
Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
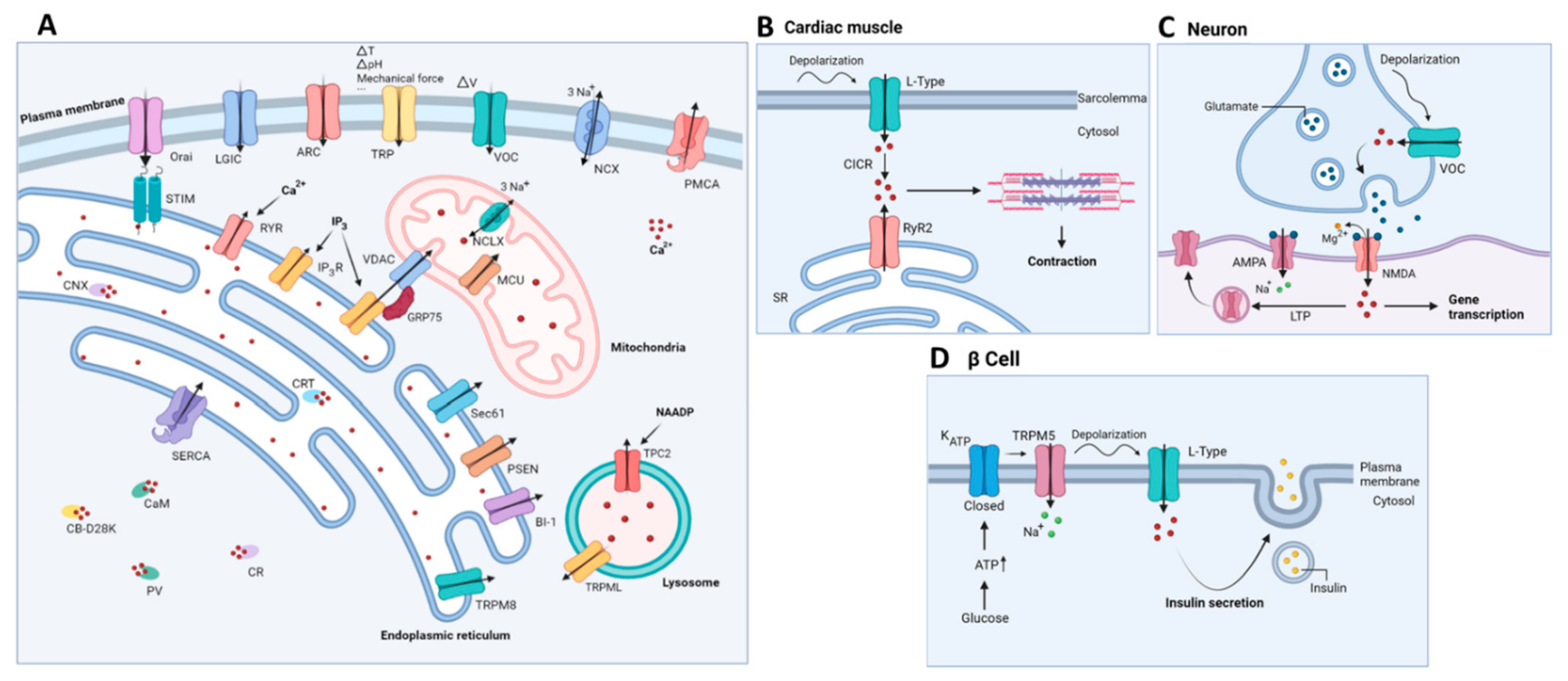
2. Ca2+ Signaling
2.1. Ca2+ Signaling: A Brief Overview
2.2. Mitochondrial Ca2+ Handling
2.3. Lysosomal Ca2+ Handling
3. Alzheimer’s Disease
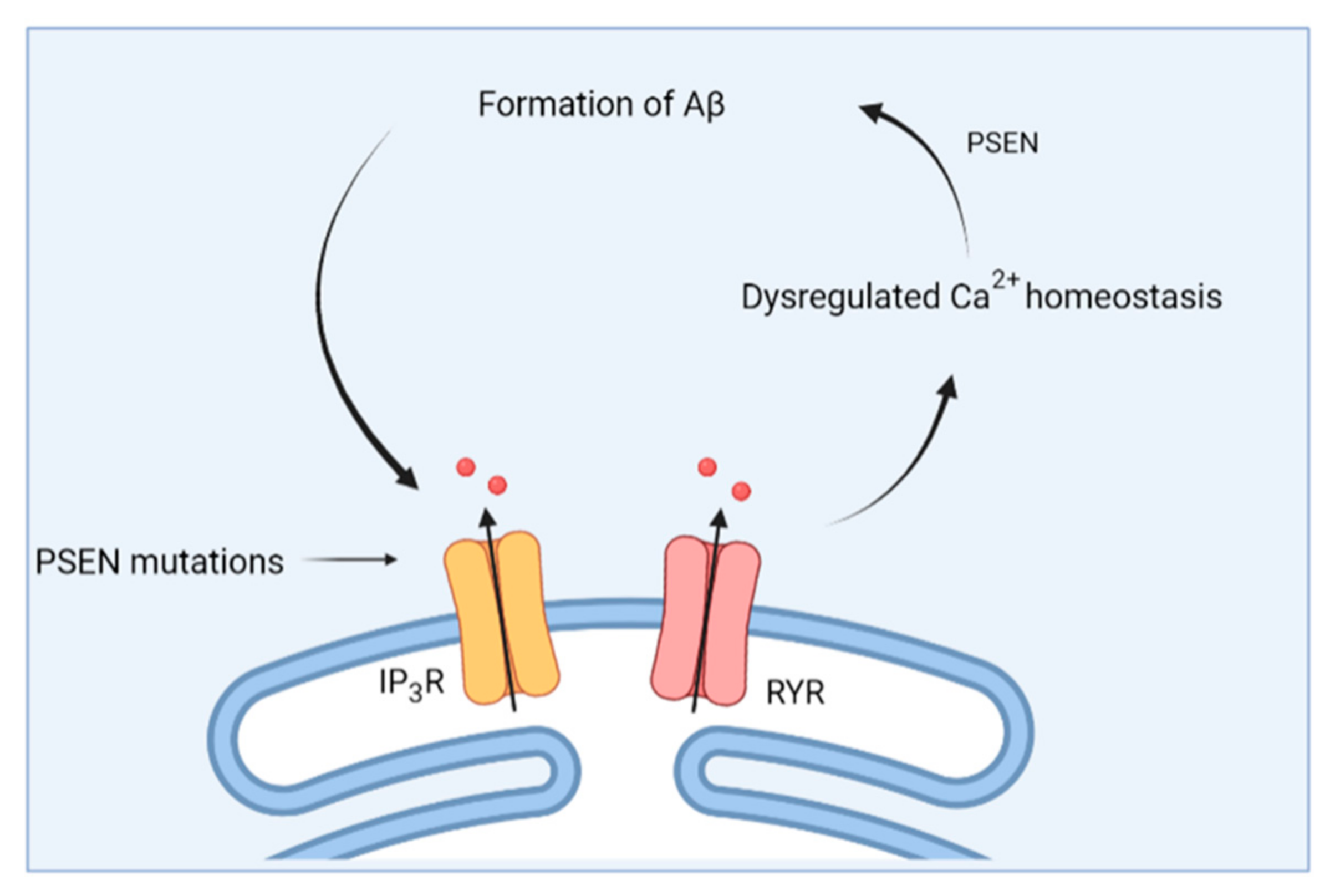
3.1. From Amyloid Hypothesis to Calcium Hypothesis
3.2. Changes in ER Ca2+ Handling in AD
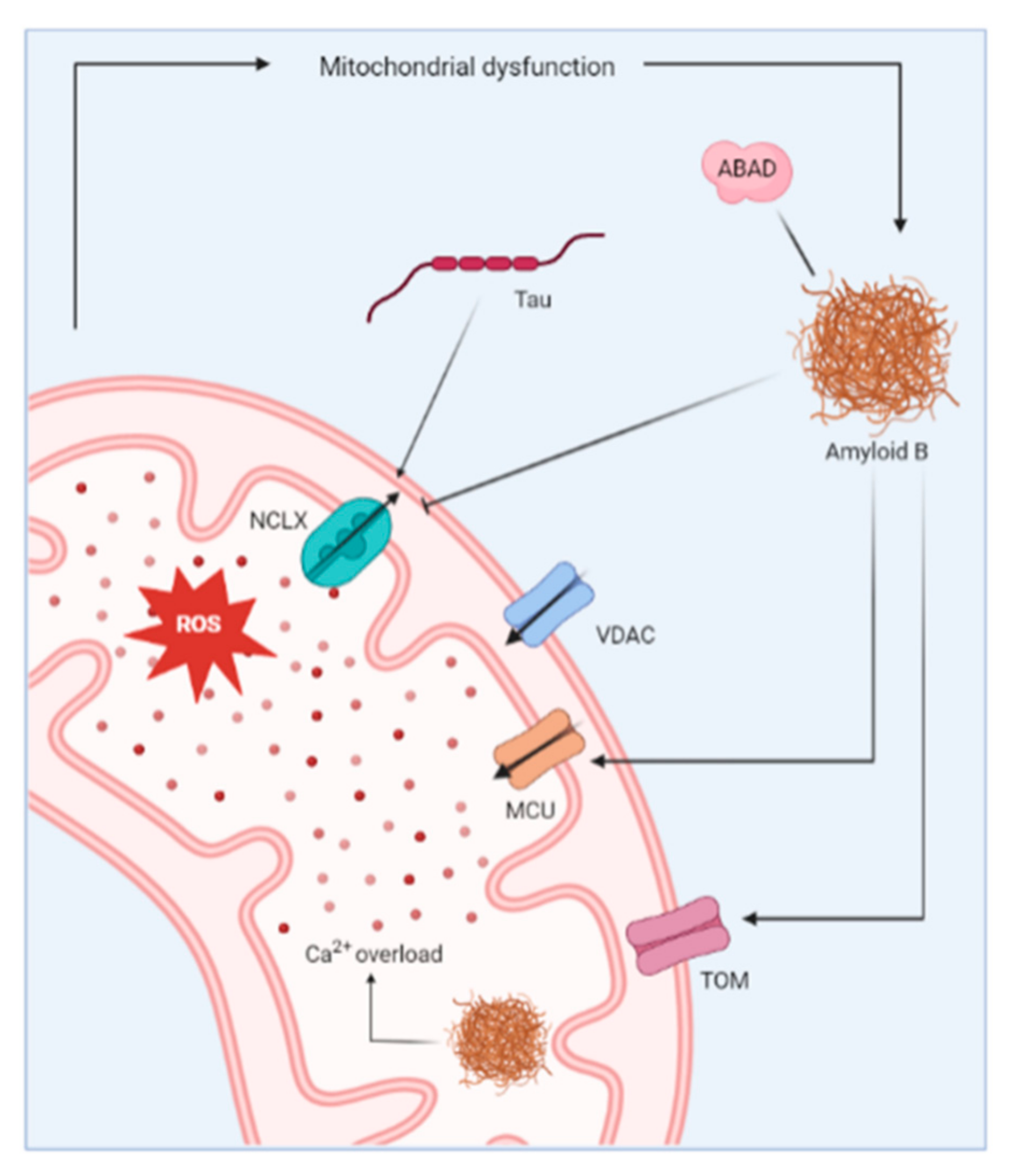
3.3. Mitochondrial Ca2+ Dysregulation in AD
3.4. MAMs and AD
4. Wolfram Syndrome
4.1. Introducing Wolfram Syndrome
4.2. The Role of Ca2+ in WS
4.3. Mitochondrial Dysfunction in WS
5. Similarities between AD and WS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Parys, J.B.; Guse, A.H. Full focus on calcium. Sci. Signal. 2019, 12, eaaz0961. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Parys, J.B.; Vervliet, T. New Insights in the IP3 Receptor and Its Regulation. Adv. Exp. Med. Biol. 2020, 1131, 243–270. [Google Scholar] [CrossRef]
- Woll, K.A.; Van Petegem, F. Calcium-release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022, 102, 209–268. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Bandara, S.; Malmersjo, S.; Meyer, T. Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci. Signal. 2013, 6, ra56. [Google Scholar] [CrossRef] [Green Version]
- Lemos, F.O.; Bultynck, G.; Parys, J.B. A comprehensive overview of the complex world of the endo- and sarcoplasmic reticulum Ca2+-leak channels. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119020. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 Is an ER Ca2+ Load-Activated Ca2+Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepulveda, M.R.; Vangheluwe, P. Primary Active Ca2+ Transport Systems in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef]
- Baraniak, J.H., Jr.; Zhou, Y.; Nwokonko, R.M.; Jennette, M.R.; Kazzaz, S.A.; Stenson, J.M.; Whitsell, A.L.; Wang, Y.; Trebak, M.; Gill, D.L. Orai channel C-terminal peptides are key modulators of STIM-Orai coupling and calcium signal generation. Cell Rep. 2021, 35, 109322. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca2+ signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef]
- Ahmad, M.; Ong, H.L.; Saadi, H.; Son, G.Y.; Shokatian, Z.; Terry, L.E.; Trebak, M.; Yule, D.I.; Ambudkar, I. Functional communication between IP3R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE. Proc. Natl. Acad. Sci. USA 2022, 119, e2114928118. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef]
- Kasri, N.N.; Bultynck, G.; Smyth, J.; Szlufcik, K.; Parys, J.B.; Callewaert, G.; Missiaen, L.; Fissore, R.A.; Mikoshiba, K.; de Smedt, H. The N-terminal Ca2+-independent calmodulin-binding site on the inositol 1,4,5-trisphosphate receptor is responsible for calmodulin inhibition, even though this inhibition requires Ca2+. Mol. Pharmacol. 2004, 66, 276–284. [Google Scholar] [CrossRef]
- Nadif Kasri, N.; Bultynck, G.; Sienaert, I.; Callewaert, G.; Erneux, C.; Missiaen, L.; Parys, J.B.; De Smedt, H. The role of calmodulin for inositol 1,4,5-trisphosphate receptor function. Biochim. Biophys. Acta 2002, 1600, 19–31. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ Buffers Are Inherently Ca2+ Signal Modulators. Cold Spring Harb. Perspect. Biol. 2020, 12, a035543. [Google Scholar] [CrossRef] [PubMed]
- Elies, J.; Yanez, M.; Pereira, T.M.C.; Gil-Longo, J.; MacDougall, D.A.; Campos-Toimil, M. An Update to Calcium Binding Proteins. Adv. Exp. Med. Biol. 2020, 1131, 183–213. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Haynes, L.P. Sense and specificity in neuronal calcium signalling. Biochim. Biophys. Acta 2015, 1853, 1921–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasri, N.N.; Holmes, A.M.; Bultynck, G.; Parys, J.B.; Bootman, M.D.; Rietdorf, K.; Missiaen, L.; McDonald, F.; De Smedt, H.; Conway, S.J.; et al. Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. EMBO J. 2004, 23, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [Green Version]
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na++/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019, 94, 59–65. [Google Scholar] [CrossRef]
- Katoshevski, T.; Ben-Kasus Nissim, T.; Sekler, I. Recent studies on NCLX in health and diseases. Cell Calcium 2021, 94, 102345. [Google Scholar] [CrossRef]
- Loncke, J.; Kaasik, A.; Bezprozvanny, I.; Parys, J.B.; Kerkhofs, M.; Bultynck, G. Balancing ER-Mitochondrial Ca2+ Fluxes in Health and Disease. Trends Cell Biol. 2021, 31, 598–612. [Google Scholar] [CrossRef]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol. 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- Hwang, M.S.; Schwall, C.T.; Pazarentzos, E.; Datler, C.; Alder, N.N.; Grimm, S. Mitochondrial Ca2+influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death Differ. 2014, 21, 1733–1745. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 2021, 81, 3866–3876.e3862. [Google Scholar] [CrossRef]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 2018, 293, 17464–17476. [Google Scholar] [CrossRef] [Green Version]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galione, A. A primer of NAADP-mediated Ca Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Brailoiu, E.; He, S.; Unterwald, E.M.; Patel, S.; Slama, J.T.; Walseth, T.F.; Marchant, J.S. Essential requirement for JPT2 in NAADP-evoked Ca Ca2+ signaling. Sci. Signal. 2021, 14, eabd5605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, X.; Shah, K.; Yan, J. Lsm12 is an NAADP receptor and a two-pore channel regulatory protein required for calcium mobilization from acidic organelles. Nat. Commun. 2021, 12, 4739. [Google Scholar] [CrossRef]
- Marchant, J.S.; Gunaratne, G.S.; Cai, X.; Slama, J.T.; Patel, S. NAADP-binding proteins find their identity. Trends Biochem. Sci. 2022, 47, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Yuan, Y.; Gunaratne, G.S.; Rahman, T.; Marchant, J.S. Activation of endo-lysosomal two-pore channels by NAADP and PI(3,5)P2. Five things to know. Cell Calcium 2022, 103, 102543. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Kruger, A.; Roggenkamp, H.G.; Alpers, R.; Lodygin, D.; Jaquet, V.; Mockl, F.; Hernandez, C.L.; Winterberg, K.; Bauche, A.; et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca Ca2+ signaling during T cell activation. Sci. Signal. 2021, 14, eabe3800. [Google Scholar] [CrossRef]
- Serge Gauthier, P.R.-N.; José, A.; Morais, C.W. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2021.pdf (accessed on 3 March 2022).
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s Disease and Parkinson’s Disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-causing mutations shift Abeta length by destabilizing gamma-secretase-Abetan interactions. Cell 2021, 184, 2257–2258. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Tichauer, J.E.; Eugenin, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef]
- Van Gassen, G.; Annaert, W. Amyloid, presenilins, and Alzheimer’s disease. Neuroscientist 2003, 9, 117–126. [Google Scholar] [CrossRef]
- Lucotte, G.; Visvikis, S.; Leininger-Muler, B.; David, F.; Berriche, S.; Reveilleau, S.; Couderc, R.; Babron, M.C.; Aguillon, D.; Siest, G. Association of apolipoprotein E allele epsilon 4 with late-onset sporadic Alzheimer’s disease. Am. J. Med. Genet. 1994, 54, 286–288. [Google Scholar] [CrossRef]
- Van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef] [PubMed]
- Lalli, G.; Schott, J.M.; Hardy, J.; De Strooper, B. Aducanumab: A new phase in therapeutic development for Alzheimer’s disease? EMBO Mol. Med. 2021, 13, e14781. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Qi, Y.; Li, Y.; Li, Z.; Yang, C.; Jia, D. Activation of the sigma-1 receptor attenuates blood-brain barrier disruption by inhibiting amyloid deposition in Alzheimer’s disease mice. Neurosci. Lett. 2022, 774, 136528. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Pike, K.E.; Chetelat, G.; Ellis, K.A.; Mulligan, R.S.; Bourgeat, P.; Ackermann, U.; Jones, G.; Szoeke, C.; Salvado, O.; et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann. Neurol. 2011, 69, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Price, J.L.; McKeel, D.W., Jr.; Buckles, V.D.; Roe, C.M.; Xiong, C.; Grundman, M.; Hansen, L.A.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W.; et al. Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 2009, 30, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflug. Arch. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- McDaid, J.; Mustaly-Kalimi, S.; Stutzmann, G.E. Ca2+ Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer’s Disease. Cells 2020, 9, 2655. [Google Scholar] [CrossRef]
- Popugaeva, E.; Chernyuk, D.; Bezprozvanny, I. Reversal of Calcium Dysregulation as Potential Approach for Treating Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 344–354. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182.e117. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling and Alzheimer’s disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Agostini, M.; Fasolato, C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035212. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Siebert, A.P.; Cheung, K.-H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Vingtdeux, V.; Marambaud, P.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, 109, E1963–E1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Tanis, J.E.; Taruno, A.; Foskett, J.K. Calcium homeostasis modulator (CALHM) ion channels. Pflug. Arch. 2016, 468, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; Laferla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef]
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [Green Version]
- Klec, C.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; Waldeck-Weiermair, M.; et al. Glycogen Synthase Kinase 3 Beta Controls Presenilin-1-Mediated Endoplasmic Reticulum Ca(2)(+) Leak Directed to Mitochondria in Pancreatic Islets and beta-Cells. Cell Physiol. Biochem. 2019, 52, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Foskett, J.K.; Daniel Mak, D.O. Regulation of IP3R Channel Gating by Ca2+ and Ca2+ Binding Proteins. Curr. Top. Membr. 2010, 66, 235–272. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, L.E.; Bultynck, G.; Luyten, T.; Amijee, H.; Bootman, M.D.; Roderick, H.L. Alzheimer’s disease-associated peptide Abeta42 mobilizes ER Ca2+ via InsP3R-dependent and -independent mechanisms. Front. Mol. Neurosci. 2013, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neurochem. 2005, 94, 1711–1718. [Google Scholar] [CrossRef]
- Kelliher, M.; Fastbom, J.; Cowburn, R.F.; Bonkale, W.; Ohm, T.G.; Ravid, R.; Sorrentino, V.; O’Neill, C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience 1999, 92, 499–513. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Herms, J.; Schneider, I.; Dewachter, I.; Caluwaerts, N.; Kretzschmar, H.; Van Leuven, F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J. Biol. Chem. 2003, 278, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Liguri, G.; Cecchi, C.; Latorraca, S.; Pieri, A.; Sorbi, S.; Degl’Innocenti, D.; Ramponi, G. Alteration of acylphosphatase levels in familial Alzheimer’s disease fibroblasts with presenilin gene mutations. Neurosci. Lett. 1996, 210, 153–156. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Querfurth, H.W.; Selkoe, D.J. Calcium Ionophore Increases Amyloid β Peptide Production by Cultured Cells. Biochemistry 1994, 33, 4550–4561. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. In vivo brain imaging of mitochondrial Ca2+ in neurodegenerative diseases with multiphoton microscopy. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118998. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Frey, W.H., 2nd. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 2007, 7, 297–310. [Google Scholar] [CrossRef]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2020, 44, 136–151. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Ottolini, D.; Brini, M. Mitochondrial Ca2+ and neurodegeneration. Cell Calcium 2012, 52, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: Current status. Peptides 2002, 23, 1311–1315. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, A.; Iyer, M.; Balasubramanian, V.; Vellingiri, B. Mitochondrial calcium uniporter as a potential therapeutic strategy for Alzheimer’s disease. Acta Neuropsychiatr. 2020, 32, 65–71. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Yan, S.D.; Stern, D.M. Mitochondrial dysfunction and Alzheimer’s disease: Role of amyloid-beta peptide alcohol dehydrogenase (ABAD). Int. J. Exp. Pathol. 2005, 86, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, T.; Resende, R.; Silva, D.F.; Marques, A.P.; Santos, A.E.; Cardoso, S.M.; Domingues, M.R.; Moreira, P.I.; Pereira, C.F. Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease. Biomedicines 2021, 9, 881. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; De Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef]
- Wolfram, D.J.; Wagener, H.P. Diabetes Mellitus and Simple Optic Atrophy among Siblings: Report on Four Cases. Mayo Clin. Proc. 1938, 13, 715–718. [Google Scholar]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Cell Death Dis. 2018, 9, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diabetes Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.T.; Ehrlich, B.E. Wolfram Syndrome: A Monogenic Model to Study Diabetes Mellitus and Neurodegeneration. Curr. Opin. Physiol. 2020, 17, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Rigoli, L.; Di Bella, C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr. Opin. Pediatr. 2012, 24, 512–517. [Google Scholar] [CrossRef]
- Rigoli, L.; Bramanti, P.; Di Bella, C.; De Luca, F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr. Res. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Tekko, T.; Lillevali, K.; Luuk, H.; Sutt, S.; Truu, L.; Ord, T.; Mols, M.; Vasar, E. Initiation and developmental dynamics of Wfs1 expression in the context of neural differentiation and ER stress in mouse forebrain. Int. J. Dev. Neurosci. 2014, 35, 80–88. [Google Scholar] [CrossRef]
- Ueda, K.; Kawano, J.; Takeda, K.; Yujiri, T.; Tanabe, K.; Anno, T.; Akiyama, M.; Nozaki, J.; Yoshinaga, T.; Koizumi, A.; et al. Endoplasmic reticulum stress induces Wfs1 gene expression in pancreatic beta-cells via transcriptional activation. Eur. J. Endocrinol. 2005, 153, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Hosoya, M.; Oishi, N.; Okano, H.; Fujioka, M.; Ogawa, K. Expression pattern of wolframin, the WFS1 (Wolfram syndrome-1 gene) product, in common marmoset (Callithrix jacchus) cochlea. Neuroreport 2016, 27, 833–836. [Google Scholar] [CrossRef]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, Y.; Sekiya, M.; Fujisaki, N.; Quan, X.; Iijima, K.M. Knockdown of wfs1, a fly homolog of Wolfram syndrome 1, in the nervous system increases susceptibility to age- and stress-induced neuronal dysfunction and degeneration in Drosophila. PLoS Genet. 2018, 14, e1007196. [Google Scholar] [CrossRef] [PubMed]
- Crouzier, L.; Richard, E.M.; Diez, C.; Alzaeem, H.; Denus, M.; Cubedo, N.; Delaunay, T.; Glendenning, E.; Baxendale, S.; Liévens, J.-C.; et al. Morphological, behavioral and cellular analyses revealed different phenotypes in Wolfram syndrome wfs1a and wfs1b zebrafish mutant lines. Hum. Mol. Genet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Loncke, J.; Vervliet, T.; Parys, J.B.; Kaasik, A.; Bultynck, G. Uniting the divergent Wolfram syndrome-linked proteins WFS1 and CISD2 as modulators of Ca2+ signaling. Sci. Signal. 2021, 14, eabc6165. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Chen, Y.F.; Wu, C.Y.; Wu, P.C.; Huang, Y.L.; Kao, C.H.; Lin, C.H.; Kao, L.S.; Tsai, T.F.; Wei, Y.H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Nechushtai, R.; Karmi, O.; Zuo, K.; Marjault, H.B.; Darash-Yahana, M.; Sohn, Y.S.; King, S.D.; Zandalinas, S.I.; Carloni, P.; Mittler, R. The balancing act of NEET proteins: Iron, ROS, calcium and metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118805. [Google Scholar] [CrossRef]
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.S.; Jennings, P.A.; et al. Interactions between mitoNEET and NAF-1 in cells. PLoS ONE 2017, 12, e0175796. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.Q.; Huang, Y.L.; Teng, Y.C.; Wang, T.W.; Kao, C.H.; Yeh, C.H.; Tsai, T.F. CISD2 maintains cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118954. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C.; Lee, S.Y.; Kao, C.H.; Chen, L.H.; Shen, Z.Q.; Lai, C.H.; Tzeng, T.Y.; Pang, J.S.; Chiu, W.T.; Tsai, T.F. Cisd2 plays an essential role in corneal epithelial regeneration. EBioMedicine 2021, 73, 103654. [Google Scholar] [CrossRef]
- Sneyers, F.; Loncke, J.; Bultynck, G. Keeping an eye on Ca2+ signalling to tackle dry eye diseases. EBioMedicine 2021, 74, 103741. [Google Scholar] [CrossRef]
- Osman, A.A.; Saito, M.; Makepeace, C.; Permutt, M.A.; Schlesinger, P.; Mueckler, M. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J. Biol. Chem. 2003, 278, 52755–52762. [Google Scholar] [CrossRef] [Green Version]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.A.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D.; et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Megy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [Green Version]
- Zatyka, M.; Da Silva Xavier, G.; Bellomo, E.A.; Leadbeater, W.; Astuti, D.; Smith, J.; Michelangeli, F.; Rutter, G.A.; Barrett, T.G. Sarco(endo)plasmic reticulum ATPase is a molecular partner of Wolfram syndrome 1 protein, which negatively regulates its expression. Hum. Mol. Genet. 2015, 24, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Kanekura, K.; Hara, T.; Mahadevan, J.; Spears, L.D.; Oslowski, C.M.; Martinez, R.; Yamazaki-Inoue, M.; Toyoda, M.; Neilson, A.; et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E5292–E5301. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.D.; Fischer, T.T.; Abreu, D.; Arroyo, A.; Urano, F.; Ehrlich, B.E. Calpain inhibitor and ibudilast rescue beta cell functions in a cellular model of Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 17389–17398. [Google Scholar] [CrossRef]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef] [Green Version]
- Abreu, D.; Stone, S.I.; Pearson, T.S.; Bucelli, R.C.; Simpson, A.N.; Hurst, S.; Brown, C.M.; Kries, K.; Onwumere, C.; Gu, H.; et al. A phase Ib/IIa clinical trial of dantrolene sodium in patients with Wolfram syndrome. JCI Insight 2021, 6, e145188. [Google Scholar] [CrossRef]
- Rouzier, C.; Moore, D.; Delorme, C.; Lacas-Gervais, S.; Ait-El-Mkadem, S.; Fragaki, K.; Burte, F.; Serre, V.; Bannwarth, S.; Chaussenot, A.; et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum. Mol. Genet. 2017, 26, 1599–1611. [Google Scholar] [CrossRef]
- Yeh, C.H.; Shen, Z.Q.; Hsiung, S.Y.; Wu, P.C.; Teng, Y.C.; Chou, Y.J.; Fang, S.W.; Chen, C.F.; Yan, Y.T.; Kao, L.S.; et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508. [Google Scholar] [CrossRef]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Teng, Y.C.; Lin, C.H.; Tsai, T.F. Cisd2 Protects the Liver from Oxidative Stress and Ameliorates Western Diet-Induced Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 10, 559. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Monaco, G.; Terry, L.E.; Rosa, N.; Baker, M.R.; Parys, J.B.; Serysheva, I.I.; Yule, D.I.; Bultynck, G. Bcl-2-protein family as modulators of IP3 receptors and other organellar Ca2+ channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035089. [Google Scholar] [CrossRef] [PubMed]
- Tamir, S.; Rotem-Bamberger, S.; Katz, C.; Morcos, F.; Hailey, K.L.; Zuris, J.A.; Wang, C.; Conlan, A.R.; Lipper, C.H.; Paddock, M.L.; et al. Integrated strategy reveals the protein interface between cancer targets Bcl-2 and NAF-1. Proc. Natl. Acad. Sci. USA 2014, 111, 5177–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ridder, I.; Kerkhofs, M.; Veettil, S.P.; Dehaen, W.; Bultynck, G. Cancer cell death strategies by targeting Bcl-2’s BH4 domain. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118983. [Google Scholar] [CrossRef]
- Bultynck, G.; Szlufcik, K.; Kasri, N.N.; Assefa, Z.; Callewaert, G.; Missiaen, L.; Parys, J.B.; De Smedt, H. Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem. J. 2004, 381, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Li, S.M.; Chen, C.H.; Chen, Y.W.; Yen, Y.C.; Fang, W.T.; Tsai, F.Y.; Chang, J.L.; Shen, Y.Y.; Huang, S.F.; Chuu, C.P.; et al. Upregulation of CISD2 augments ROS homeostasis and contributes to tumorigenesis and poor prognosis of lung adenocarcinoma. Sci. Rep. 2017, 7, 11893. [Google Scholar] [CrossRef] [Green Version]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.F.; Tsai, T.F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Lin, C.H.; Tsai, T.F. Cisd2 slows down liver aging and attenuates age-related metabolic dysfunction in male mice. Aging Cell 2021, 20, e13523. [Google Scholar] [CrossRef] [PubMed]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouzier, L.; Danese, A.; Yasui, Y.; Richard, E.M.; Lievens, J.C.; Patergnani, S.; Couly, S.; Diez, C.; Denus, M.; Cubedo, N.; et al. Activation of the sigma-1 receptor chaperone alleviates symptoms of Wolfram syndrome in preclinical models. Sci. Transl. Med. 2022, 14, eabh3763. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.H.; Wu, C.Y.; Tsai, C.Y.; Liu, F.C.; Yang, C.W.; Wei, Y.H.; Hsu, M.T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [Green Version]
- Danielpur, L.; Sohn, Y.S.; Karmi, O.; Fogel, C.; Zinger, A.; Abu-Libdeh, A.; Israeli, T.; Riahi, Y.; Pappo, O.; Birk, R.; et al. GLP-1-RA Corrects Mitochondrial Labile Iron Accumulation and Improves beta-Cell Function in Type 2 Wolfram Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 3592–3599. [Google Scholar] [CrossRef]
- Holt, S.H.; Darash-Yahana, M.; Sohn, Y.S.; Song, L.; Karmi, O.; Tamir, S.; Michaeli, D.; Luo, Y.; Paddock, M.L.; Jennings, P.A.; et al. Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells. J. Cell Sci. 2016, 129, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Bonnet Wersinger, D.; Benkafadar, N.; Jagodzinska, J.; Hamel, C.; Tanizawa, Y.; Lenaers, G.; Delettre, C. Impairment of visual function and retinal ER stress activation in Wfs1-deficient mice. PLoS ONE 2014, 9, e97222. [Google Scholar] [CrossRef] [Green Version]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef]
- Chen, S.; Acosta, D.; Li, L.; Liang, J.; Chang, Y.; Wang, C.; Fitzgerald, J.; Morrison, C.; Goulbourne, C.N.; Nakano, Y.; et al. Wolframin is a novel regulator of tau pathology and neurodegeneration. Acta Neuropathol. 2022, 143, 547–569. [Google Scholar] [CrossRef]
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Yao, J.; Liu, Y.; Sun, B.; Zhan, X.; Estillore, J.P.; Turner, R.W.; Chen, S.R.W. Increased RyR2 open probability induces neuronal hyperactivity and memory loss with or without Alzheimer’s disease-causing gene mutations. Alzheimer’s Dement. 2022. [Google Scholar] [CrossRef]
- Iwamoto, M.; Nakamura, Y.; Takemura, M.; Hisaoka-Nakashima, K.; Morioka, N. TLR4-TAK1-p38 MAPK pathway and HDAC6 regulate the expression of sigma-1 receptors in rat primary cultured microglia. J. Pharmacol. Sci. 2020, 144, 23–29. [Google Scholar] [CrossRef]
- Prause, J.; Goswami, A.; Katona, I.; Roos, A.; Schnizler, M.; Bushuven, E.; Dreier, A.; Buchkremer, S.; Johann, S.; Beyer, C.; et al. Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 1581–1600. [Google Scholar] [CrossRef]
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M.; et al. Function of sigma1 receptors in Parkinson’s disease. Acta Neurol. Scand. 2005, 112, 103–107. [Google Scholar] [CrossRef]
- Ryskamp, D.; Wu, L.; Wu, J.; Kim, D.; Rammes, G.; Geva, M.; Hayden, M.; Bezprozvanny, I. Pridopidine stabilizes mushroom spines in mouse models of Alzheimer’s disease by acting on the sigma-1 receptor. Neurobiol. Dis. 2019, 124, 489–504. [Google Scholar] [CrossRef]
- Callens, M.; Kraskovskaya, N.; Derevtsova, K.; Annaert, W.; Bultynck, G.; Bezprozvanny, I.; Vervliet, T. The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118997. [Google Scholar] [CrossRef]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Venkataraman, L.; Chen, S.; Fu, H. Function of WFS1 and WFS2 in the Central Nervous System: Implications for Wolfram Syndrome and Alzheimer’s disease. Neurosci. Biobehav. Rev. 2020, 118, 775–783. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callens, M.; Loncke, J.; Bultynck, G. Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome. Cells 2022, 11, 1963. https://doi.org/10.3390/cells11121963
Callens M, Loncke J, Bultynck G. Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome. Cells. 2022; 11(12):1963. https://doi.org/10.3390/cells11121963
Chicago/Turabian StyleCallens, Manon, Jens Loncke, and Geert Bultynck. 2022. "Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome" Cells 11, no. 12: 1963. https://doi.org/10.3390/cells11121963