
Formation of Porous Structures and Crystalline Phases in Poly(vinylidene fluoride) Membranes Prepared with Nonsolvent-Induced Phase Separation—Roles of Solvent Polarity

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PVDF Solutions
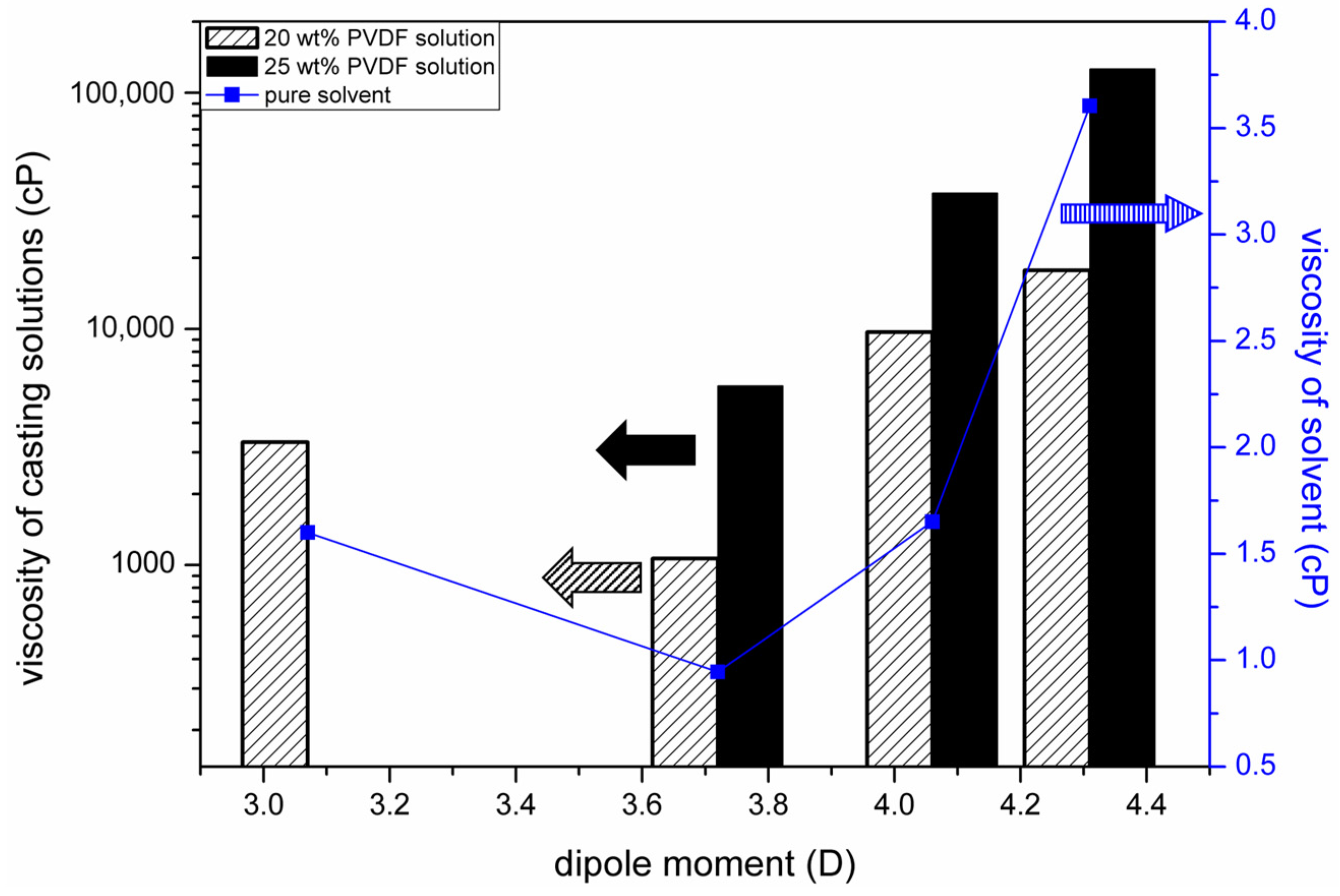
2.3. Measurement of Viscosities of PVDF Solutions
2.4. Preparation of PVDF Membranes
2.5. Characterization of Membrane Morphologies
2.6. Characterization of Crystalline Phases
2.6.1. X-ray Diffraction (XRD)
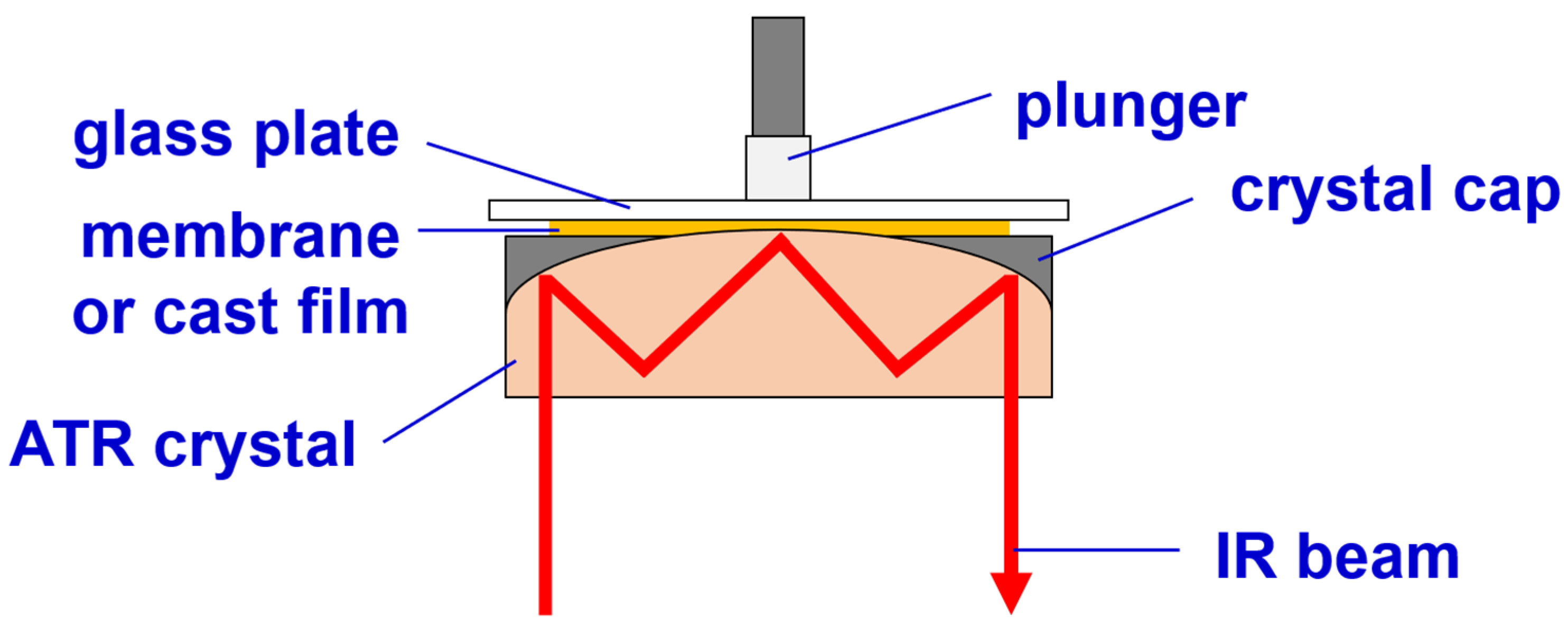
2.6.2. Infrared (IR) Spectroscopy
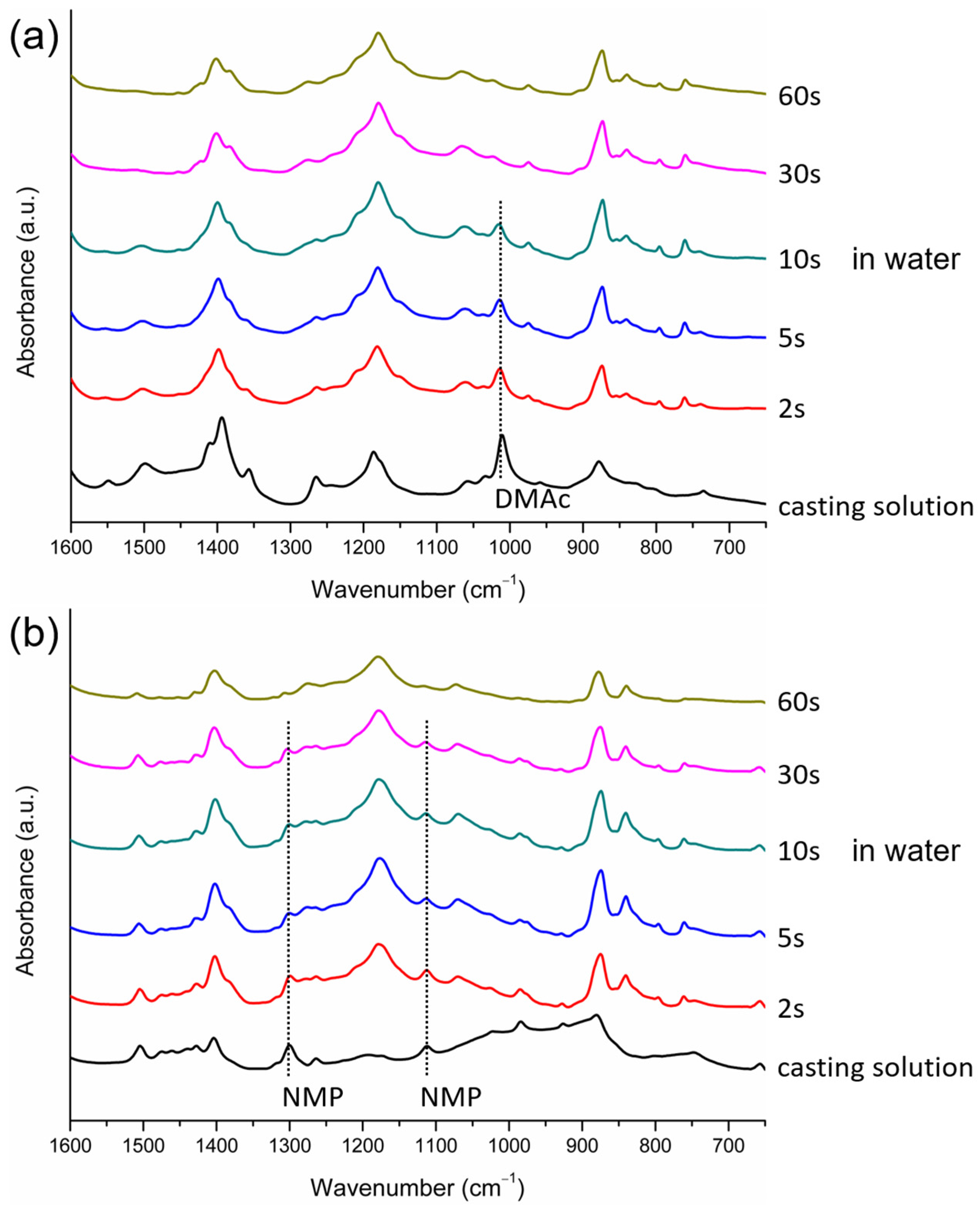
2.7. Characterization of the Evolution of Crystalline Phases during Membrane Formation
2.8. Measurement of Degrees of Crystallinity of Membranes
2.9. Characterization of Water Permeabilities and PVP Retentions of Membranes
3. Results and Discussion
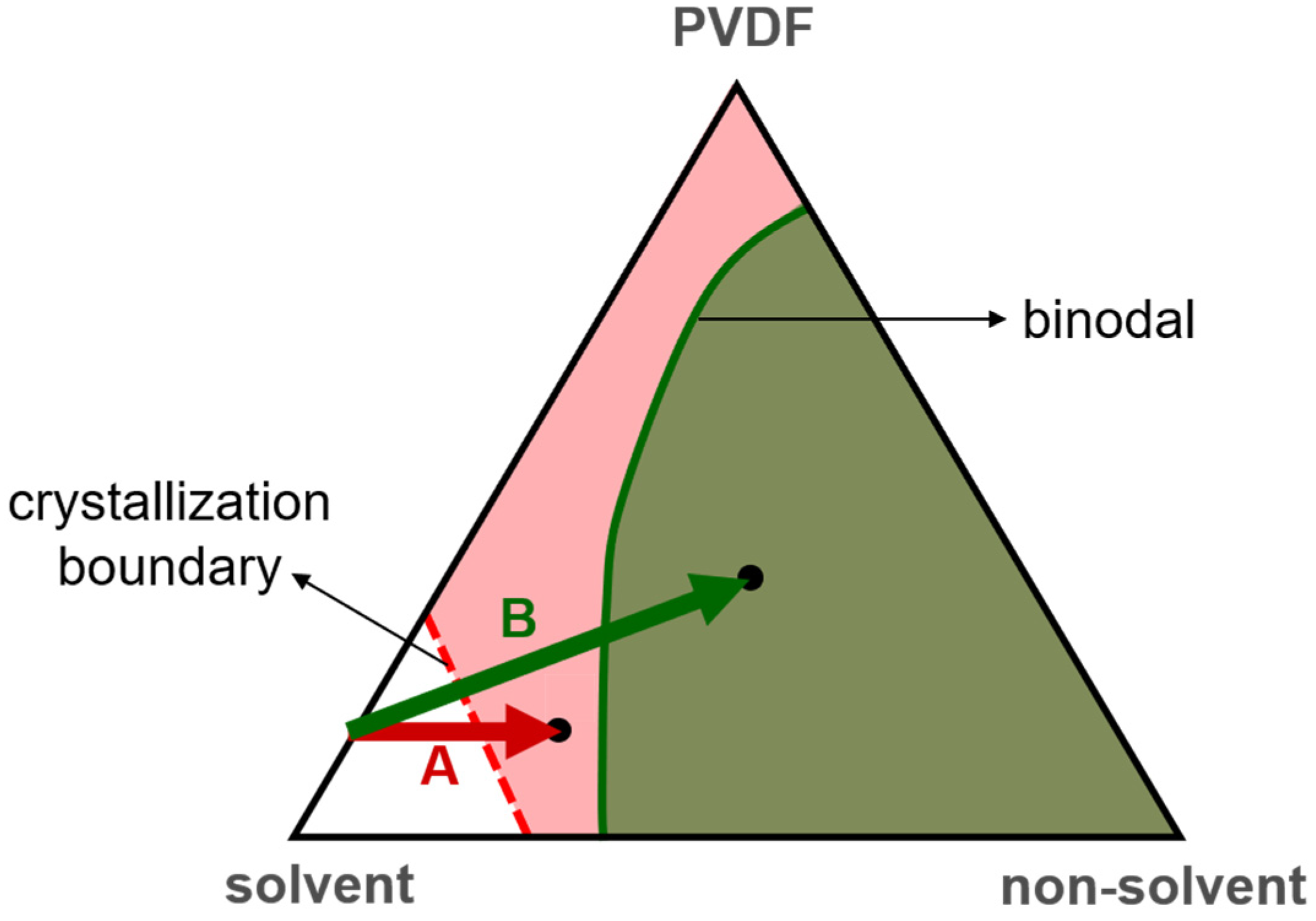
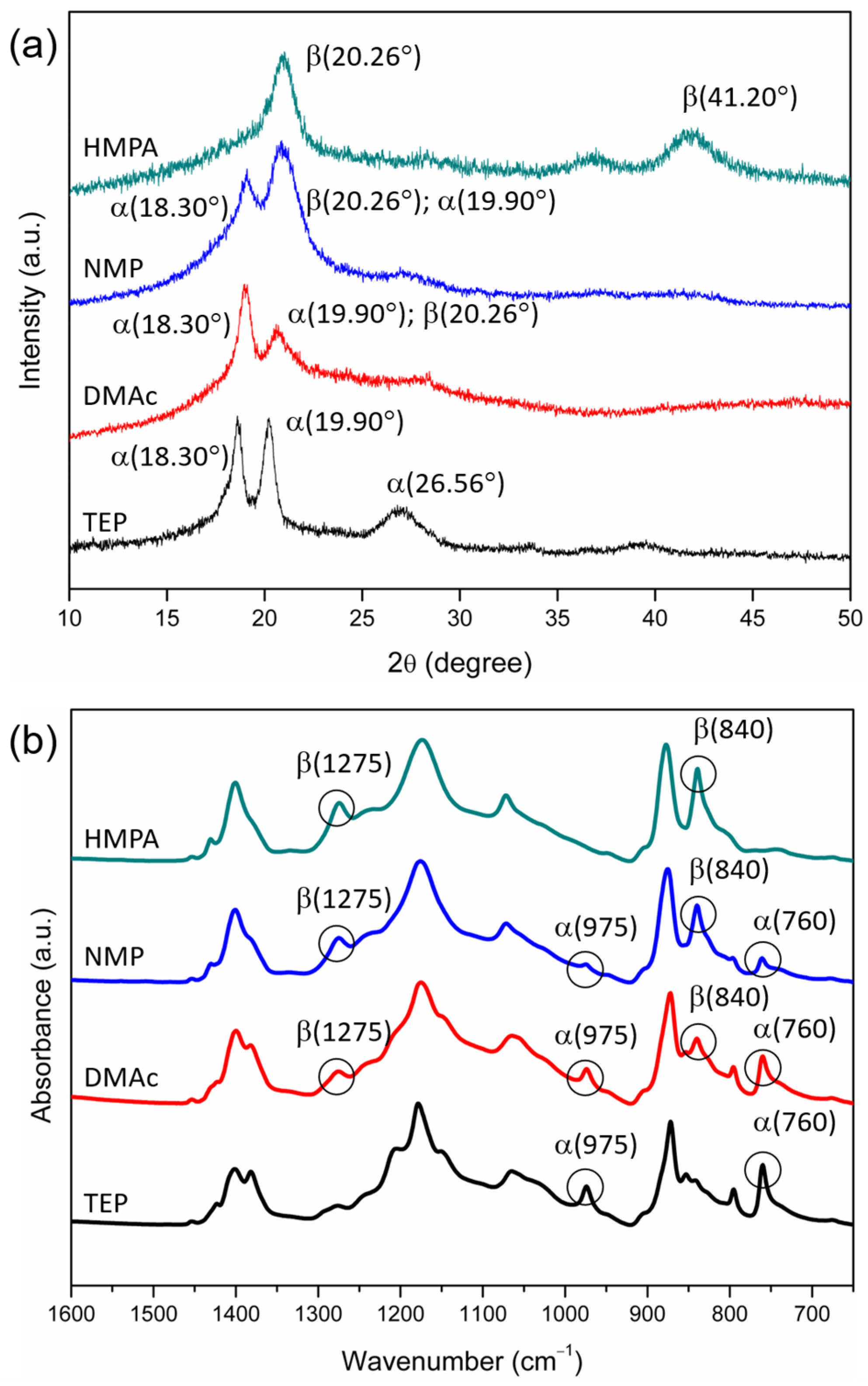
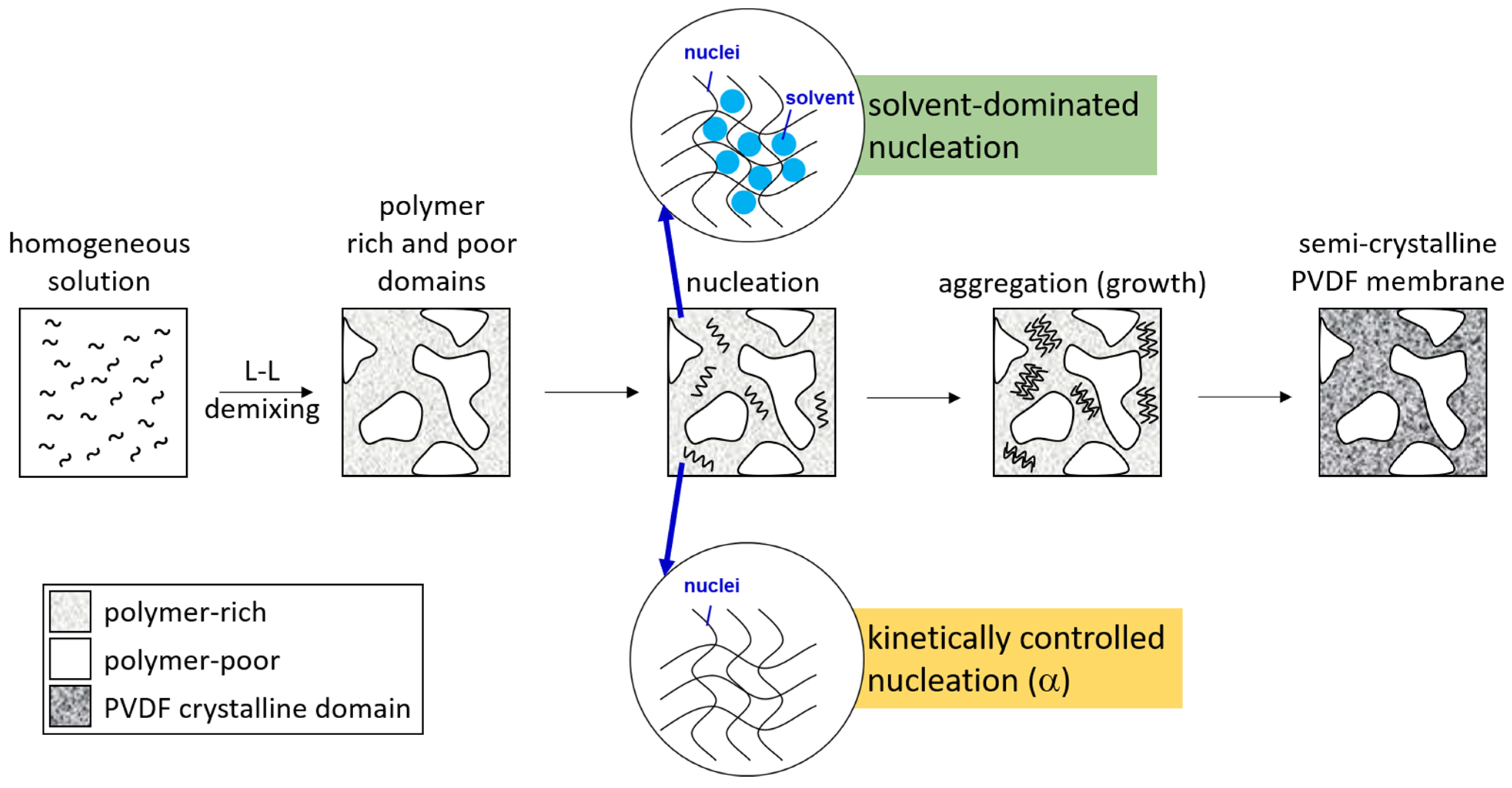
3.1. Crystalline Phases of PVDF Membranes Prepared with NIPS—Crystallization in Solvent-Rich and Solvent-Poor Environments
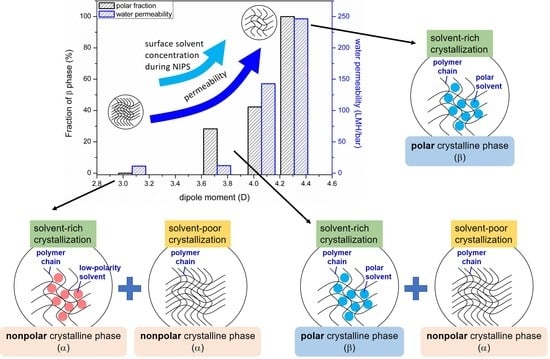
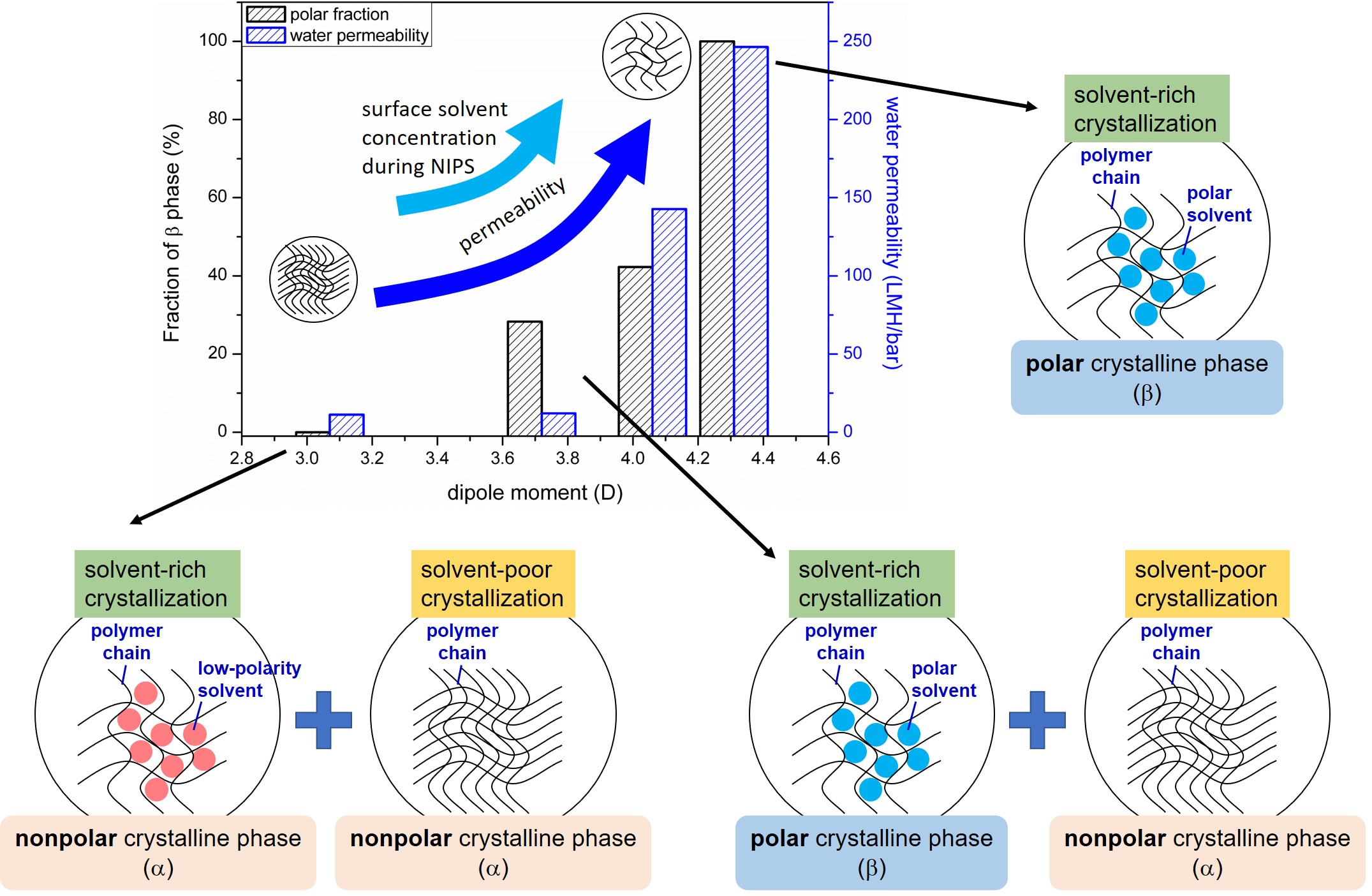
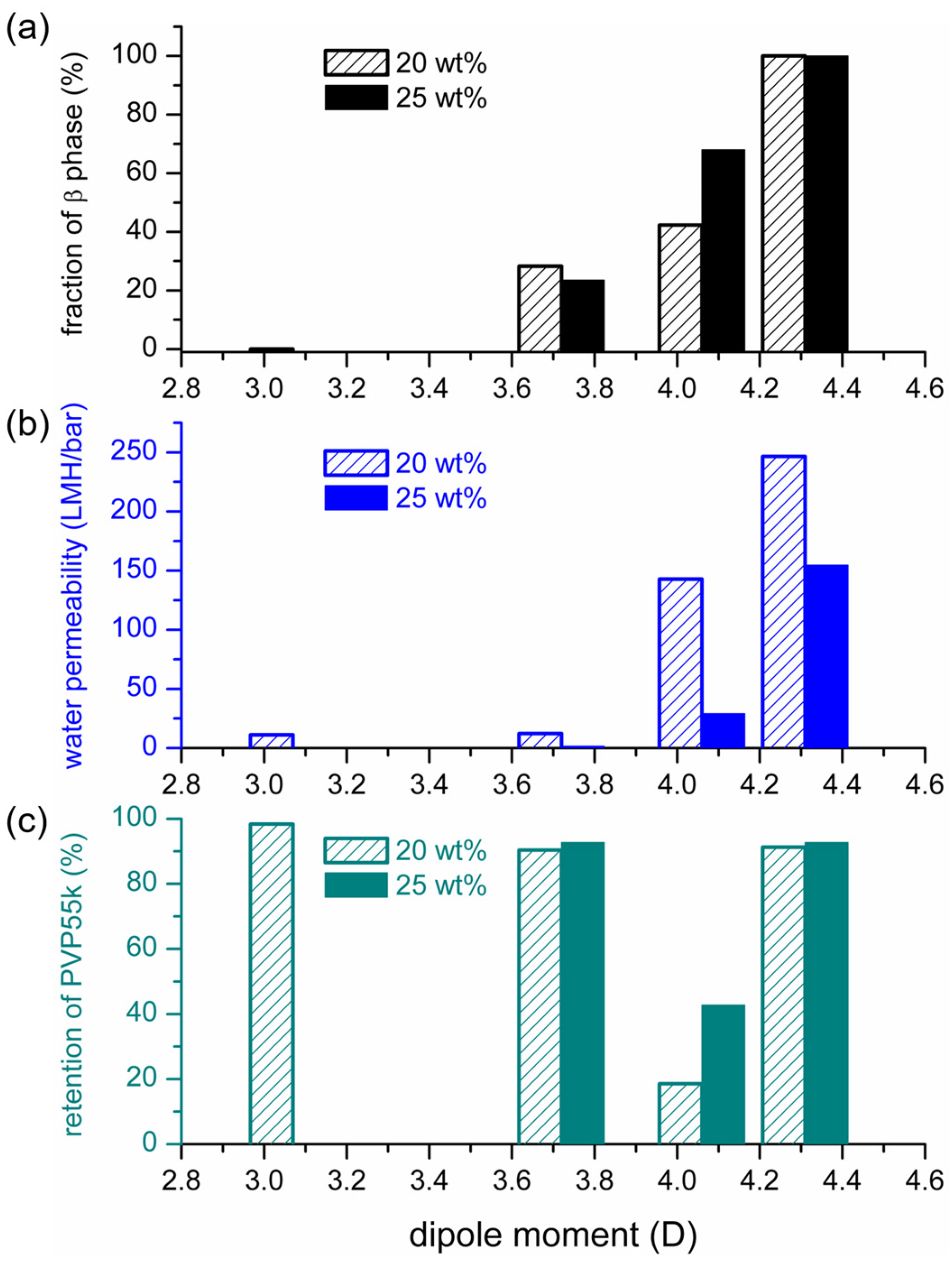
3.2. Effects of Solvent Dipole Moments on Formation of Crystalline Phases for PVDF Membranes Prepared with NIPS
3.3. Effects of Solvent Dipole Moments on Water Permeabilities of PVDF Membranes Prepared with NIPS
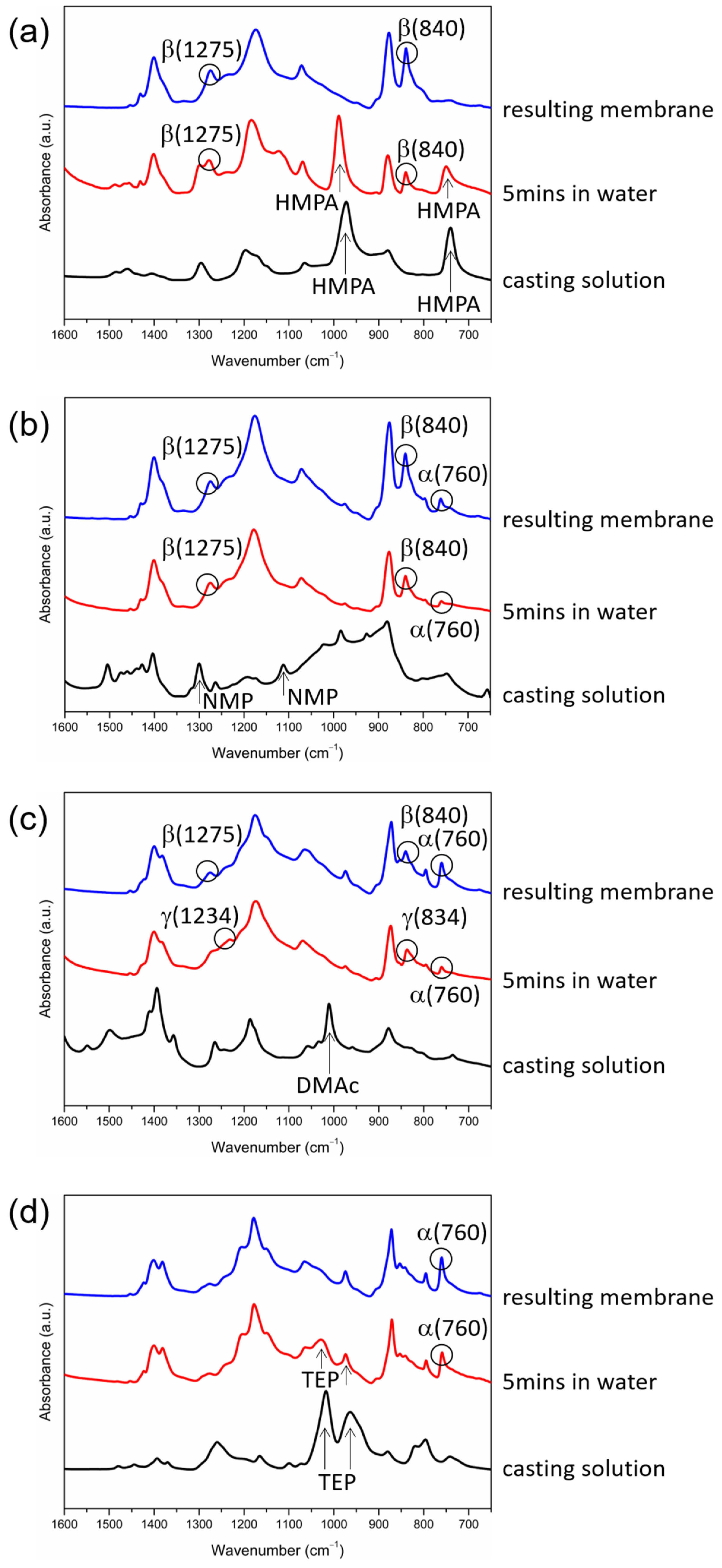
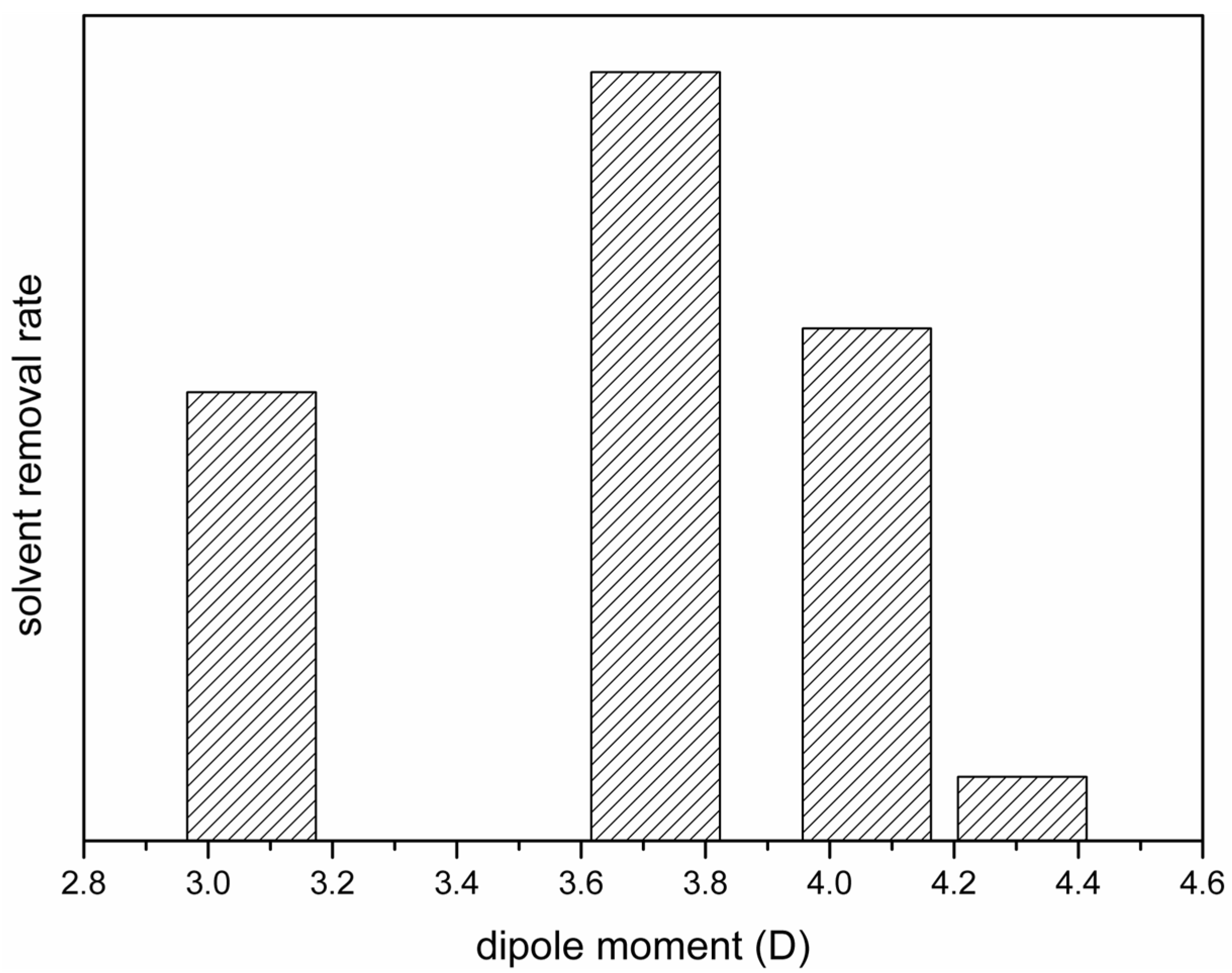
3.4. Mechanism Responsible for the Dependence of Solvent Removal Rates on Solvent Dipole Moments
4. Conclusions
- Both the fraction of the polar crystalline phase (β) and the water permeability of the prepared membrane increased monotonously with the increasing solvent dipole moment.
- The increase in membrane permeability with the increasing solvent dipole moment can be explained by the change in the solvent concentration at the surface of the cast film. A higher solvent concentration at the film surface led to a more porous surface with higher permeability.
- The solvent content in the crystallization environment played a critical role in the formation of PVDF crystalline phases. A high solvent content allowed the solvent polarity to govern the formation of crystalline phases. With a low solvent content, the dominant crystalline phase was α, the kinetically favored one.
- The solvent removal rate from a cast film strongly influenced the fraction of the β phase and the water permeability of the prepared PVDF membrane. The removal rate was dependent on the dipole moment of the solvent, and the dependence can be well explained by the change in the viscosity of the casting solution.
- FTIR/ATR analyses were performed at the surfaces of the cast films at different time points during membrane formation to provide information on when crystallization occurred, how crystalline phases evolved, and whether or not the solvents were present in cast films as PVDF crystallized. The technique was shown to be a useful tool to obtain insight into the formation of membranes with semi-crystalline polymers.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Marshall, J.E.; Zhenova, A.; Roberts, S.; Petchey, T.; Zhu, P.; Dancer, C.E.; McElroy, C.R.; Kendrick, E.; Goodship, V. On the solubility and stability of polyvinylidene fluoride. Polymers 2021, 13, 1354. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hashim, N.A.; Liu, Y.; Abed, M.M.; Li, K. Progress in the production and modification of PVDF membranes. J. Membr. Sci. 2011, 375, 1–27. [Google Scholar] [CrossRef]
- Cui, Z.; Hassankiadeh, N.T.; Zhuang, Y.; Drioli, E.; Lee, Y.M. Crystalline polymorphism in poly (vinylidenefluoride) membranes. Prog. Polym. Sci. 2015, 51, 94–126. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, Y.; Ma, W.; Wang, X. A review on microporous polyvinylidene fluoride membranes fabricated via thermally induced phase separation for MF/UF application. J. Membr. Sci. 2021, 639, 119759. [Google Scholar] [CrossRef]
- Tan, X.; Rodrigue, D. A review on porous polymeric membrane preparation. Part I: Production techniques with polysulfone and poly (vinylidene fluoride). Polymers 2019, 11, 1160. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.P.; Lin, D.J.; Shih, C.H.; Dwan, A.H.; Gryte, C.C. PVDF membrane formation by diffusion-induced phase separation-morphology prediction based on phase behavior and mass transfer modeling. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 2079–2092. [Google Scholar] [CrossRef]
- Li, C.-L.; Wang, D.-M.; Deratani, A.; Quémener, D.; Bouyer, D.; Lai, J.-Y. Insight into the preparation of poly (vinylidene fluoride) membranes by vapor-induced phase separation. J. Membr. Sci. 2010, 361, 154–166. [Google Scholar] [CrossRef]
- Jung, J.T.; Kim, J.F.; Wang, H.H.; Di Nicolo, E.; Drioli, E.; Lee, Y.M. Understanding the non-solvent induced phase separation (NIPS) effect during the fabrication of microporous PVDF membranes via thermally induced phase separation (TIPS). J. Membr. Sci. 2016, 514, 250–263. [Google Scholar] [CrossRef]
- Meringolo, C.; Mastropietro, T.F.; Poerio, T.; Fontananova, E.; De Filpo, G.; Curcio, E.; Di Profio, G. Tailoring PVDF membranes surface topography and hydrophobicity by a sustainable two-steps phase separation process. ACS Sustain. Chem. Eng. 2018, 6, 10069–10077. [Google Scholar] [CrossRef]
- Mulder, M. Basic Principles of Membrane Technology; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1991. [Google Scholar]
- Wang, D.-M.; Venault, A.; Lai, J.-Y. Fundamentals of nonsolvent-induced phase separation. In Hollow Fiber Membranes; Chung, T.-S., Feng, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 13–56. [Google Scholar]
- Bottino, A.; Camera-Roda, G.; Capannelli, G.; Munari, S. The formation of microporous polyvinylidene difluoride membranes by phase separation. J. Membr. Sci. 1991, 57, 1–20. [Google Scholar] [CrossRef]
- Young, T.-H.; Cheng, L.-P.; Lin, D.-J.; Fane, L.; Chuang, W.-Y. Mechanisms of PVDF membrane formation by immersion-precipitation in soft (1-octanol) and harsh (water) nonsolvents. Polymer 1999, 40, 5315–5323. [Google Scholar] [CrossRef]
- Martins, P.; Lopes, A.; Lanceros-Mendez, S. Electroactive phases of poly (vinylidene fluoride): Determination, processing and applications. Prog. Polym. Sci. 2014, 39, 683–706. [Google Scholar] [CrossRef]
- Hirschinger, J.; Schaefer, D.; Spiess, H.W.; Lovinger, A.J. Chain dynamics in the crystalline α-phase of poly (vinylidene fluoride) by two-dimensional exchange deuteron NMR. Macromolecules 1991, 24, 2428–2433. [Google Scholar] [CrossRef]
- Lovinger, A.J. Annealing of poly (vinylidene fluoride) and formation of a fifth phase. Macromolecules 1982, 15, 40–44. [Google Scholar] [CrossRef]
- Boccaccio, T.; Bottino, A.; Capannelli, G.; Piaggio, P. Characterization of PVDF membranes by vibrational spectroscopy. J. Membr. Sci. 2002, 210, 315–329. [Google Scholar] [CrossRef]
- Darestani, M.T.; Coster, H.; Chilcott, T.C.; Fleming, S.; Nagarajan, V.; An, H. Piezoelectric membranes for separation processes: Fabrication and piezoelectric properties. J. Membr. Sci. 2013, 434, 184–192. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, A.-Q.; Zhu, B.-K.; Du, C.-H.; Xu, Y.-Y. Polymorphism in porous poly (vinylidene fluoride) membranes formed via immersion precipitation process. J. Membr. Sci. 2008, 319, 169–175. [Google Scholar] [CrossRef]
- Kalimuldina, G.; Turdakyn, N.; Abay, I.; Medeubayev, A.; Nurpeissova, A.; Adair, D.; Bakenov, Z. A review of piezoelectric PVDF film by electrospinning and its applications. Sensors 2020, 20, 5214. [Google Scholar] [CrossRef]
- Chen, D.; Pomalaza-Ráez, C. A self-cleaning piezoelectric PVDF membrane system for filtration of kaolin suspension. Sep. Purif. Technol. 2019, 215, 612–618. [Google Scholar] [CrossRef]
- Sukumaran, S.; Chatbouri, S.; Rouxel, D.; Tisserand, E.; Thiebaud, F.; Ben Zineb, T. Recent advances in flexible PVDF based piezoelectric polymer devices for energy harvesting applications. J. Intell. Mater. Syst. Struct. 2021, 32, 746–780. [Google Scholar] [CrossRef]
- Silva, M.; Sencadas, V.; Botelho, G.; Machado, A.; Rolo, A.G.; Rocha, J.G.; Lanceros-Méndez, S. α-and γ-PVDF: Crystallization kinetics, microstructural variations and thermal behaviour. Mater. Chem. Phys. 2010, 122, 87–92. [Google Scholar] [CrossRef]
- Gregorio Jr, R.; Cestari, M. Effect of crystallization temperature on the crystalline phase content and morphology of poly (vinylidene fluoride). J. Polym. Sci. Part B Polym. Phys. 1994, 32, 859–870. [Google Scholar] [CrossRef]
- Song, D.; Yang, D.; Feng, Z. Formation of β-phase microcrystals from the melt of PVF2-PMMA blends induced by quenching. J. Mater. Sci. 1990, 25, 57–64. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tashiro, K.; Tadokoro, H. Molecular vibrations of three crystal forms of poly (vinylidene fluoride). Macromolecules 1975, 8, 158–171. [Google Scholar] [CrossRef]
- Chang, H.-H.; Chang, L.-K.; Yang, C.-D.; Lin, D.-J.; Cheng, L.-P. Effect of solvent on the dipole rotation of poly (vinylidene fluoride) during porous membrane formation by precipitation in alcohol baths. Polymer 2017, 115, 164–175. [Google Scholar] [CrossRef]
- Salimi, A.; Yousefi, A.A. Conformational changes and phase transformation mechanisms in PVDF solution-cast films. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 3487–3495. [Google Scholar] [CrossRef]
- Nishiyama, T.; Sumihara, T.; Sato, E.; Horibe, H. Effect of solvents on the crystal formation of poly (vinylidene fluoride) film prepared by a spin-coating process. Polym. J. 2017, 49, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, T.; Sumihara, T.; Sasaki, Y.; Sato, E.; Yamato, M.; Horibe, H. Crystalline structure control of poly (vinylidene fluoride) films with the antisolvent addition method. Polym. J. 2016, 48, 1035–1038. [Google Scholar] [CrossRef]
- Gregorio Jr, R.; Borges, D.S. Effect of crystallization rate on the formation of the polymorphs of solution cast poly (vinylidene fluoride). Polymer 2008, 49, 4009–4016. [Google Scholar] [CrossRef]
- Chinaglia, D.L.; Gregorio Jr, R.; Stefanello, J.C.; Pisani Altafim, R.A.; Wirges, W.; Wang, F.; Gerhard, R. Influence of the solvent evaporation rate on the crystalline phases of solution-cast poly (vinylidene fluoride) films. J. Appl. Polym. Sci. 2010, 116, 785–791. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, J.; Chen, S.; Wang, X. Crystalline phase formation of poly (vinylidene fluoride) from tetrahydrofuran/N, N-dimethylformamide mixed solutions. J. Macromol. Sci. Phys. 2008, 47, 434–449. [Google Scholar] [CrossRef]
- Tao, M.-m.; Liu, F.; Ma, B.-r.; Xue, L.-x. Effect of solvent power on PVDF membrane polymorphism during phase inversion. Desalination 2013, 316, 137–145. [Google Scholar] [CrossRef]
- Haponska, M.; Trojanowska, A.; Nogalska, A.; Jastrzab, R.; Gumi, T.; Tylkowski, B. PVDF membrane morphology—Influence of polymer molecular weight and preparation temperature. Polymers 2017, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Normant, H. Hexamethylphosphoramide. Angew. Chem. Int. Ed. 1967, 6, 1046–1067. [Google Scholar] [CrossRef]
- Lee, C.M.; Kumler, W. The dipole moment and structure of five-and six-membered lactams. J. Am. Chem. Soc. 1961, 83, 4593–4596. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B. Organic Solvents, 3rd ed.; Wiley-Interscience: New York, NY, USA, 1970. [Google Scholar]
- Svirbely, W.J.; Lander, J.J. The Dipole Moments of Diethyl Sulfite, Triethyl Phosphate and Tetraethyl Silicate. J. Am. Chem. Soc. 1948, 70, 4121–4123. [Google Scholar] [CrossRef]
- Lo, C.-C.; Chao, P.-M. Replacement of carcinogenic solvent HMPA by DMI in insect sex pheromone synthesis. J. Chem. Ecol. 1990, 16, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Epelle, E.; Otaru, A.; Zubair, Y.; Okolie, J. Improving the viscosity index of used lubricating oil by solvent extraction. Int. Res. J. Eng. Technol. 2017, 4, 1581–1585. [Google Scholar]
- Iloukhani, H.; Khanlarzadeh, K. Densities, viscosities, and refractive indices for binary and ternary mixtures of N, N-dimethylacetamide (1)+ 2-methylbutan-2-ol (2)+ ethyl acetate (3) at 298.15 K for the liquid region and at ambient pressure. J. Chem. Eng. Data 2006, 51, 1226–1231. [Google Scholar] [CrossRef]
- Lalia, B.S.; Yoshimoto, N.; Egashira, M.; Morita, M. A mixture of triethylphosphate and ethylene carbonate as a safe additive for ionic liquid-based electrolytes of lithium ion batteries. J. Power Sources 2010, 195, 7426–7431. [Google Scholar] [CrossRef]
- Scholer, R.; Merbach, A. Raman and infrared study of hexamethylphosphoramide complexes of lanthanide perchlorates. Inorg. Chim. Acta 1975, 15, 15–20. [Google Scholar] [CrossRef]
- Kumar, D.B.K.; Reddy, K.R.; Rao, G.S.; Sandhyasri, P.; Begum, Z.; Rambabu, C. Measurements of some physical properties of binary liquid mixtures (N-methyl-2-pyrrolidone+ an aliphatic ester) at several temperatures and data processing of viscosity and ultrasonic speed. J. Mol. Liq. 2013, 183, 31–44. [Google Scholar] [CrossRef]
- Verbovy, D.M.; Smagala, T.G.; Brynda, M.A.; Fawcett, W.R. A FTIR study of ion-solvent interactions in N,N-dimethylacetamide. J. Mol. Liq. 2006, 129, 13–17. [Google Scholar] [CrossRef]
- Ruan, C.; Huang, H.; Rodgers, M. Modeling Metal Cation−Phosphate Interactions in Nucleic Acids in the Gas Phase via Alkali Metal Cation− Triethyl Phosphate Complexes. J. Phys. Chem. A 2007, 111, 13521–13527. [Google Scholar] [CrossRef]
- Teixeira, J.; Cardoso, V.F.; Botelho, G.; Morão, A.M.; Nunes-Pereira, J.; Lanceros-Mendez, S. Effect of polymer dissolution temperature and conditioning time on the morphological and physicochemical characteristics of poly (Vinylidene fluoride) membranes prepared by non-solvent induced phase separation. Polymers 2021, 13, 4062. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, R., Jr. Determination of the α, β, and γ crystalline phases of poly (vinylidene fluoride) films prepared at different conditions. J. Appl. Polym. Sci. 2006, 100, 3272–3279. [Google Scholar] [CrossRef]
- Kochervinskii, V.V. The structure and properties of block poly (vinylidene fluoride) and systems based on it. Russ. Chem. Rev. 1996, 65, 865. [Google Scholar] [CrossRef]
- Yang, D.; Tornga, S.; Orler, B.; Welch, C. Aging of poly (vinylidene fluoride) hollow fibers in light hydrocarbon environments. J. Membr. Sci. 2012, 409, 302–317. [Google Scholar] [CrossRef]
- Mohammadi, B.; Yousefi, A.A.; Bellah, S.M. Effect of tensile strain rate and elongation on crystalline structure and piezoelectric properties of PVDF thin films. Polym. Test. 2007, 26, 42–50. [Google Scholar] [CrossRef]
- Lin, D.-J.; Chang, H.-H.; Chen, T.-C.; Lee, Y.-C.; Cheng, L.-P. Formation of porous poly (vinylidene fluoride) membranes with symmetric or asymmetric morphology by immersion precipitation in the water/TEP/PVDF system. Eur. Polym. J. 2006, 42, 1581–1594. [Google Scholar] [CrossRef]
- Yeow, M.; Liu, Y.; Li, K. Morphological study of poly (vinylidene fluoride) asymmetric membranes: Effects of the solvent, additive, and dope temperature. J. Appl. Polym. Sci. 2004, 92, 1782–1789. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Sun, D.; An, Q.; Chen, H. Formation mechanism and crystallization of poly (vinylidene fluoride) membrane via immersion precipitation method. Desalination 2009, 236, 170–178. [Google Scholar] [CrossRef]
- Lin, D.-J.; Chang, C.-L.; Chen, T.-C.; Cheng, L.-P. Microporous PVDF membrane formation by immersion precipitation from water/TEP/PVDF system. Desalination 2002, 145, 25–29. [Google Scholar] [CrossRef]
- Ruan, L.; Yao, X.; Chang, Y.; Zhou, L.; Qin, G.; Zhang, X. Properties and applications of the β phase poly (vinylidene fluoride). Polymers 2018, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- Zha, D.-A.; Mei, S.; Wang, Z.; Li, H.; Shi, Z.; Jin, Z. Superhydrophobic polyvinylidene fluoride/graphene porous materials. Carbon 2011, 49, 5166–5172. [Google Scholar] [CrossRef]
- Wilke, C.R.; Chang, P. Correlation of diffusion coefficients in dilute solutions. AIChE J. 1955, 1, 264–270. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | HMPA | NMP | DMAc | TEP |
|---|---|---|---|---|
| Dipole moment (D) | 4.31 [36] | 4.06 [37] | 3.72 [38] | 3.07 [39] |
| Viscosity (cP) at 25 °C | 3.605 [40] | 1.65 [41] | 0.945 [42] | 1.6 [43] |
| IR characteristic peaks (cm−1) | 744, 992 [44] | 1112, 1300 [45] | 1011 [46] | 966, 1021 [47] |
| FTIR | XRD | |
|---|---|---|
| Crystalline Phase | Wavenumber (cm−1) | 2θ (°) |
| α | 760 | 18.30 |
| 975 | 19.90 | |
| 26.56 | ||
| β | 840 | 20.26 |
| 1275 | 41.20 | |
| γ | 834 | 20.04 |
| 1234 |
| Solvent | PVDF Concentration (wt%) | Degree of Crystallinity (%) | Ratio of β Phase (%) | Water Permeability (LMH/bar) | PVP Retention (%) |
|---|---|---|---|---|---|
| TEP | 20 | 54.3 | 0 | 11.19 | 98.36 |
| DMAc | 20 | 51.6 | 28.3 | 12.10 | 90.39 |
| DMAc | 25 | 47.4 | 23.5 | 1.59 | 92.72 |
| NMP | 20 | 47.5 | 42.3 | 142.72 | 18.52 |
| NMP | 25 | 53.5 | 68.0 | 28.98 | 42.74 |
| HMPA | 20 | 58.6 | 100 | 246.47 | 91.28 |
| HMPA | 25 | 57.4 | 100 | 154.68 | 92.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, K.-Y.; Li, C.-L.; Wang, D.-M.; Lai, J.-Y. Formation of Porous Structures and Crystalline Phases in Poly(vinylidene fluoride) Membranes Prepared with Nonsolvent-Induced Phase Separation—Roles of Solvent Polarity. Polymers 2023, 15, 1314. https://doi.org/10.3390/polym15051314
Chan K-Y, Li C-L, Wang D-M, Lai J-Y. Formation of Porous Structures and Crystalline Phases in Poly(vinylidene fluoride) Membranes Prepared with Nonsolvent-Induced Phase Separation—Roles of Solvent Polarity. Polymers. 2023; 15(5):1314. https://doi.org/10.3390/polym15051314
Chicago/Turabian StyleChan, Kuan-Ying, Chia-Ling Li, Da-Ming Wang, and Juin-Yih Lai. 2023. "Formation of Porous Structures and Crystalline Phases in Poly(vinylidene fluoride) Membranes Prepared with Nonsolvent-Induced Phase Separation—Roles of Solvent Polarity" Polymers 15, no. 5: 1314. https://doi.org/10.3390/polym15051314