iino, T.; Yamano, T.; Yasumoto, A.; Oguchi, Y.; Suzuki, N.; et al. A practical guide to intelligent image-activated cell sorting. Nat. Protoc. 2019, 14, 2370–2415. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.A.; Probst, C.E.; Zayats, A.; Davidson, B.; Riedel, M.; Li, Y.; Venkatachalam, V. The in vitro micronucleus assay using imaging flow cytometry and deep learning. NPJ Syst. Biol. Appl. 2021, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.; Zayats, A.; Venkatachalam, V.; Davidson, B. Advanced Characterization of Silicone Oil Droplets in Protein Therapeutics Using Artificial Intelligence Analysis of Imaging Flow Cytometry Data. J. Pharm. Sci. 2020, 109, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Furrer, R.; Jauch, A.J.; Rao, T.N.; Dilbaz, S.; Rhein, P.; Steurer, S.A.; Recher, M.; Skoda, R.C.; Handschin, C. Remodeling of metabolism and inflammation by exercise ameliorates tumor-associated anemia. Sci. Adv. 2021, 7, eabi4852. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, C.A.; Bill, M.; Rodrigues, M.A.; Hauerslev, M.; Kerndrup, G.B.; Hokland, P.; Ludvigsen, M. Exploring dyserythropoiesis in patients with myelodysplastic syndrome by imaging flow cytometry and machine-learning assisted morphometrics. Cytom. B Clin. Cytom. 2021, 100, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, M.; Rhein, P.; Czaczyk, K.; Białas, W.; Juzwa, W. Imaging Flow Cytometry to Study Biofilm-Associated Microbial Aggregates. Molecules 2021, 26, 7096. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Cell Type (Senescence Inducing Stress) | Expected Range of Senescent Cells Estimated by SA-β-Gal Assays | SI | % LAF |
|---|---|---|---|---|
| In vitro | MearF (DOX) | 70–90% | +++ | +++ |
| In vitro | HuDe (MMC) | 60–80% | +++ | +++ |
| In vitro | MearF (MMC) | 60–80% | +++ | ++ |
| In vitro | HUVEC (replicative stress) | 70–90% | ++ | +++ |
| In vitro | MearF (Irradiated, 8 days) | 30–60% | ++ | ++ |
| In vitro | MSC (replicative stress) | 60–80% | ++ | ++ |
| Ex vivo | Cardiac pericytes (heart failure) | 25–40% | ++ | + |
| In vitro | MearF (H2O2) | 10–40% | + | + |
| In vitro | MearF (replicative stress) | 10–40% | + | + |
| In vivo | Ear skin biopsies (geriatric age) | 1–3% | + | − |
| Any | Non senescent cells | 0% | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malavolta, M.; Giacconi, R.; Piacenza, F.; Strizzi, S.; Cardelli, M.; Bigossi, G.; Marcozzi, S.; Tiano, L.; Marcheggiani, F.; Matacchione, G.; et al. Simple Detection of Unstained Live Senescent Cells with Imaging Flow Cytometry. Cells 2022, 11, 2506. https://doi.org/10.3390/cells11162506
Malavolta M, Giacconi R, Piacenza F, Strizzi S, Cardelli M, Bigossi G, Marcozzi S, Tiano L, Marcheggiani F, Matacchione G, et al. Simple Detection of Unstained Live Senescent Cells with Imaging Flow Cytometry. Cells. 2022; 11(16):2506. https://doi.org/10.3390/cells11162506
Chicago/Turabian StyleMalavolta, Marco, Robertina Giacconi, Francesco Piacenza, Sergio Strizzi, Maurizio Cardelli, Giorgia Bigossi, Serena Marcozzi, Luca Tiano, Fabio Marcheggiani, Giulia Matacchione, and et al. 2022. "Simple Detection of Unstained Live Senescent Cells with Imaging Flow Cytometry" Cells 11, no. 16: 2506. https://doi.org/10.3390/cells11162506