
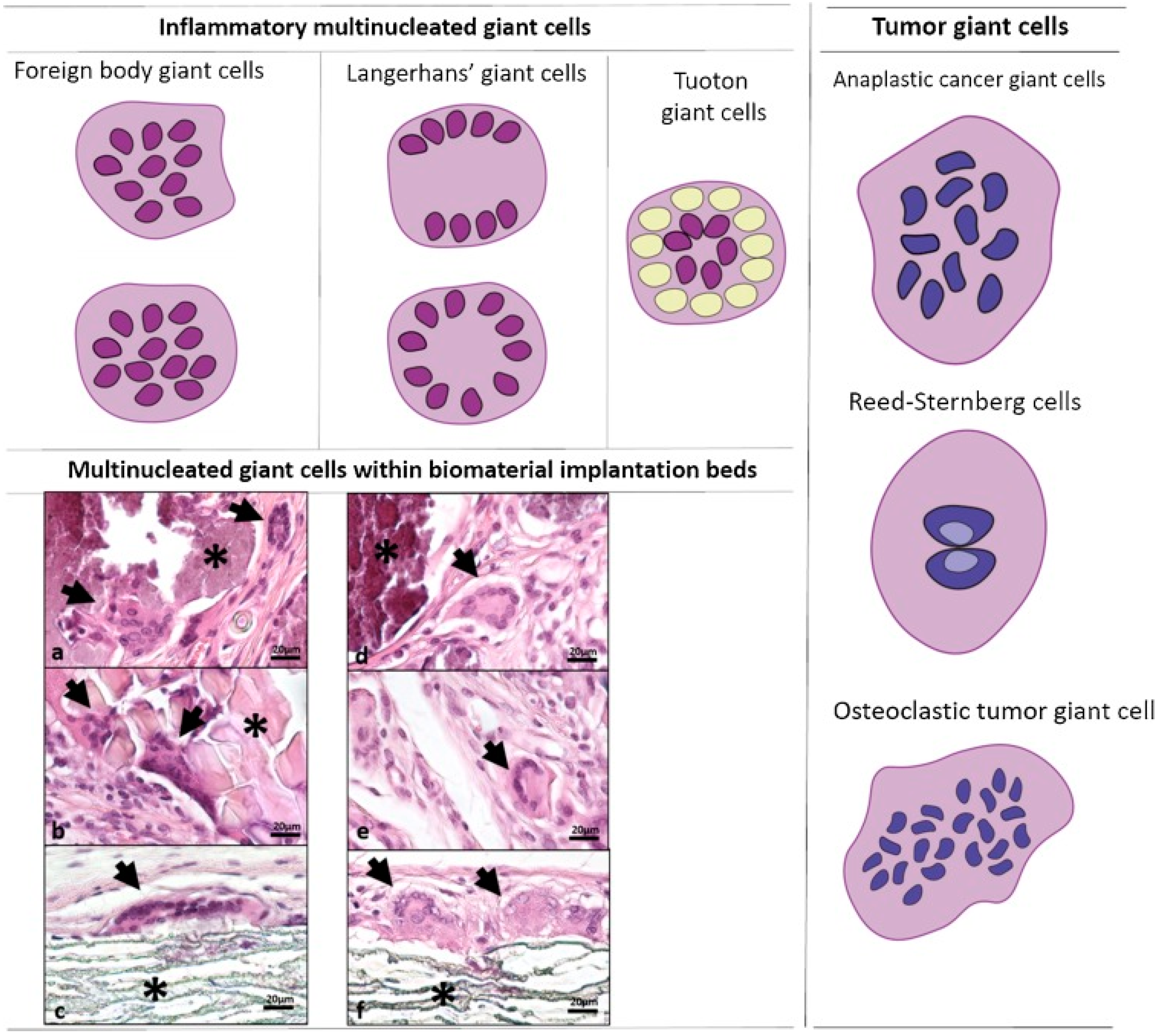
Mechanisms of Foreign Body Giant Cell Formation in Response to Implantable Biomaterials

 and
and
Abstract
:1. Introduction
2. Macrophages Fusion Competency
2.1. Exogenous Stimulus for Macrophage Fusion
2.2. Endogenous Stimulus for Macrophage Fusion
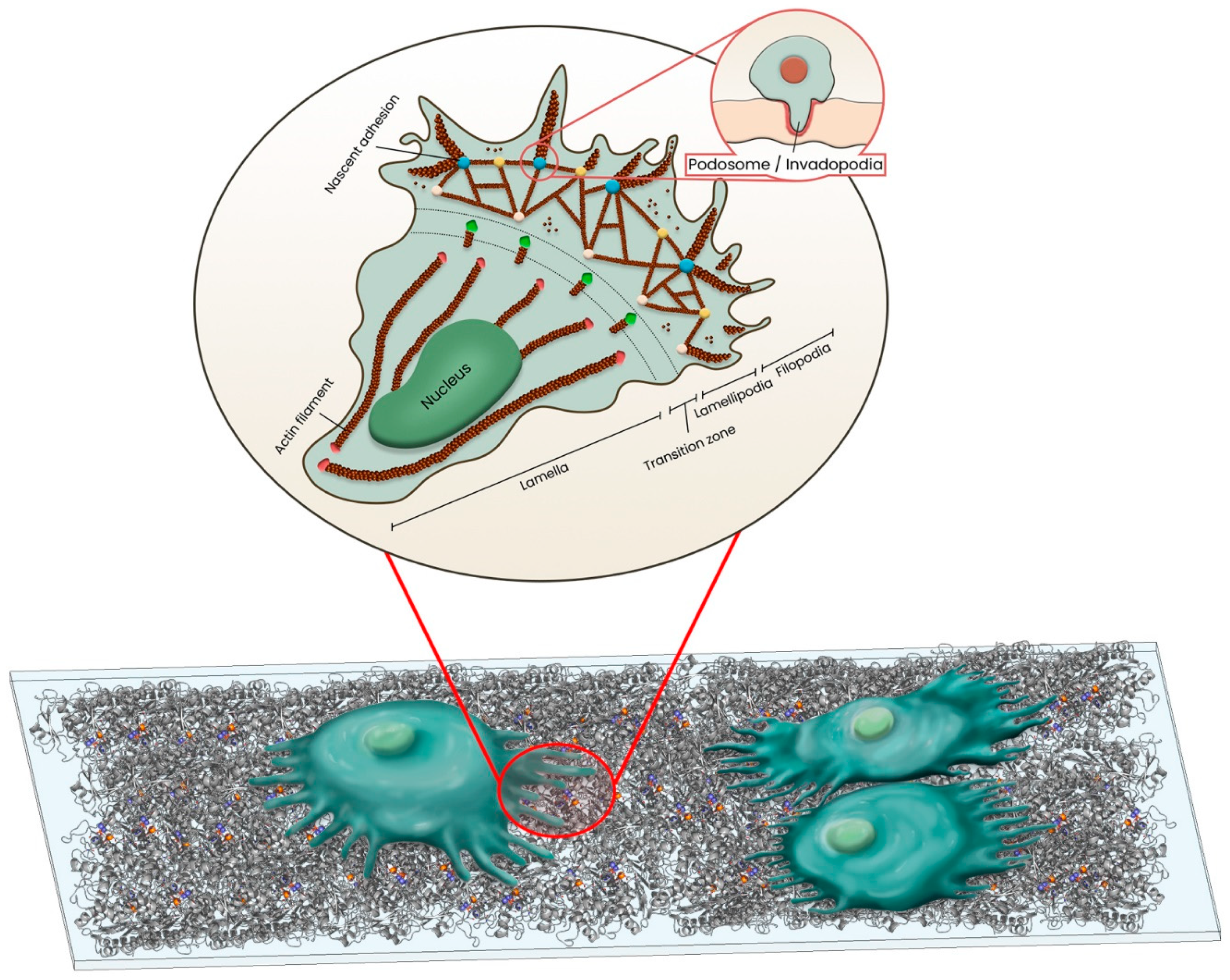
3. Cell Migration on Biomaterial’s Surface
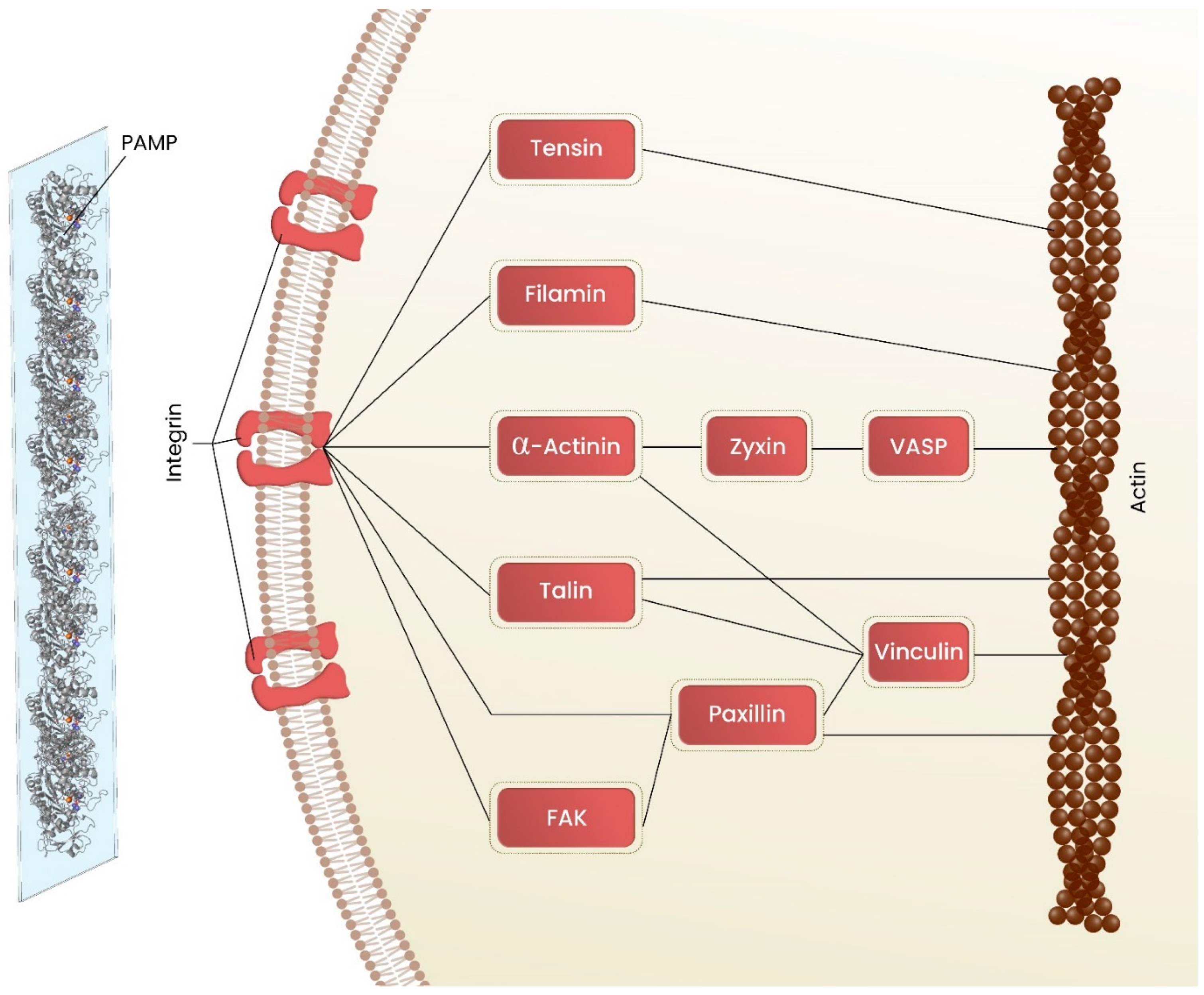
3.1. Cell Adhesion on Biomaterial
3.2. Cell Protrusions
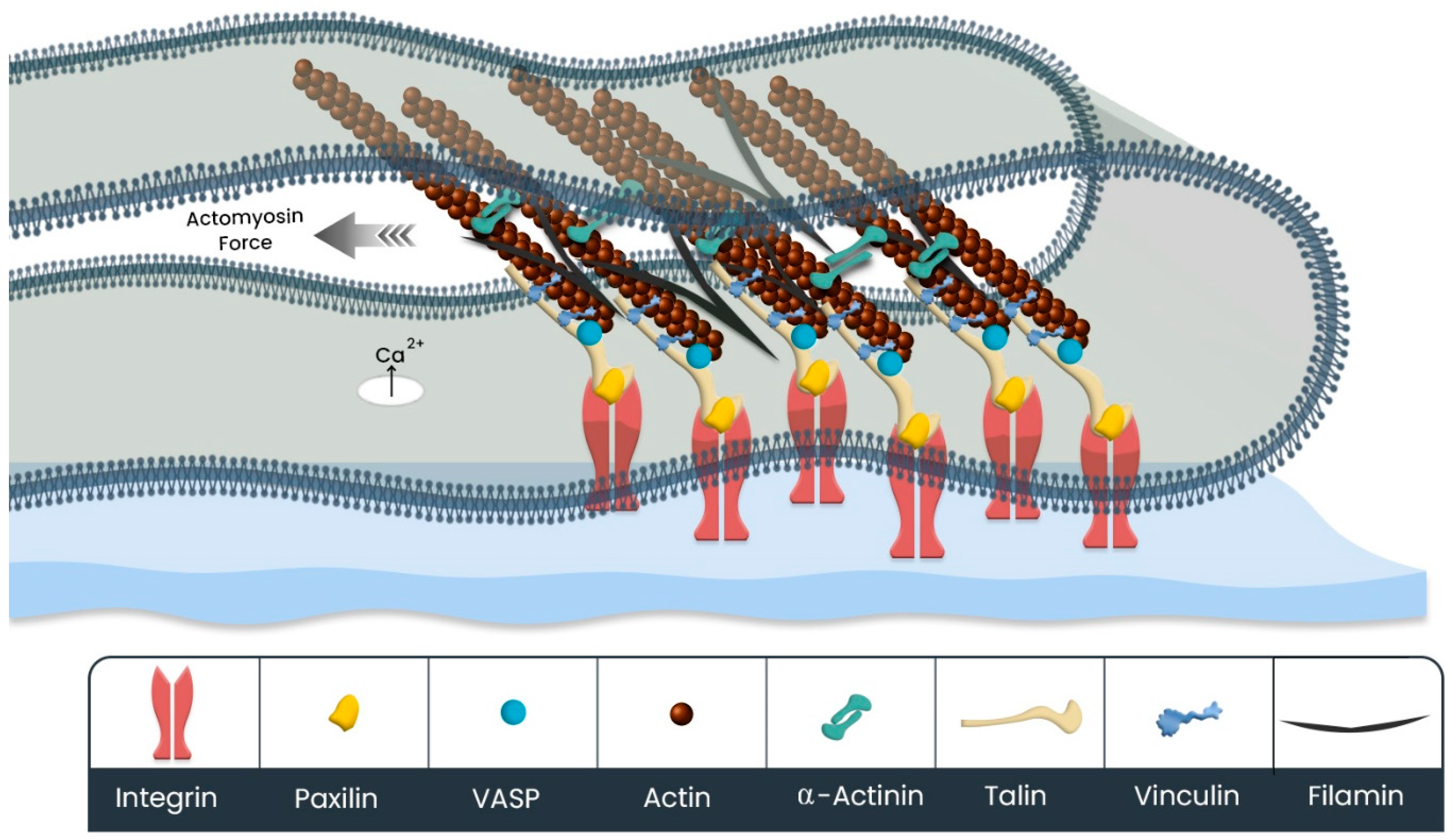
3.2.1. Actin Cytoskeleton Rearrangement
3.2.2. Mechanosensing/Mechanotransduction in Cell Protrusions Mediates the Cell Migration
4. Cell Migration and Fusion
Fusogens Are Involved in Macrophage Fusion
5. Consequences of Macrophages Fusion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomaterial | FBGCs/No of Patients | Ref | Case Report |
|---|---|---|---|
| Polymethylmethacrylate (PMMA) microsphere | 15 in 587 | [105] | Requena et al. observed Strong FBGCs formation after injection of (PMMA) microsphere in 4 patients. Time of appearance of FBGCs vary between 6 to 14 months [106]. |
| Poly-lactic acid (PLA) microsphere | 5 in 722 | [107] | PLA-related FBGCs appear 6–24 months after injection [105] |
| 3 in 2131 | [108] | ||
| Poly-hydroxyethyl-methacrylate (pHEMA) | 9 in 455 | [109] | _ |
| Silicone oil | 5 in 608 | [110] | Arin MD et al. observed granulomas composed of multinucleated giant cells after 18 months of silicone oil injection in a patient [111]. |
| 1 in 500 | [112] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mohammadi, M.; Luong, J.C.; Rodriguez, S.M.; Cao, R.; Wheeler, A.E.; Lau, H.; Li, S.; Shabestari, S.K.; Chadarevian, J.P.; Alexander, M.; et al. Controlled release of stem cell secretome attenuates inflammatory response against implanted biomaterials. Adv. Healthc. Mater. 2020, 9, 1901874. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.R.; Rodrigez, S.; Cao, R.; Alexander, M.; Lakey, J.R.T. Immune response to subcutaneous implants of alginate microcapsules. Mater. Today Proc. 2018, 5, 15580–15585. [Google Scholar] [CrossRef]
- Eslami-kaliji, F.; Sarafbidabad, M.; Kiani-Esfahani, A.; Mirahmadi-Zare, S.Z.; Dormiani, K. 10-hydroxy-2-decenoic acid a bio-immunomodulator in tissue engineering; generates tolerogenic dendritic cells by blocking the toll-like receptor 4. J. Biomed. Mater. Res. Part A 2021, 2021 109, 1575–1587. [Google Scholar] [CrossRef]
- Veiseh, O.; Vegas, A.J. Domesticating the foreign body response: Recent advances and applications. Adv. Drug Deliv. Rev. 2019, 144, 148–161. [Google Scholar] [CrossRef]
- Eslami-Kaliji, F.; Sarafbidabad, M.; Rajadas, J.; Mohammadi, M.R. Dendritic cells as targets for biomaterial-based immunomodulation. ACS Biomater. Sci. Eng. 2020, 6, 2726–2739. [Google Scholar] [CrossRef]
- Brooks, P.J.; Glogauer, M.; McCulloch, C.A. An overview of the derivation and function of multinucleated giant cells and their role in pathologic processes. Am. J. Pathol. 2019, 189, 1145–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Maawi, S.; Orlowska, A.; Sader, R.; Kirkpatrick, C.J.; Ghanaati, S. In vivo cellular reactions to different biomaterials—Physiological and pathological aspects and their consequences. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Lay, G.; Poquet, Y.; Salek-Peyron, P.; Puissegur, M.-P.; Botanch, C.; Bon, H.; Levillain, F.; Duteyrat, J.-L.; Emile, J.-F.; Altare, F. Langhans giant cells from M. tuberculosis-induced human granulomas cannot mediate mycobacterial uptake. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 76–85. [Google Scholar]
- Stephenson, T. General and Systemic Pathology, 3rd ed.; Underwood, J.C.E., Cross, S.S., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2000; pp. 202–221. [Google Scholar]
- Aterman, K.; Remmele, W.; Smith, M. Karl Touton and his “xanthelasmatic giant cell.” A selective review of multinucleated giant cells. Am. J. Dermatopathol. 1988, 10, 257–269. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Rodriguez, S.M.; Luong, J.C.; Li, S.; Cao, R.; Alshetaiwi, H.; Lau, H.; Davtyan, H.; Jones, M.B.; Jafari, M.; et al. Exosome loaded immunomodulatory biomaterials alleviate local immune response in immunocompetent diabetic mice post islet xenotransplantation. Commun. Biol. 2021, 4, 685. [Google Scholar] [CrossRef]
- McNally, A.K.; Anderson, J.M. Macrophage fusion and multinucleated giant cells of inflammation. Cell Fusion Health Dis. 2011, 713, 97–111. [Google Scholar]
- Mcinnes, A.; Rennick, D.M. Interleukin 4 induces cultured monocytes/macrophages to form giant multinucleated cells. J. Exp. Med. 1988, 167, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Enelow, R.I.; Sullivan, G.W.; Carper, H.T.; Mandell, G.L. Induction of multinucleated giant cell formation from in vitro culture of human monocytes with interleukin-3 and interferon-c: Comparison with other stimulating factors. Am. J. Resp. Cell Mol. Biol. 1992, 6, 57–62. [Google Scholar]
- Takashima, T.; Ohnishi, K.; Tsuyuguchi, I.; Kishimoto, S. Differential regulation of formation of multinucleated giant cells from concanavalin A-stimulated human blood monocytes by IFN-gamma and IL-4. J. Immunol. 1993, 150, 3002–3010. [Google Scholar] [CrossRef]
- Elliott, M.J.; Gamble, J.R.; Park, L.S.; Vadas, M.A.; Lopez, A.F. Inhibition of human monocyte adhesion by interleukin-4. Blood 1991, 77, 2739–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, A.K.; Anderson, J.M. Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am. J. Pathol. 1995, 147, 1487. [Google Scholar] [PubMed]
- Anderson, J.; Defife, K.; McNally, A. Monocyte, macrophage and foreign body giant cell interactions with molecularly engineered surfaces. J. Mater. Sci. Mater. Med. 1999, 10, 579–588. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur. J. Immunol. 2007, 37, 33–42. [Google Scholar] [CrossRef]
- DeFife, K.M.; Jenney, C.R.; McNally, A.K.; Colton, E.; Anderson, J.M. Interleukin-13 induces human monocyte/macrophage fusion and macrophage mannose receptor expression. J. Immunol. 1997, 158, 3385–3390. [Google Scholar] [CrossRef]
- Moreno, J.L.; Mikhailenko, I.; Tondravi, M.M.; Keegan, A.D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: Contribution of E-cadherin. J. Leukoc. Biol. 2007, 82, 1542–1553. [Google Scholar] [CrossRef]
- Belo, M.A.; Oliveira, M.F.; Oliveira, S.L.; Aracati, M.F.; Rodrigues, L.F.; Costa, C.C.; Conde, G.; Gomes, J.M.; Prata, M.N.; Barra, A.; et al. Zebrafish as a model to study inflammation: A tool for drug discovery. Biomed. Pharmacother. 2021, 144, 112310. [Google Scholar] [CrossRef]
- Schubert, M.A.; Wiggins, M.J.; DeFife, K.M.; Hiltner, A.; Anderson, J.M. Vitamin E as an antioxidant for poly (etherurethane urea): In vivo studies. J. Biomed. Mater. Res. Off. J. Soc. Biomater. Jpn. Soc. Biomater. 1996, 32, 493–504. [Google Scholar] [CrossRef]
- McNally, A.K.; Anderson, J.M. Foreign body-type multinucleated giant cell formation is potently induced by α-tocopherol and prevented by the diacylglycerol kinase inhibitor R59022. Am. J. Pathol. 2003, 163, 1147–1156. [Google Scholar] [CrossRef]
- Azzi, A.; Stocker, A. Vitamin E: Non-antioxidant roles. Prog. Lipid Res. 2000, 39, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Tomasello, E.; Kyriakides, T.R.; Martinez, F.O.; Takai, T.; Gordon, S.; Vivier, E. Essential role of DAP12 signaling in macrophage programming into a fusion-competent state. Sci. Signal. 2008, 1, ra11. [Google Scholar] [CrossRef] [Green Version]
- Curnock, A.P.; Logan, M.K.; Ward, S.G. Chemokine signalling: Pivoting around multiple phosphoinositide 3-kinases. Immunology 2002, 105, 125–136. [Google Scholar] [CrossRef]
- Hauck, C.R.; Klingbeil, C.K.; Schlaepfer, D.D. Focal adhesion kinase functions as a receptor-proximal signaling component required for directed cell migration. Immunol. Res. 2000, 21, 293–303. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Bysani, S.; Mummidi, S. CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, IκB kinase, and nuclear factor-κB and induces cell-cell adhesion and aortic smooth muscle cell proliferation. J. Biol. Chem. 2004, 279, 3188–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, J.; Raman, D.; Liu, Y.; Wikswo, J.; Richmond, A. Parallel phosphatidylinositol 3-kinase (PI3K)-dependent and Src-dependent pathways lead to CXCL8-mediated Rac2 activation and chemotaxis. J. Biol. Chem. 2008, 283, 26538–26547. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Garcia, A.J.; Keselowsky, B.G. Biomimetic surfaces for control of cell adhesion to facilitate bone formation. Crit. Rev. ™ Eukaryot. Gene Expr. 2002, 12, 151–162. [Google Scholar] [CrossRef]
- Jung, D.; Kapur, R.; Adams, T.; Giuliano, K.A.; Mrksich, M.; Craighead, H.G.; Taylor, D.L. Topographical and physicochemical modification of material surface to enable patterning of living cells. Crit. Rev. Biotechnol. 2001, 21, 111–154. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- SenGupta, S.; Parent, C.A.; Bear, J.E. The principles of directed cell migration. Nat. Rev. Mol. Cell Biol. 2021, 22, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Wu, C.; Yang, W.; Liang, W.; Yu, H.; Liu, L. Recent advance in surface modification for regulating cell adhesion and behaviors. Nanotechnol. Rev. 2020, 9, 971–989. [Google Scholar] [CrossRef]
- Keselowsky, B.G.; Collard, D.M.; García, A.J. Surface chemistry modulates fibronectin conformation and directs integrin binding and specificity to control cell adhesion. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2003, 66, 247–259. [Google Scholar] [CrossRef]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernández, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface functionalization of nanoparticles with polyethylene glycol: Effects on protein adsorption and cellular uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef]
- Puleo, D.A.; Bizios, R. Biological Interactions on Materials Surfaces: Understanding and Controlling Protein, Cell, and Tissue Responses; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Schmidt, D.R.; Waldeck, H.; Kao, W.J. Protein Adsorption to Biomaterials. In Biological Interactions on Materials Surfaces; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–18. [Google Scholar]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMP s and DAMP s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M. In vitro and in vivo monocyte, macrophage, foreign body giant cell, and lymphocyte interactions with biomaterials. In Biological Interactions on Materials Surfaces; Springer: Berlin/Heidelberg, Germany, 2009; pp. 225–244. [Google Scholar]
- Lv, L.; Xie, Y.; Li, K.; Hu, T.; Lu, X.; Cao, Y.; Zheng, X. Unveiling the mechanism of surface hydrophilicity-modulated macrophage polarization. Adv. Healthc. Mater. 2018, 7, 1800675. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, T.D.; Lewis, J.S.; Dolgova, N.V.; Clare-Salzler, M.J.; Keselowsky, B.G. Integrin-directed modulation of macrophage responses to biomaterials. Biomaterials 2014, 35, 3504–3515. [Google Scholar] [CrossRef] [Green Version]
- Damsky, C.H.; Ilić, D. Integrin signaling: It’s where the action is. Curr. Opin. Cell Biol. 2002, 14, 594–602. [Google Scholar] [CrossRef]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439. [Google Scholar] [CrossRef]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Calderwood, D.A. Organization, dynamics and mechanoregulation of integrin-mediated cell–ECM adhesions. Nat. Rev. Mol. Cell Biol. 2022, 24, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Sokabe, M.; Lim, C.T. Molecular mechanisms underlying the force-dependent regulation of actin-to-ECM linkage at the focal adhesions. Prog. Mol. Biol. Transl. Sci. 2014, 126, 135–154. [Google Scholar] [PubMed]
- Chhabra, E.S.; Higgs, H.N. The many faces of actin: Matching assembly factors with cellular structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef]
- Giannone, G.; Dubin-Thaler, B.J.; Rossier, O.; Cai, Y.; Chaga, O.; Jiang, G.; Beaver, W.; Döbereiner, H.-G.; Freund, Y.; Borisy, G.; et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 2007, 128, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balabiyev, A.; Podolnikova, N.P.; Mursalimov, A.; Lowry, D.; Newbern, J.M.; Roberson, R.W.; Ugarova, T.P. Transition of podosomes into zipper-like structures in macrophage-derived multinucleated giant cells. Mol. Biol. Cell 2020, 31, 2002–2020. [Google Scholar] [CrossRef]
- Flevaris, P.; Stojanovic, A.; Gong, H.; Chishti, A.; Welch, E.; Du, X. A molecular switch that controls cell spreading and retraction. J. Cell Biol. 2007, 179, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Albiges-Rizo, C.; Destaing, O.; Fourcade, B.; Planus, E.; Block, M.R. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J. Cell Sci. 2009, 122, 3037–3049. [Google Scholar] [CrossRef] [Green Version]
- Buccione, R.; Orth, J.D.; McNiven, M.A. Foot and mouth: Podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 2004, 5, 647–657. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The’ins’ and’outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.R.; Owens, T.W.; Naylor, M.J. Structural and mechanical functions of integrins. Biophys. Rev. 2014, 6, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faix, J.; Rottner, K. The making of filopodia. Curr. Opin. Cell Biol. 2006, 18, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.F.; Sauser, R.; Ambrosi, D.; Meister, J.-J.; Verkhovsky, A.B. Force transmission in migrating cells. J. Cell Biol. 2010, 188, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Hall, A.; Small, J. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 1999, 9, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Kiosses, W.B.; Shattil, S.J.; Pampori, N.; Schwartz, M.A. Rac recruits high-affinity integrin αvβ3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 2001, 3, 316–320. [Google Scholar] [CrossRef]
- Ballestrem, C.; Hinz, B.; Imhof, B.A.; Wehrle-Haller, B. Marching at the front and dragging behind: Differential αVβ3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 2001, 155, 1319–1332. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar] [CrossRef]
- Bachir, A.I.; Horwitz, A.R.; Nelson, W.J.; Bianchini, J.M. Actin-based adhesion modules mediate cell interactions with the extracellular matrix and neighboring cells. Cold Spring Harb. Perspect. Biol. 2017, 9, a023234. [Google Scholar] [CrossRef] [Green Version]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Jacquemet, G. Cell matrix adhesion in cell migration. Essays Biochem. 2019, 63, 535–551. [Google Scholar] [PubMed]
- Rantala, J.K.; Pouwels, J.; Pellinen, T.; Veltel, S.; Laasola, P.; Mattila, E.; Potter, C.S.; Duffy, T.; Sundberg, J.P.; Kallioniemi, O.; et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 2011, 13, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, H.-N.; Dupé-Manet, S.; Bouvard, D.; Lacombe, M.-L.; Marie, C.; Block, M.R.; Albiges-Rizo, C. Integrin cytoplasmic domain-associated protein 1α (ICAP-1α) interacts directly with the metastasis suppressor nm23-H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J. Biol. Chem. 2002, 277, 20895–20902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Savinko, T.; Guenther, C.; Uotila, L.M.; Asens, M.L.; Yao, S.; Tojkander, S.; Fagerholm, S.C. Filamin A is required for optimal T cell integrin-mediated force transmission, flow adhesion, and T cell trafficking. J. Immunol. 2018, 200, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Case, L.B.; Waterman, C.M. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 2015, 17, 955–963. [Google Scholar] [CrossRef]
- Alexandrova, A.Y.; Arnold, K.; Schaub, S.; Vasiliev, J.M.; Meister, J.-J.; Bershadsky, A.D.; Verkhovsky, A.B. Comparative dynamics of retrograde actin flow and focal adhesions: Formation of nascent adhesions triggers transition from fast to slow flow. PLoS ONE 2008, 3, e3234. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.K.; Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.A.; Mogilner, A.; Horwitz, A.R. Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008, 10, 1039–1050. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Schneider, I.C.; Rericha, E.; Schlaepfer, D.D.; Waterman, C.M. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 2010, 188, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Choquet, D.; Felsenfeld, D.P.; Sheetz, M.P. Extracellular matrix rigidity causes strengthening of integrin–cytoskeleton linkages. Cell 1997, 88, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Giannone, G.; Critchley, D.R.; Fukumoto, E.; Sheetz, M.P. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 2003, 424, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.D.; Mullins, R.D. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J. Cell Biol. 2010, 191, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Vinzenz, M.; Stradal, T.E.B.; Small, J.V.; Curth, U.; Dickinson, R.B.; Faix, J. Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 2011, 30, 456–467. [Google Scholar] [CrossRef]
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 2015, 8, 5671–5701. [Google Scholar] [CrossRef] [Green Version]
- Mukai, A.; Hashimoto, N. Localized cyclic AMP-dependent protein kinase activity is required for myogenic cell fusion. Exp. Cell Res. 2008, 314, 387–397. [Google Scholar] [CrossRef]
- Mukai, A.; Kurisaki, T.; Sato, S.B.; Kobayashi, T.; Kondoh, G.; Hashimoto, N. Dynamic clustering and dispersion of lipid rafts contribute to fusion competence of myogenic cells. Exp. Cell Res. 2009, 315, 3052–3063. [Google Scholar] [CrossRef]
- Barry, A.K.; Tabdili, H.; Muhamed, I.; Wu, J.; Shashikanth, N.; Gomez, G.A.; Yap, A.S.; Gottardi, C.J.; de Rooij, J.; Wang, N.; et al. α-catenin cytomechanics–role in cadherin-dependent adhesion and mechanotransduction. J. Cell Sci. 2014, 127, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.; Sarpal, R.; Ishiyama, N.; Pellikka, M.; Ikura, M.; Tepass, U. Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat. Cell Biol. 2013, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nelson, W.J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell–cell adhesion. J. Cell Biol. 2007, 178, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Vignery, A. Macrophage fusion: The making of osteoclasts and giant cells. J. Exp. Med. 2005, 202, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.R.; Martinez-Pomares, L.; Stacey, M.; Lin, H.-H.; Brown, G.; Gordon, S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005, 23, 901–944. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.M.; Pavlath, G.K. Mannose receptor regulates myoblast motility and muscle growth. J. Cell Biol. 2006, 174, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Cervero, P.; Eddy, R.; Condeelis, J. Mechanisms and roles of podosomes and invadopodia. Nat. Rev. Mol. Cell Biol. 2022, 24, 86–106. [Google Scholar] [CrossRef] [PubMed]
- McNally, A.K.; DeFife, K.M.; Anderson, J.M. Interleukin-4-induced macrophage fusion is prevented by inhibitors of mannose receptor activity. Am. J. Pathol. 1996, 149, 975. [Google Scholar]
- Han, X.; Sterling, H.; Chen, Y.; Saginario, C.; Brown, E.J.; Frazier, W.A.; Lindberg, F.P.; Vignery, A. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 2000, 275, 37984–37992. [Google Scholar] [CrossRef] [Green Version]
- Oldenborg, P.-A.; Gresham, H.D.; Lindberg, F.P. Cd47-signal regulatory protein α (Sirpα) regulates Fcγ and complement receptor–mediated phagocytosis. J. Exp. Med. 2001, 193, 855–862. [Google Scholar] [CrossRef]
- Xu, Z.; Jin, B. A novel interface consisting of homologous immunoglobulin superfamily members with multiple functions. Cell. Mol. Immunol. 2010, 7, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, C.; Persson, E.; Baldock, P.; Stenberg, Å.; Boström, I.; Matozaki, T.; Oldenborg, P.-A.; Lundberg, P. Lack of CD47 impairs bone cell differentiation and results in an osteopenic phenotype in vivo due to impaired signal regulatory protein α (SIRPα) signaling. J. Biol. Chem. 2013, 288, 29333–29344. [Google Scholar] [CrossRef] [Green Version]
- Jeremic, A.; Kelly, M.; Cho, J.A.; Cho, S.-J.; Hörber, J.; Jena, B.P. Calcium drives fusion of SNARE-apposed bilayers. Cell Biol. Int. 2004, 28, 19–31. [Google Scholar] [CrossRef]
- MacLauchlan, S.; Skokos, E.A.; Meznarich, N.; Zhu, D.H.; Raoof, S.; Shipley, J.M.; Senior, R.M.; Bornstein, P.; Kyriakides, T.R. Macrophage fusion, giant cell formation, and the foreign body response require matrix metalloproteinase 9. J. Leukoc. Biol. 2009, 85, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, E.; Ziyyat, A.; Prenant, M.; Wrobel, E.; Wolf, J.-P.; Levy, S.; Le Naour, F.; Boucheix, C. Reduced fertility of female mice lacking CD81. Dev. Biol. 2006, 290, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; He, P.; Tachibana, I.; Zhou, B.; Miyado, K.; Kaneko, H.; Suzuki, M.; Minami, S.; Iwasaki, T.; Goya, S.; et al. Double deficiency of tetraspanins CD9 and CD81 alters cell motility and protease production of macrophages and causes chronic obstructive pulmonary disease-like phenotype in mice. J. Biol. Chem. 2008, 283, 26089–26097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.E.; García, A.J. Macrophage phenotypes in tissue repair and the foreign body response: Implications for biomaterial-based regenerative medicine strategies. Acta Biomater. 2021, 133, 4–16. [Google Scholar] [CrossRef]
- Dondossola, E.; Holzapfel, B.M.; Alexander, S.; Filippini, S.; Hutmacher, D.W.; Friedl, P. Examination of the foreign body response to biomaterials by nonlinear intravital microscopy. Nat. Biomed. Eng. 2016, 1, 0007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmati, M.; Silva, E.A.; Reseland, J.E.; Heyward, C.A.; Haugen, H.J. Biological responses to physicochemical properties of biomaterial surface. Chem. Soc. Rev. 2020, 49, 5178–5224. [Google Scholar] [CrossRef]
- Lemperle, G.; Romano, J.J.; Busso, M. Soft tissue augmentation with Artecoll: 10-year history, indications, techniques, and complications. Dermatol. Surg. 2003, 29, 573–587. [Google Scholar]
- Requena, C.; Izquierdo, M.J.; Navarro, M.; Martínez, A.; Vilata, J.J.; Botella, R.; Amorrortu, J.; Sabater, V.; Aliaga, A.; Requena, L. Adverse reactions to injectable aesthetic microimplants. Am. J. Dermatopathol. 2001, 23, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.M.; Soyer, H.P.; Schuller-Petrovic, S.; Kerl, H. Foreign body granulomas due to injectable aesthetic microimplants. Am. J. Surg. Pathol. 1999, 23, 113–117. [Google Scholar] [CrossRef]
- Vleggaar, D. Soft-tissue augmentation and the role of poly-L-lactic acid. Plast. Reconstr. Surg. 2006, 118, 46S–54S. [Google Scholar] [CrossRef] [PubMed]
- Laglenne, S.; Lalanne, B.; Laglenne, B.; Asius, J. Un nouveau produit de comblement des rides, entirement resorbable. Dermatologie 2000, 54, 30. [Google Scholar]
- Fulton, J.E., Jr.; Porumb, S.; Caruso, J.C.; Shitabata, P.K. Lip augmentation with liquid silicone. Dermatol. Surg. 2005, 31, 1577–1586. [Google Scholar] [CrossRef]
- Arin, M.J.; Bäte, J.; Krieg, T.; Hunzelmann, N. Silicone granuloma of the face treated with minocycline. J. Am. Acad. Dermatol. 2005, 52, S53–S56. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Carruthers, A.; Orentreich, D.; Brody, H.J.; Lai, M.-Y.; Azen, S.; Van Dyke, G.S. Highly purified 1000-cSt silicone oil for treatment of human immunodeficiency virus-associated facial lipoatrophy: An open pilot trial. Dermatol. Surg. 2004, 30, 1279–1286. [Google Scholar] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eslami-Kaliji, F.; Hedayat Nia, N.; Lakey, J.R.T.; Smink, A.M.; Mohammadi, M. Mechanisms of Foreign Body Giant Cell Formation in Response to Implantable Biomaterials. Polymers 2023, 15, 1313. https://doi.org/10.3390/polym15051313
Eslami-Kaliji F, Hedayat Nia N, Lakey JRT, Smink AM, Mohammadi M. Mechanisms of Foreign Body Giant Cell Formation in Response to Implantable Biomaterials. Polymers. 2023; 15(5):1313. https://doi.org/10.3390/polym15051313
Chicago/Turabian StyleEslami-Kaliji, Farshid, Niloufar Hedayat Nia, Jonathan R. T. Lakey, Alexandra M. Smink, and Mohammadreza Mohammadi. 2023. "Mechanisms of Foreign Body Giant Cell Formation in Response to Implantable Biomaterials" Polymers 15, no. 5: 1313. https://doi.org/10.3390/polym15051313