Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments


{kind=link}
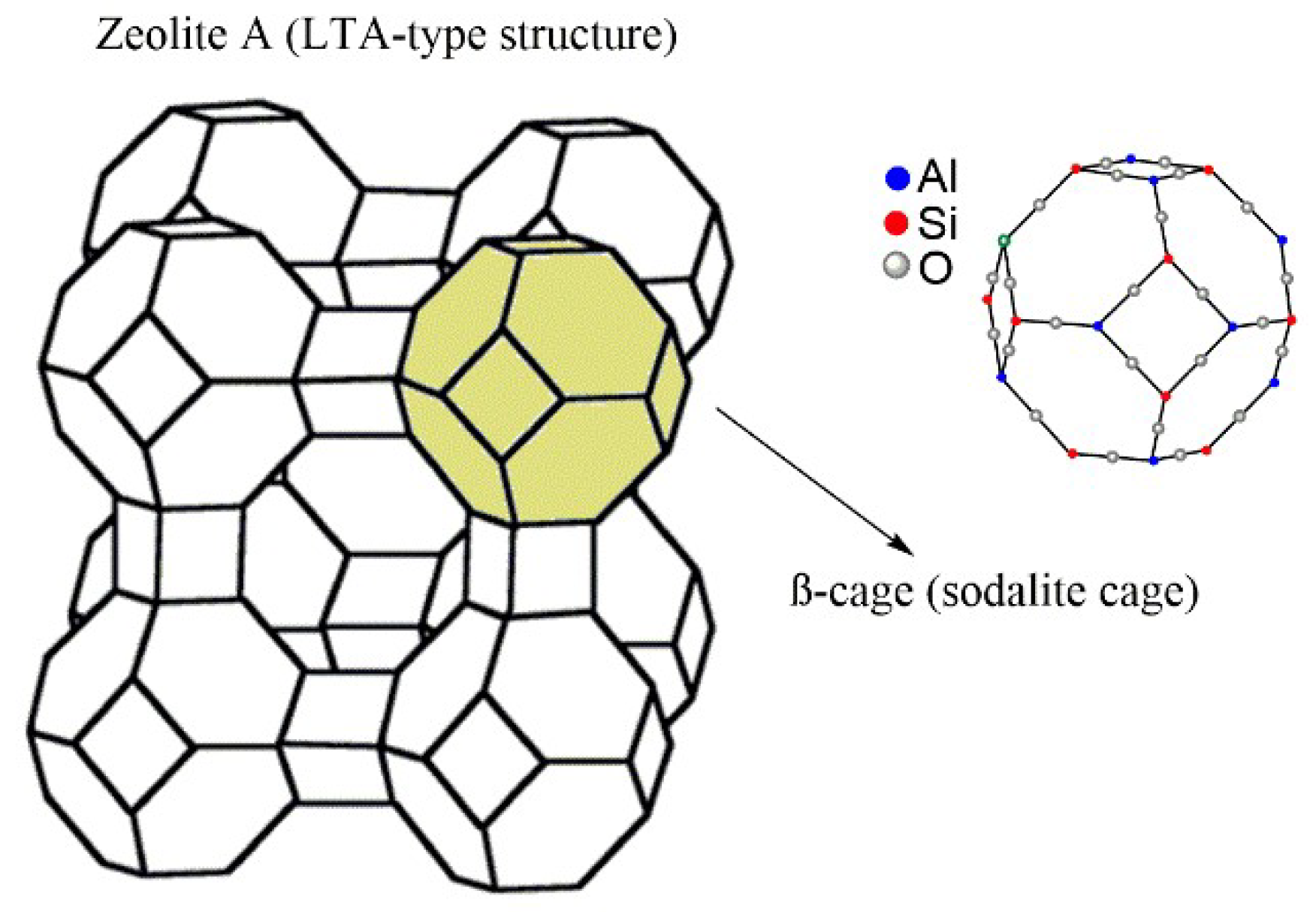{kind=link}
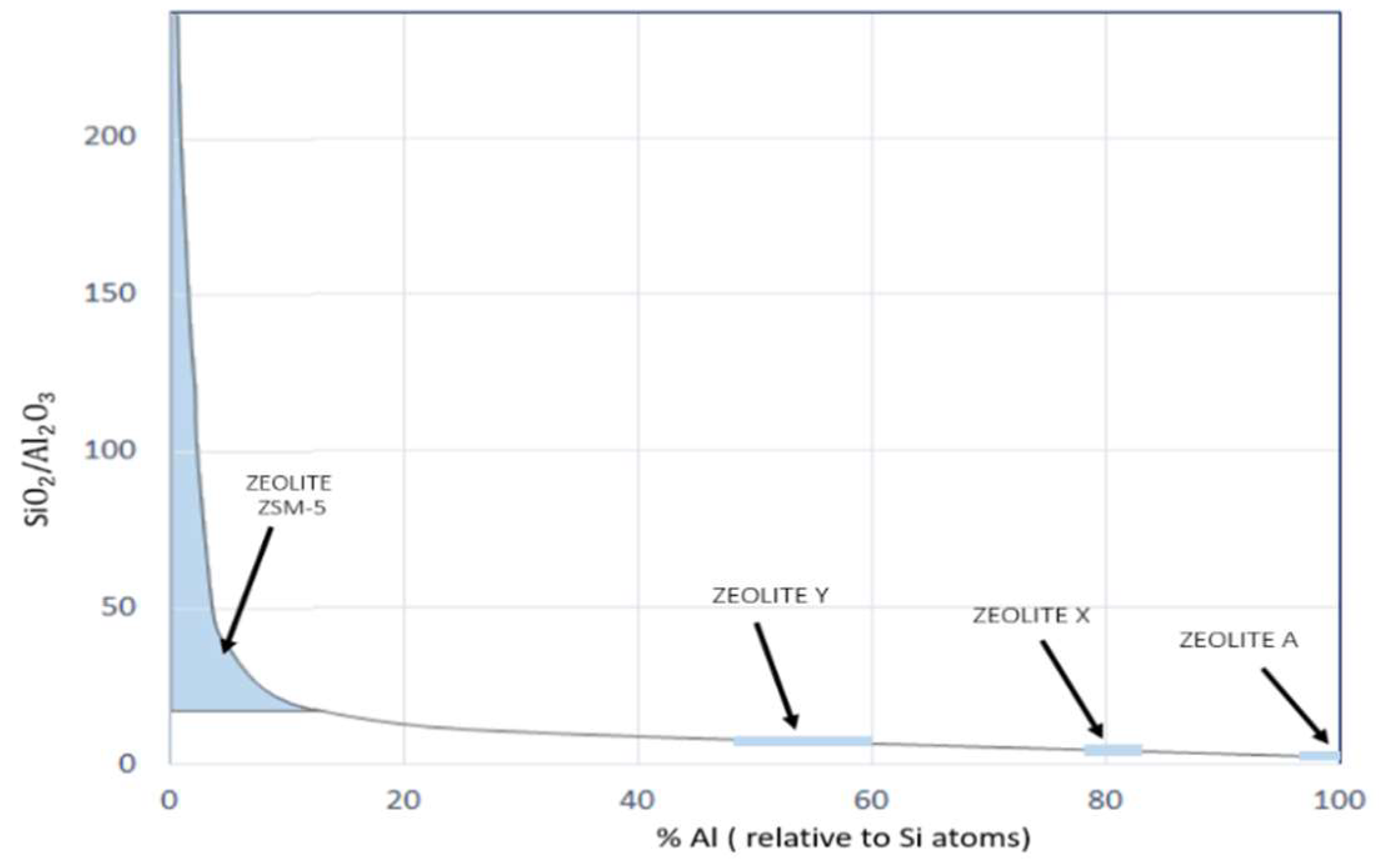{kind=link}
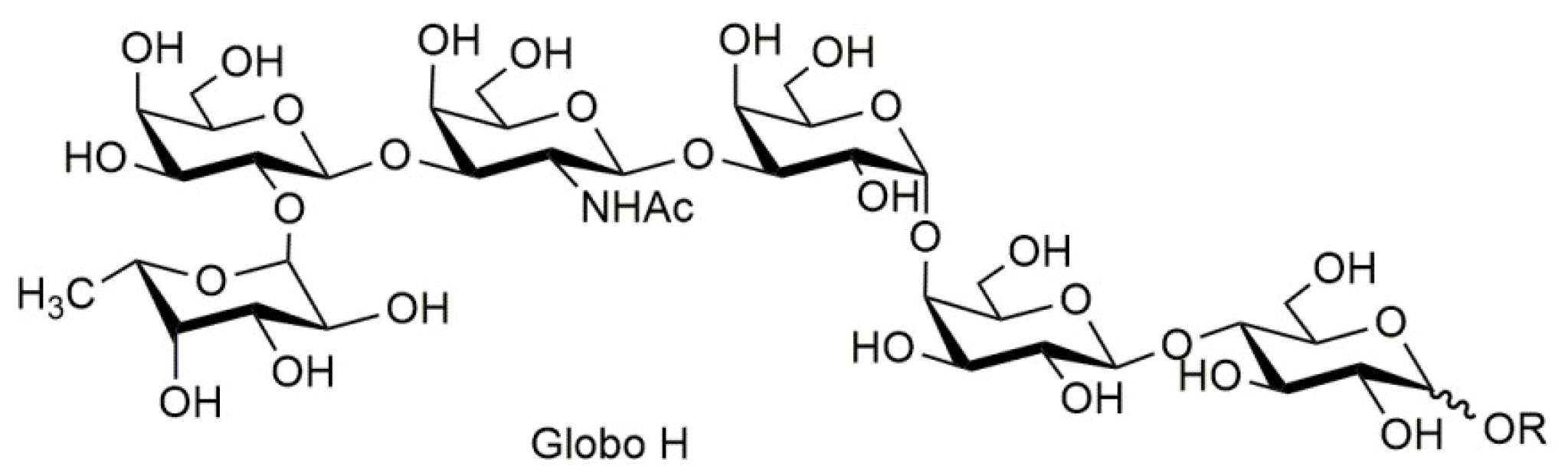{kind=link}
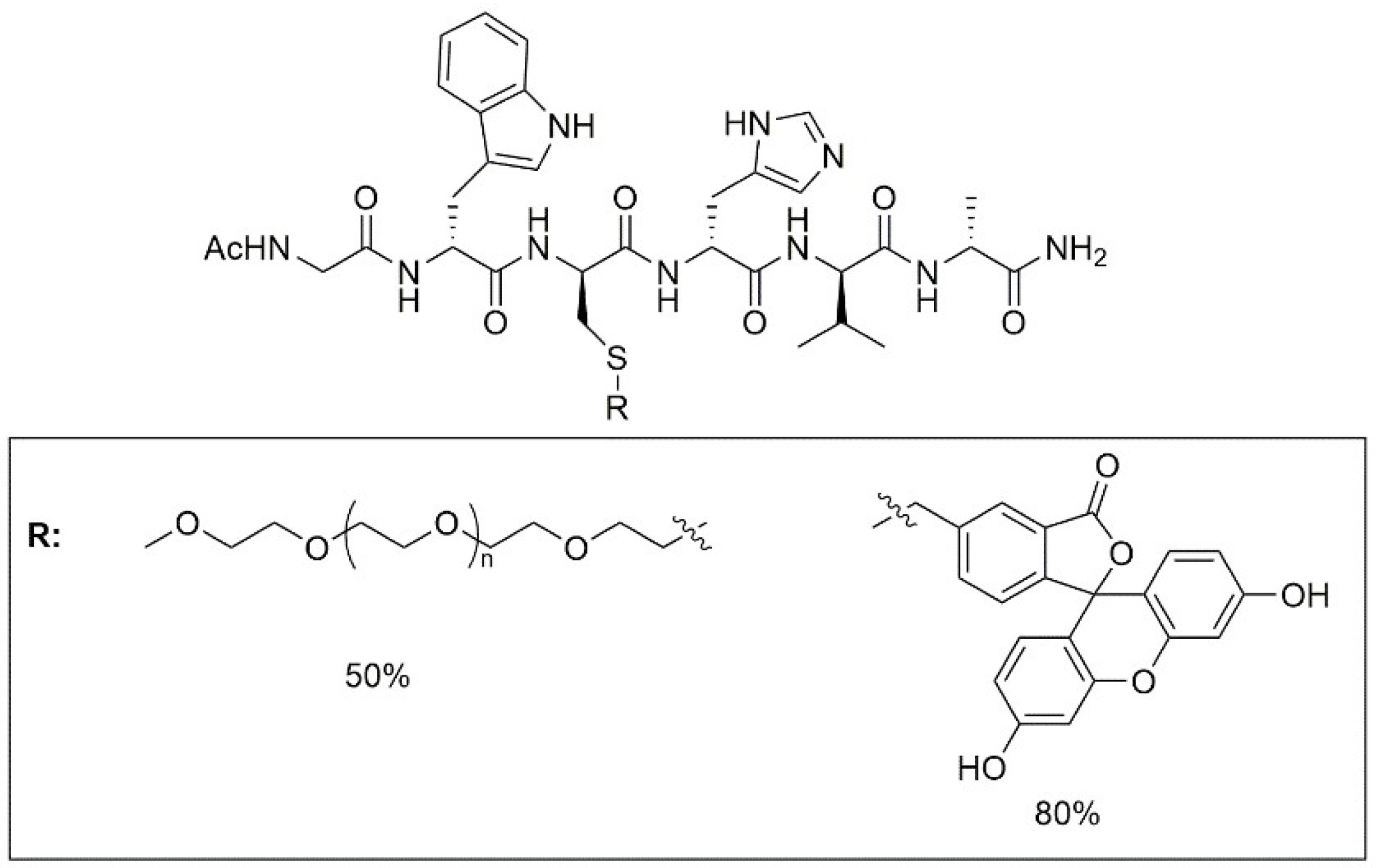{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
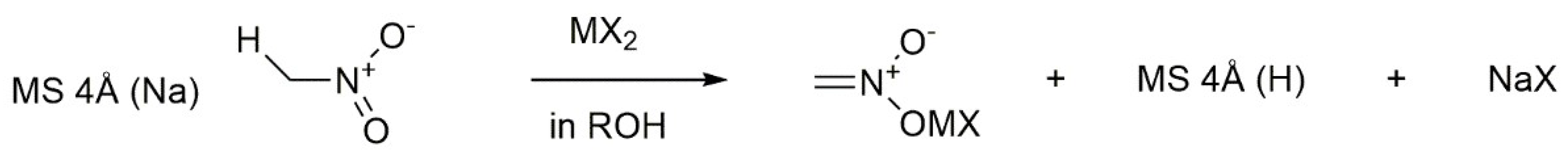{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glycosidation Reactions Promoted by Molecular Sieves
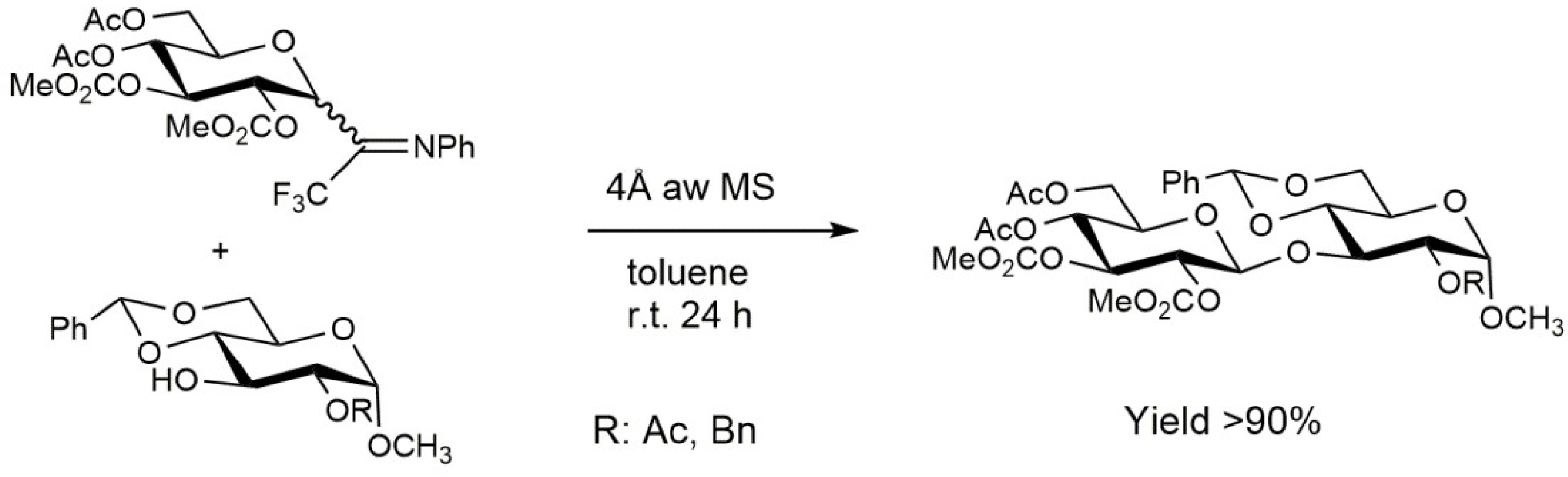
2.1. Glycosidation Reaction via Activation of Glycosyl Trichloro- and N-Phenyltrifluoroacetimidates
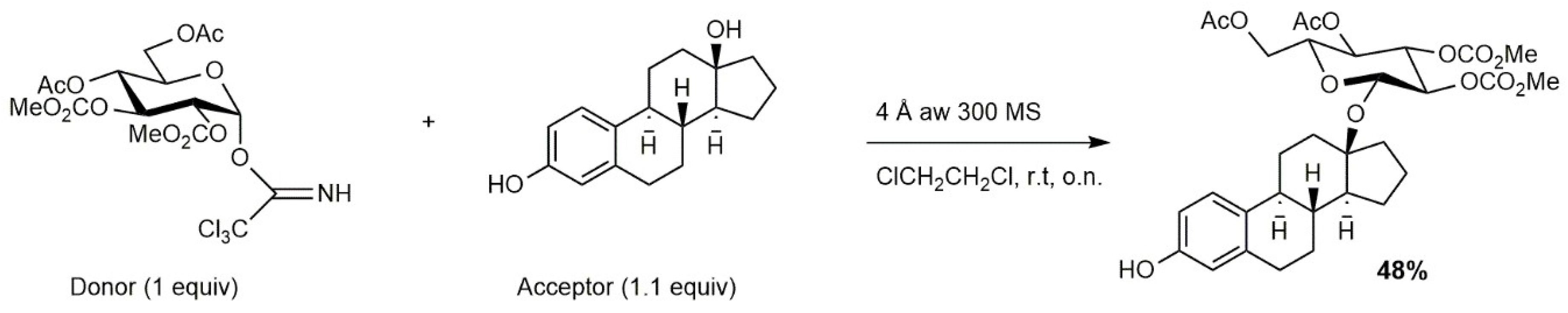
2.2. Carbinol Glycosidation Reaction of the 17-β-Estradiol
2.3. Selective α-Fucosylation for Preparing the Antigenic Trisaccharide Sequence of Lewis X
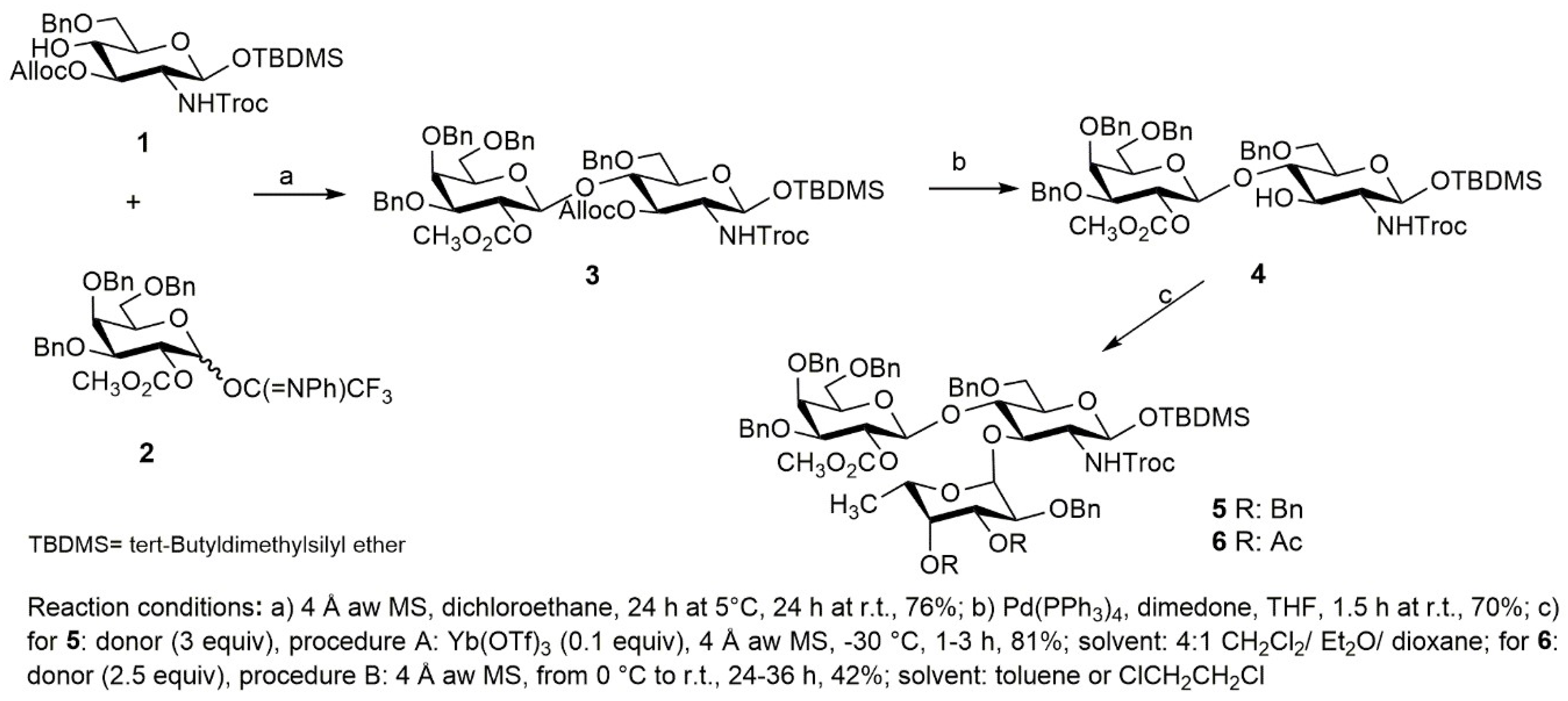
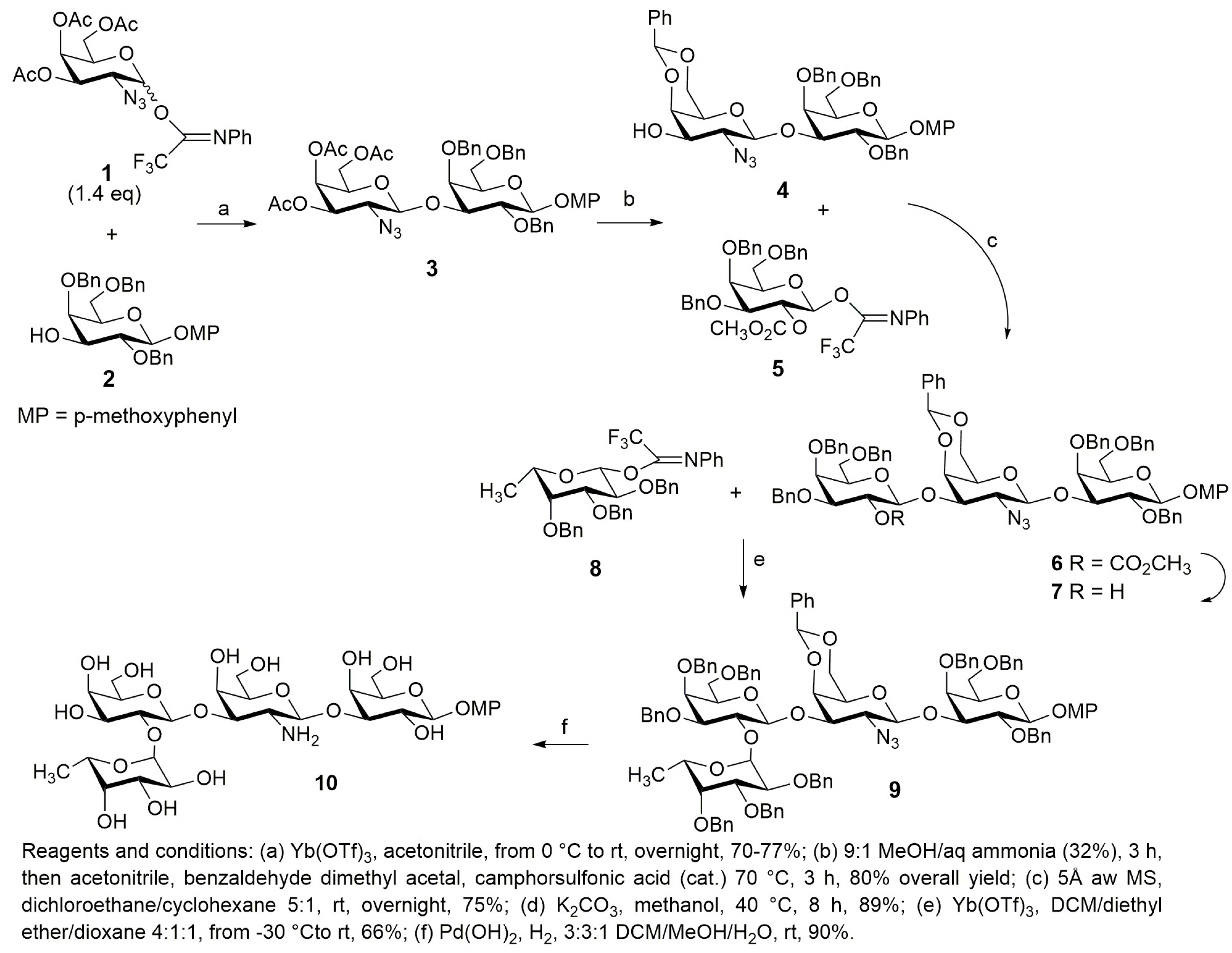
2.4. Synthesis of a Tetrasaccharide Sequence of Globo H
3. O-Protection of Sugars Promoted by Molecular Sieves
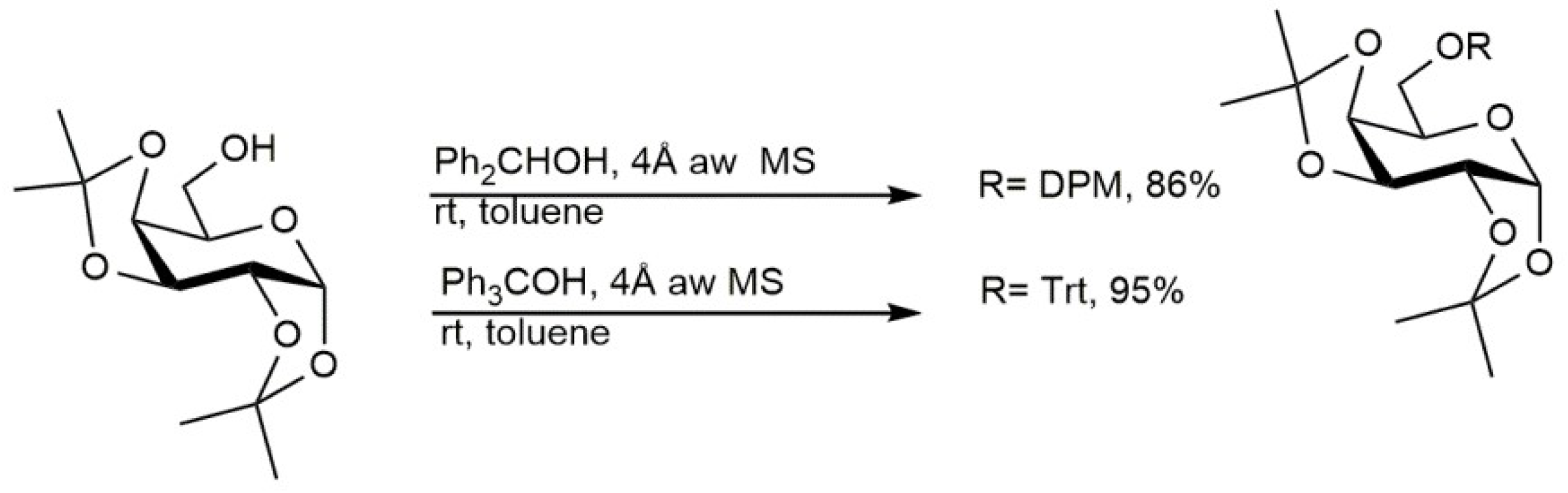
3.1. Protection Procedure of Sugar Alcoholic Functions
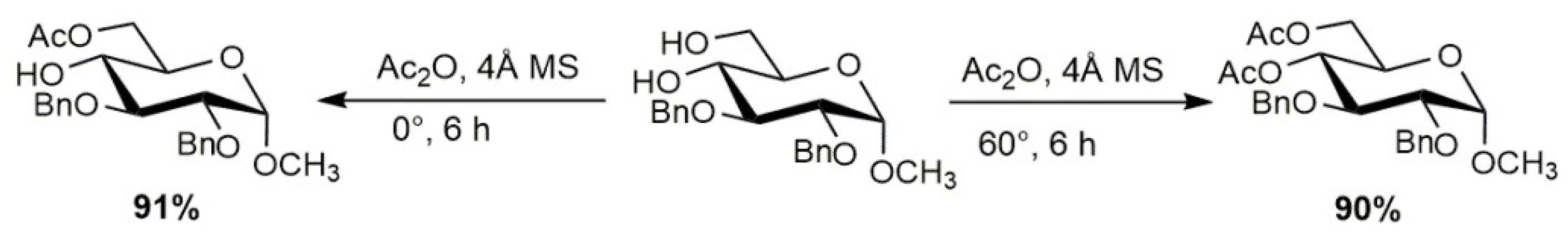
3.2. O-Acetylation and De-O-Acetylation of Carbohydrates Activated by Molecular Sieves
4. Alkylation Reactions Promoted by Molecular Sieves
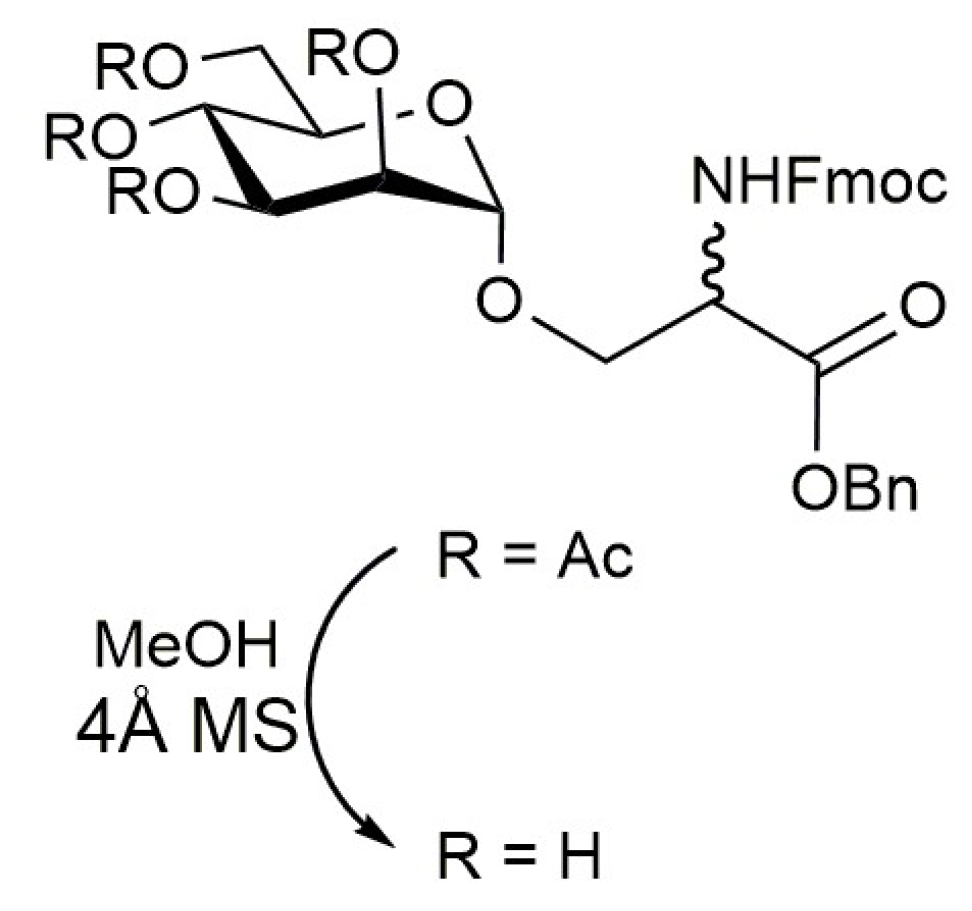
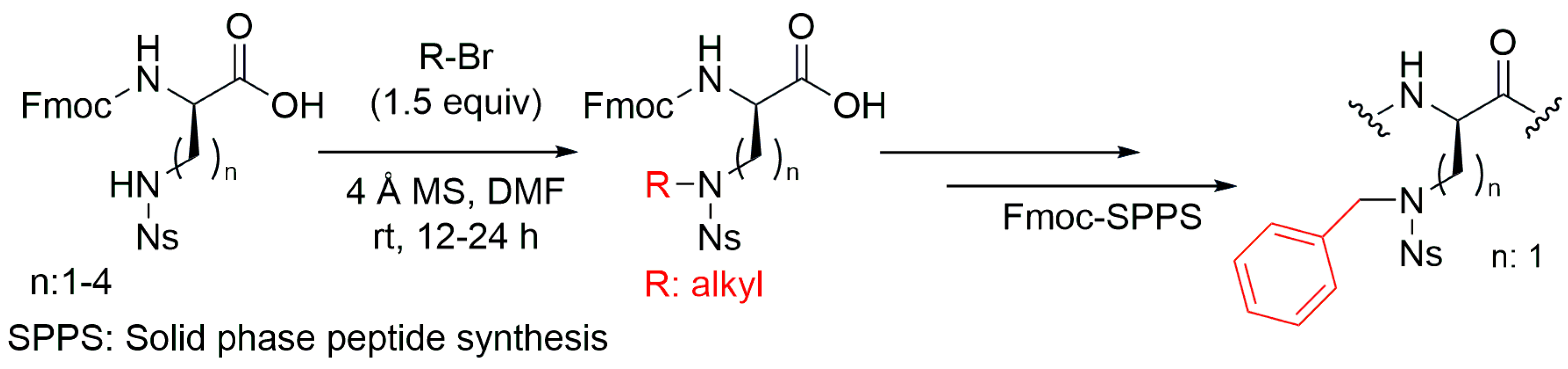
4.1. Side Chain Mono-N-Alkylation of Fmoc-Amino Acids and Peptide Sequences
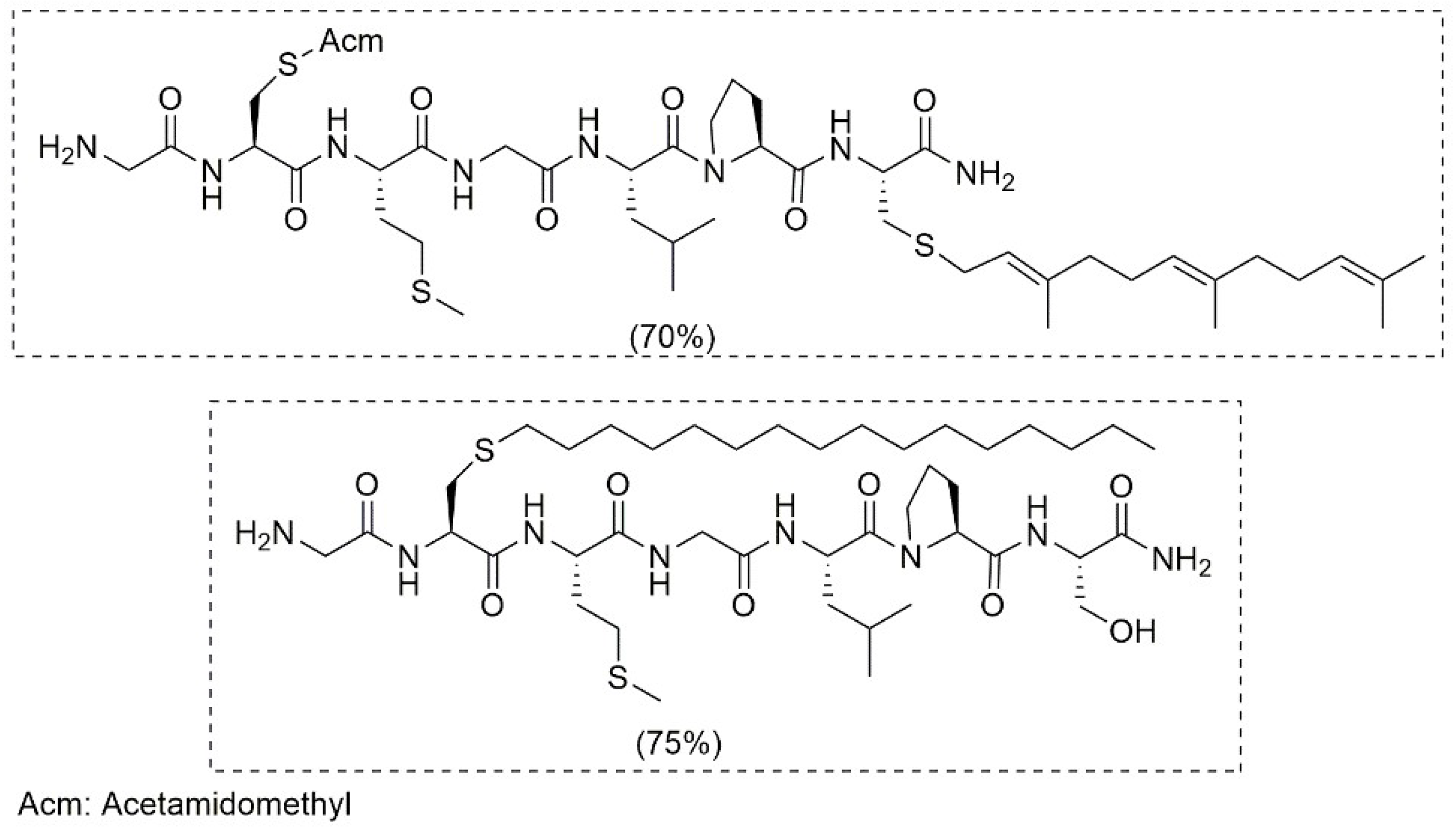
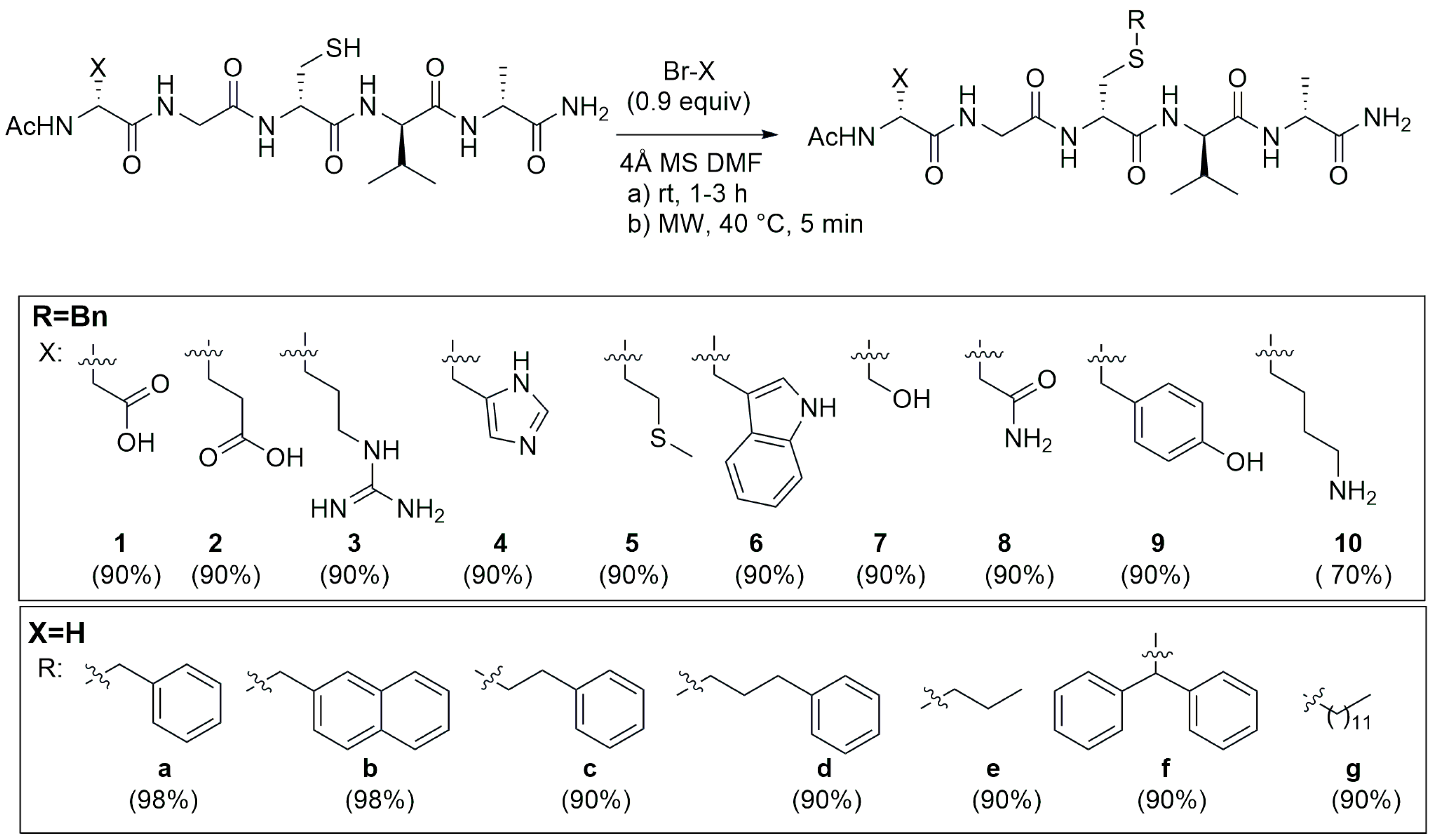
4.2. S-Alkylation Reaction for Introducing Peptide Modification
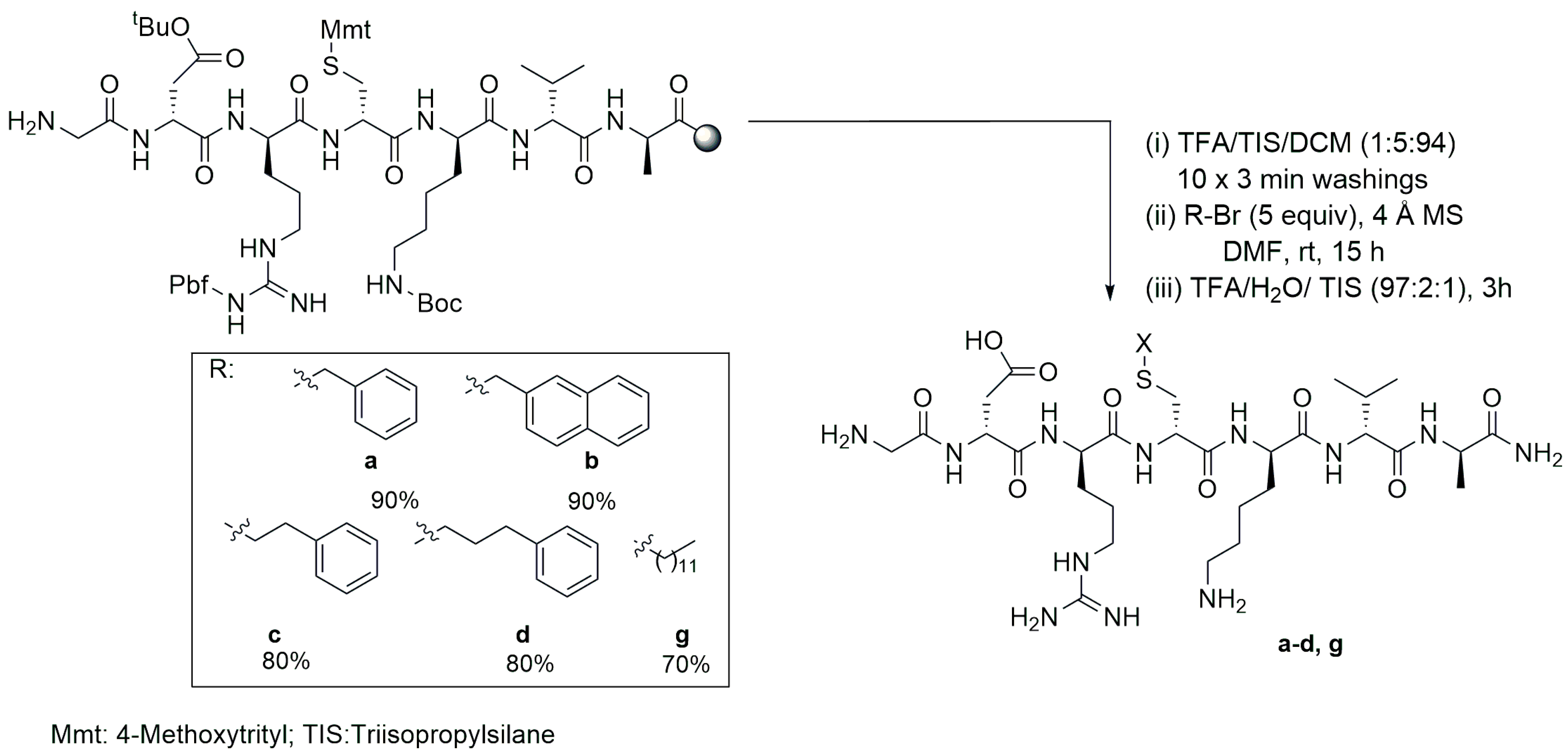
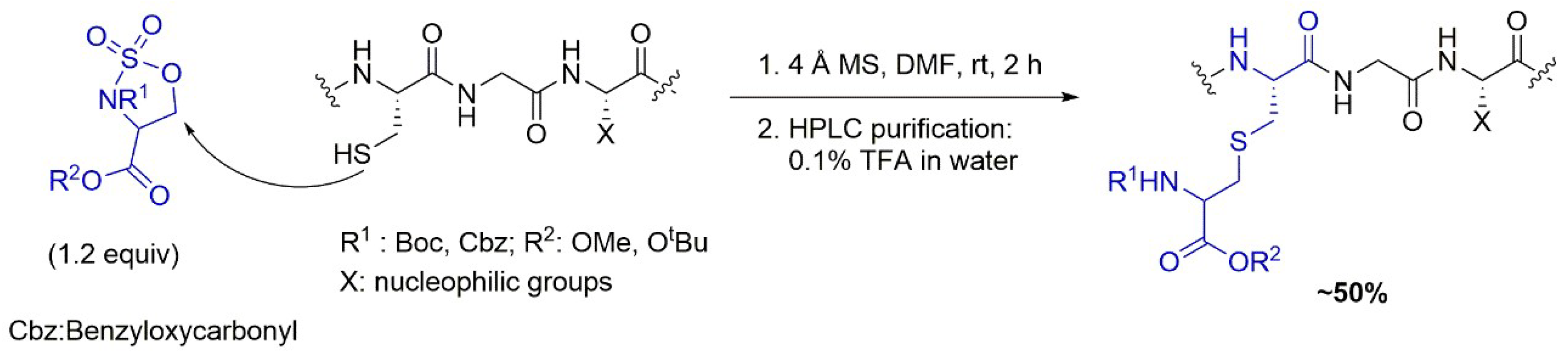
4.3. Solid-Phase S-Alkylation Reaction for Introducing Peptide Modification
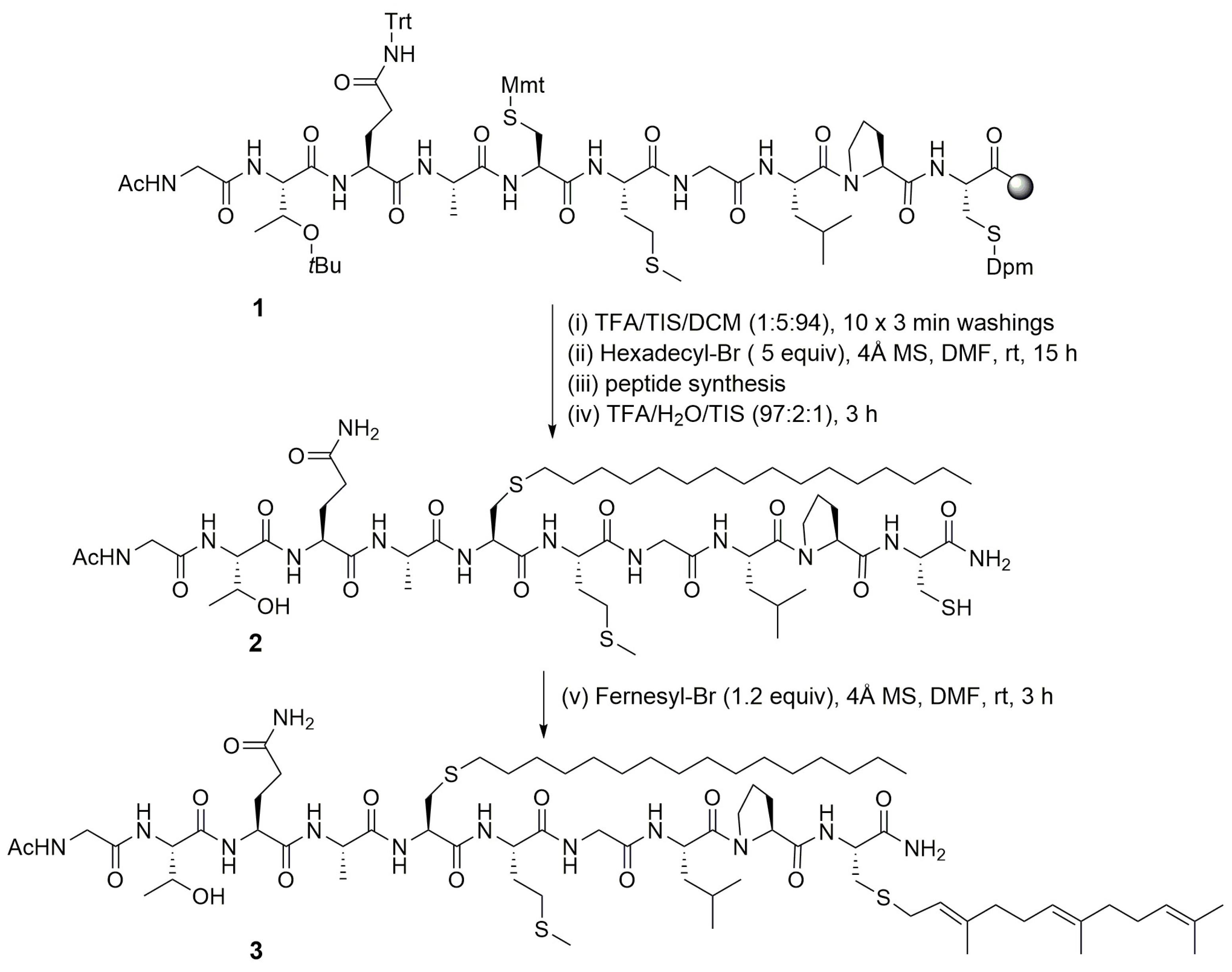
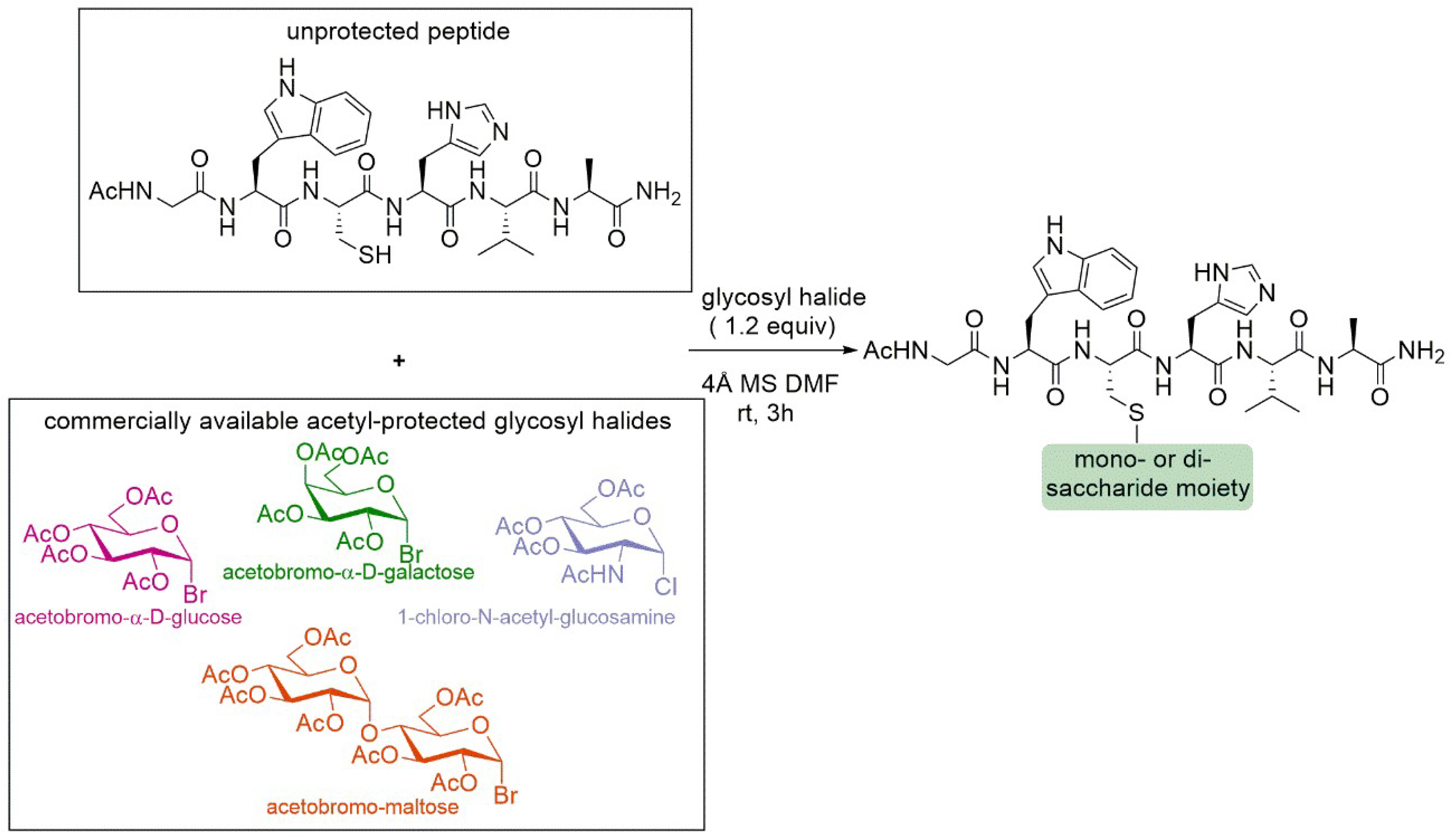
4.4. Chemoselective Glycosylation of Peptides through S-Alkylation Reaction
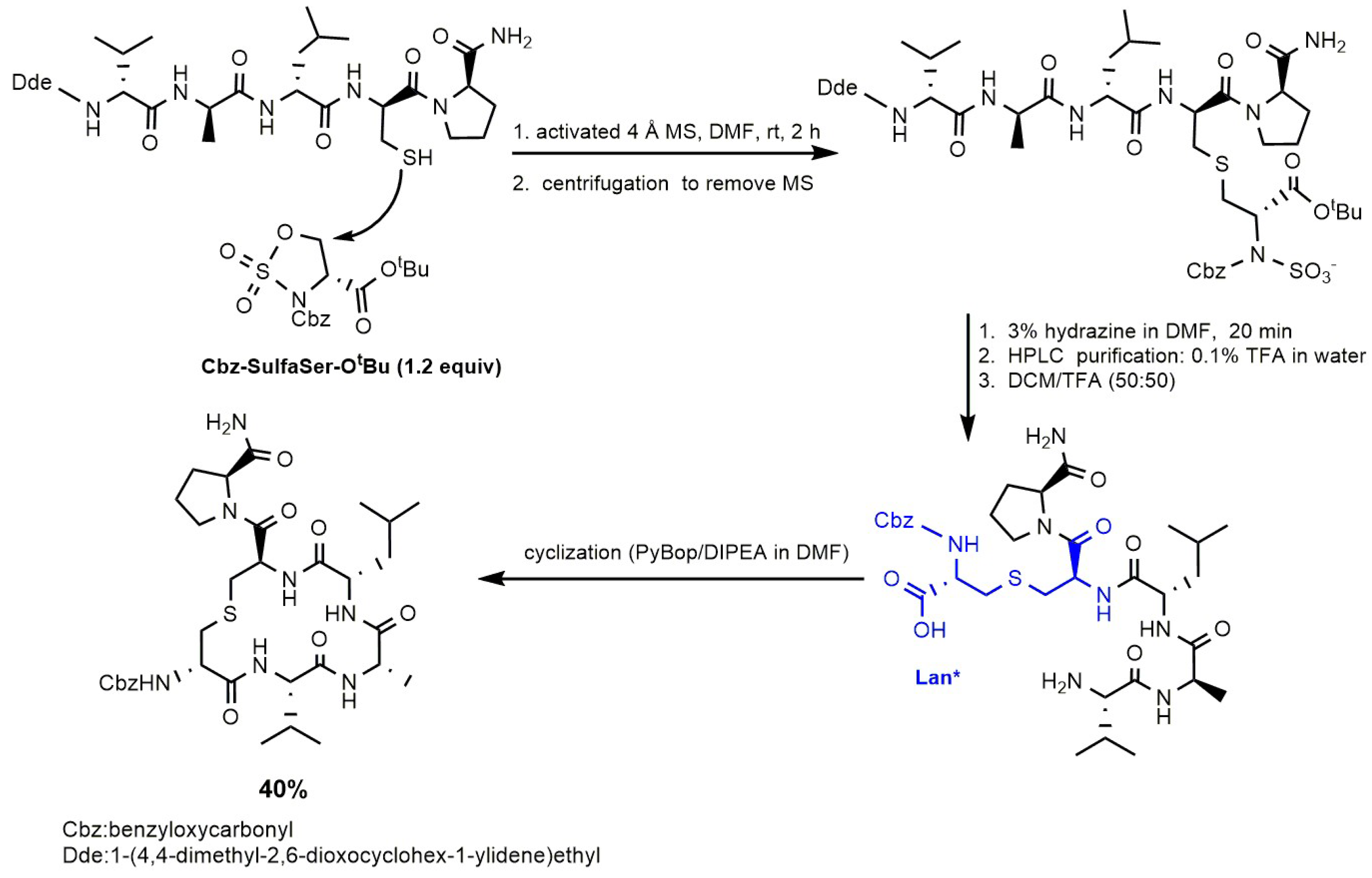
4.5. Lanthionine-Containing Peptide Obtained via Cysteine S-Alkylation on Cyclic Sulfamidates
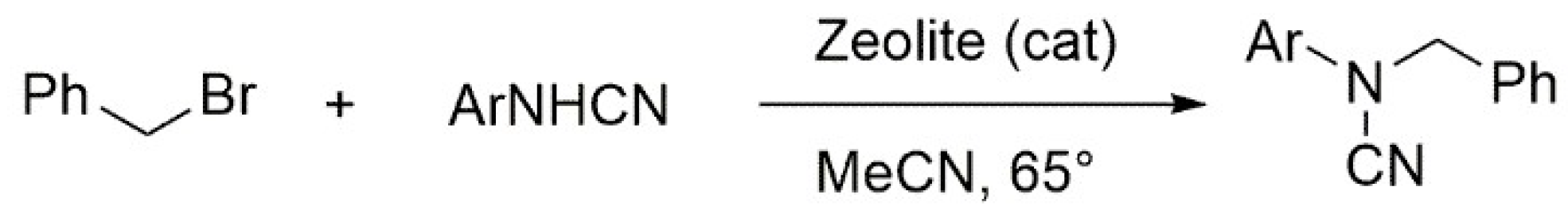
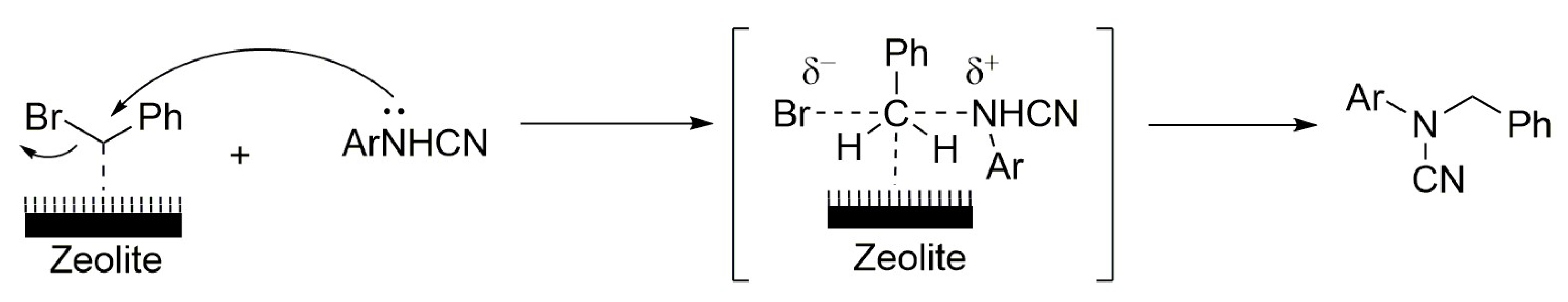
4.6. Benzylation of Arylcyanamides Performed via Acid Catalysis of Zeolites
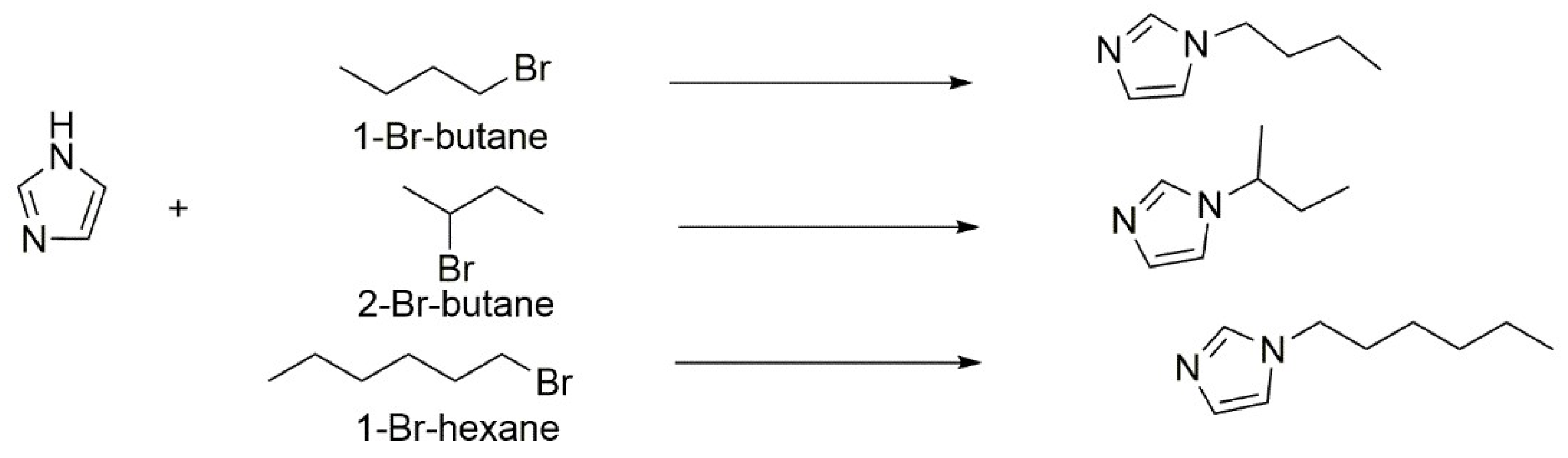
4.7. Microwave Irradiation Promotes the N-Alkylation of Imidazole in Presence of a Basic Zeolite as Catalyst
5. Useful Examples of Broad Scope Reactions Promoted by Molecular Sieves
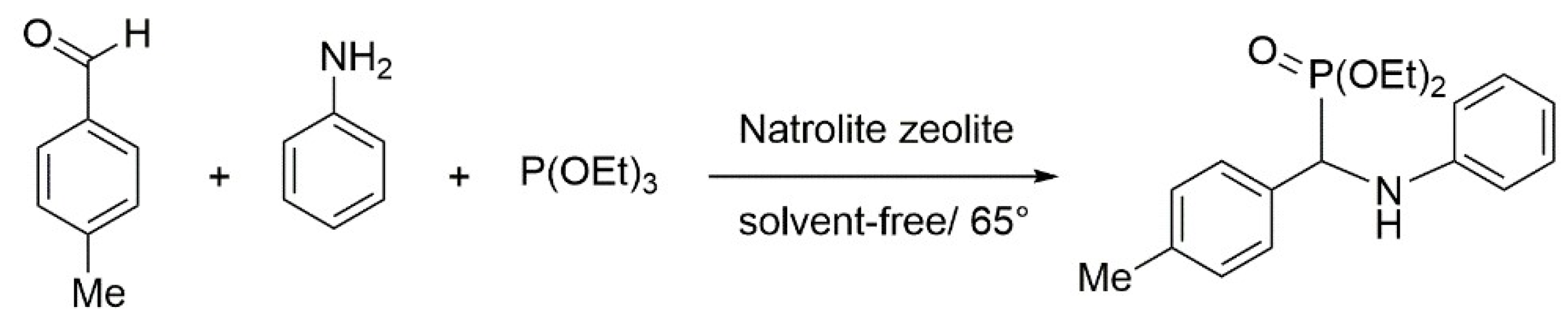
5.1. Synthesis of α-Aminophosphonates by Using Natural Natrolite as Reusable and Efficient Catalyst
5.2. Metal-Catalyzed Enolization of Nucleophiles Precursors
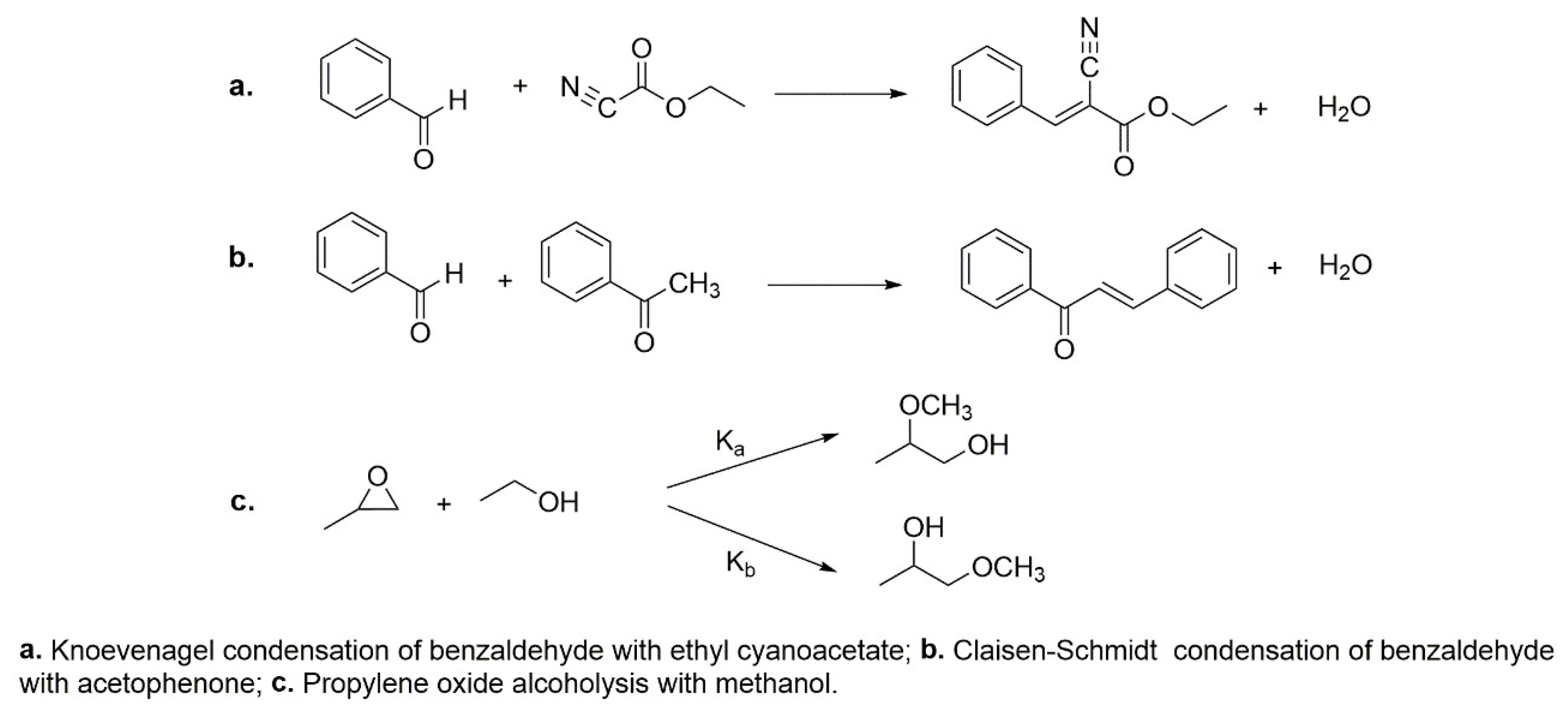
5.3. Different Base-Catalyzed Reactions Promoted by Methylammonium- Faujasite
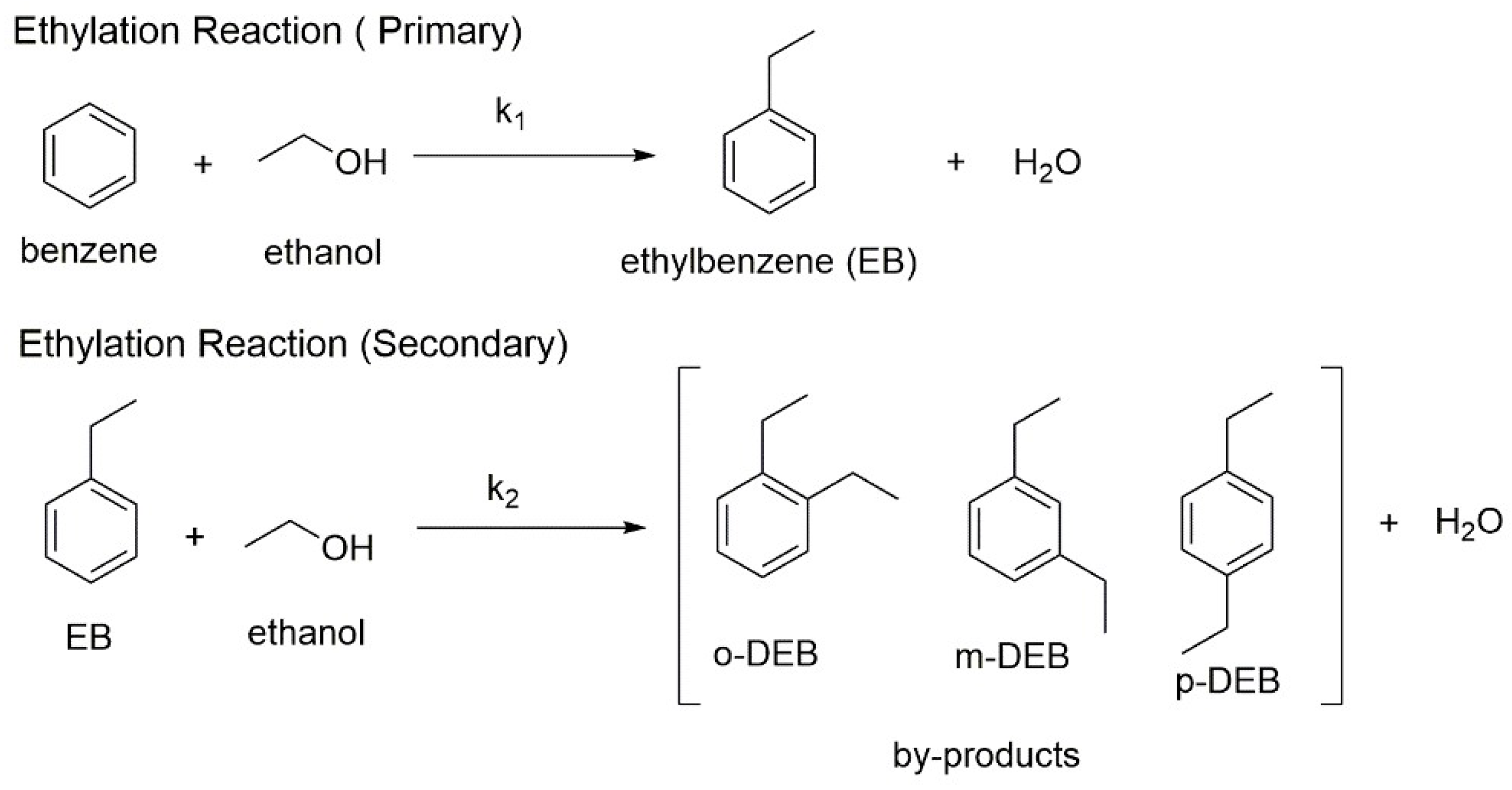
5.4. Benzene Ethylation Performed with Ethanol in Presence of Modified Acidic Zeolite
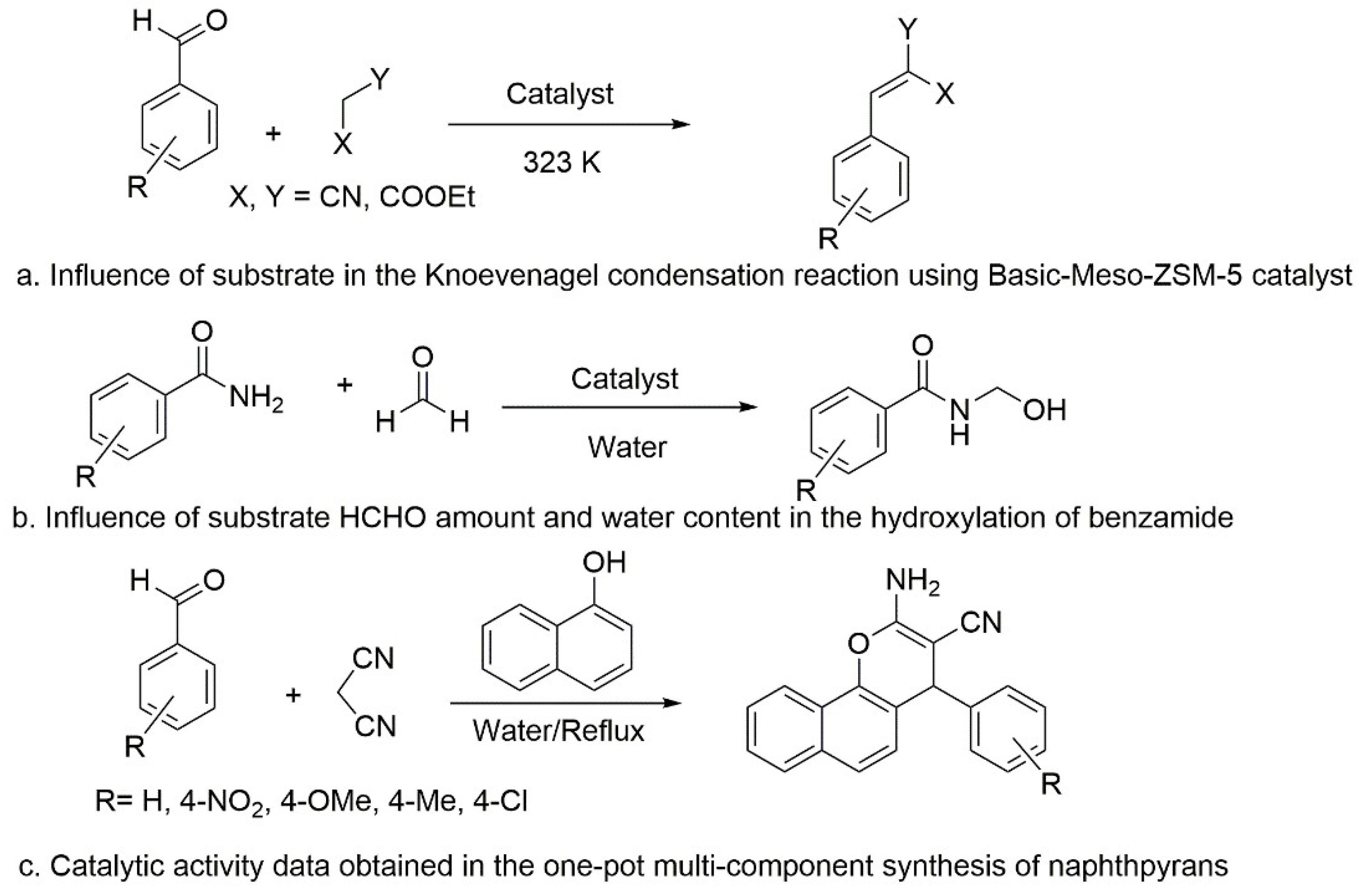
5.5. Basic Mesoporous Zeolite Proved to Be an Efficent Catalyst for Several Reactions: Condensation, Hydroxylation, and Cycloaddition Reactions
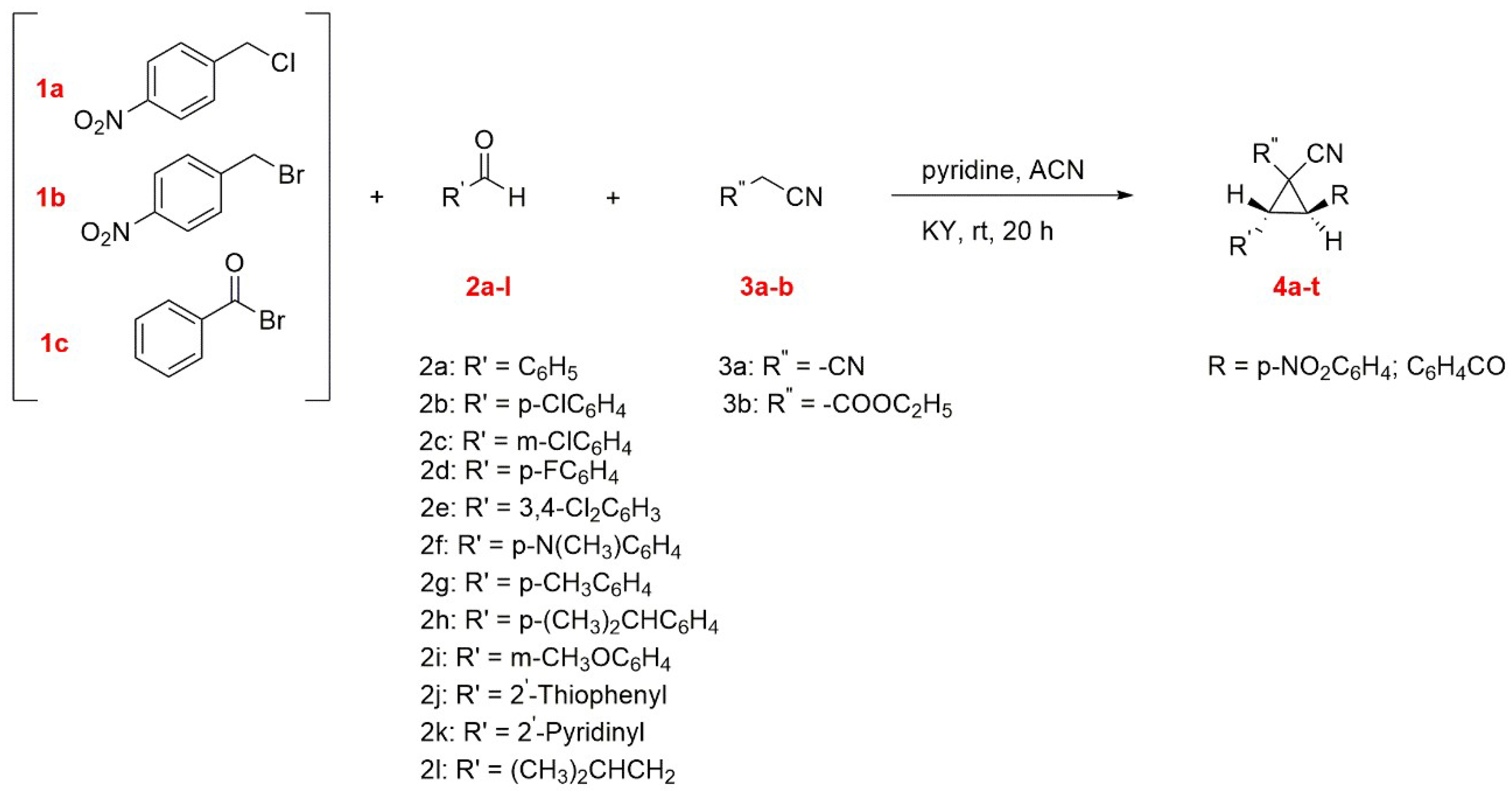
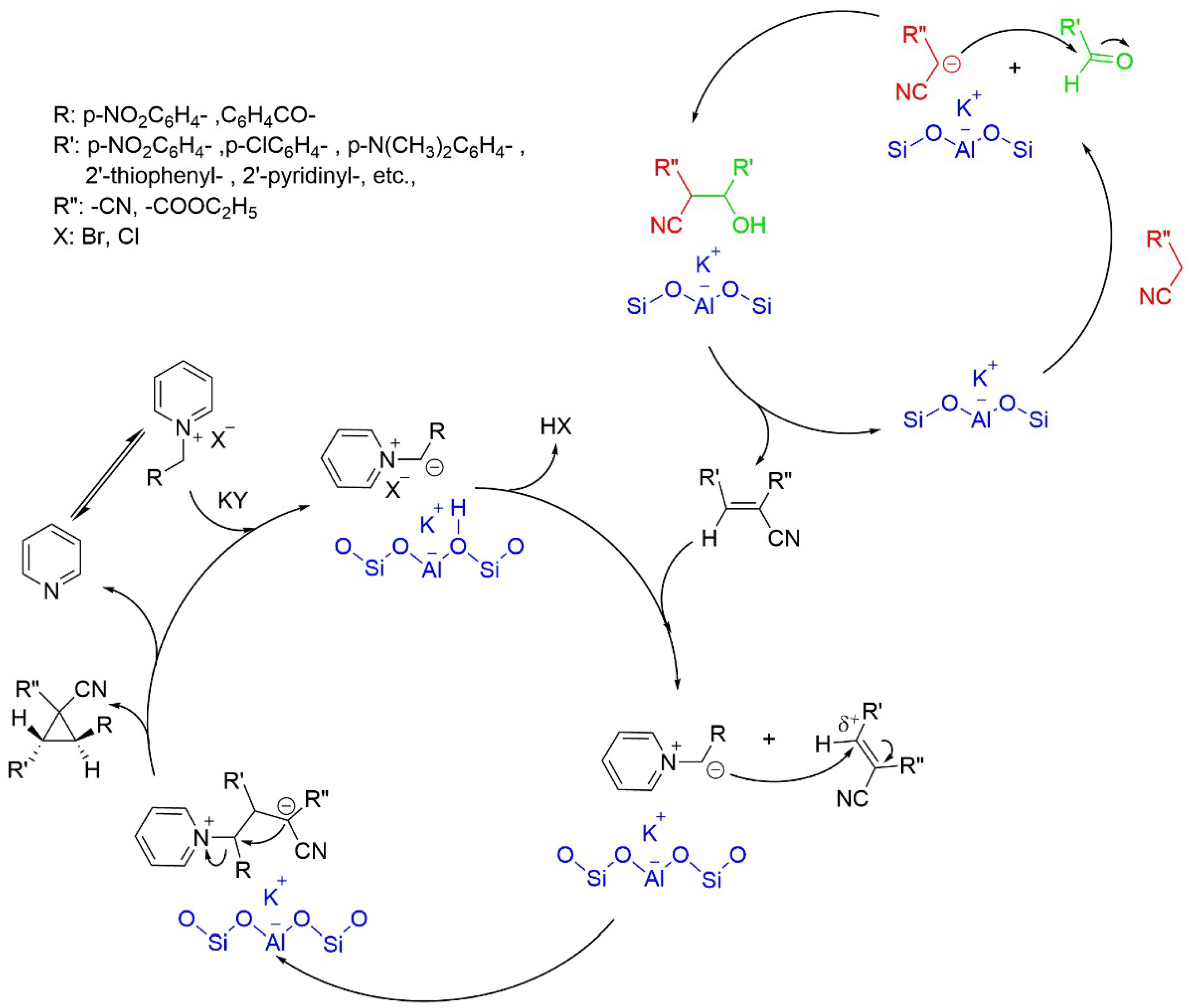
5.6. Synthesis of Polysubstituted Cyclopropanes Catalyzed by Basic Zeolite
6. Final Remarks
Funding
Conflicts of Interest
References
- Pines, H. Acid-Catalyzed Reactions. In The Chemistry of Catalytic Hydrocarbon Conversions, 1st ed.; Academic Press: Evanston, IL, USA, 1981; pp. 1–304. ISBN 9780323155922. [Google Scholar]
- Weitkamp, J. Zeolites and catalysis. Solid State Ion. 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Primo, A.; Garcia, H. Zeolites as catalysts in oil refining. Chem. Soc. Rev. 2014, 43, 7548–7561. [Google Scholar] [CrossRef] [PubMed]
- Busca, G. Acidity and basicity of zeolites: A fundamental approach. Microporous Mesoporous Mater. 2017, 254, 3–16. [Google Scholar] [CrossRef]
- Ugal, J.R.; Hassan, K.H.; Ali, I.H. Preparation of type 4Å zeolite from Iraqi Kaolin: Characterization and properties measurements. J. Assoc. Arab Univ. Basic Appl. Sci. 2010, 9, 2–5. [Google Scholar] [CrossRef]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Corma, A. From Microporous to Mesoporous Molecular Sieves Materials and Their Use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef] [PubMed]
- Busca, G. Base and Basic Materials in Chemical and Environmental Processes. Liquid Versus Solid Basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef]
- Haag, W.O.; Lago, R.M.; Weisz, P.B. The active site of acidic aluminosilicate catalysts. Nature 1984, 309, 589–591. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Update 1 of: Use of Solid Catalysts in Friedel-Crafts Acylation Reactions. Chem. Rev. 2011, 111, PR181–PR214. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Protection (and Deprotection) of Functional Groups in Organic Synthesis by Heterogeneous Catalysis. Chem. Rev. 2010, 113, PR1–PR54. [Google Scholar] [CrossRef]
- Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B.F. Potential and challenges of zeolite chemistry in the catalytic conversion of biomass. Chem. Soc. Rev. 2016, 45, 584–611. [Google Scholar] [CrossRef] [PubMed]
- Louis, B.; Gomes, E.S.; Losch, P.; Lutzweiler, G.; Coelho, T.; Faro, A., Jr.; Pinto, J.F.; Cardoso, C.S.; Silva, A.V.; Pereira, M.M. Biomass-assisted Zeolite Syntheses as a Tool for Designing New Acid Catalysts. ChemCatChem 2017, 9, 2065–2079. [Google Scholar] [CrossRef]
- Kloetstra, K.R.; van Bekkum, H. Base and Acid Catalysis by the Alkali-containing MCM-41 Mesoporous Molecular Sieve. J. Chem. Soc. Chem. Commun. 1995, 1005–1006. [Google Scholar] [CrossRef]
- Barthomeuf, D. Conjugate Acid-Base Pairs in Zeolites. J. Phys. Chem. 1984, 88, 42–45. [Google Scholar] [CrossRef]
- Romero, M.D.; Rodrìguez, A.; Gòmez, J.M. Basicity in zeolite. In New Topics in Catalysis Research; McReynolds, D.K., Ed.; Nova Science Pub Inc.: Madrid, Spain, 2007; pp. 197–220. ISBN 1-60021-286-7. [Google Scholar]
- Guo, P.; Yan, N.; Wang, L.; Zou, X. Database Mining of Zeolite Structures. Cryst. Growth Des. 2017, 17, 6821–6835. [Google Scholar] [CrossRef]
- Titiloye, J.O.; Tschaufeser, P.; Parker, S.C. Recent Advances in Computational Studies of Zeolites. In Spectroscopic and Computational Studies of Supramolecular Systems. Topics in Inclusion Science; Davies, J.E.D., Ed.; Springer: Dordrecht, The Netherlands, 1992; Volume 4, pp. 137–185. ISBN 978-94-015-7989-6. [Google Scholar]
- Lima, C.G.S.; Moreina, N.M.; Paixão, M.W.; Corrêa, A.G. Heterogenous green catalysis: Application of zeolites on multicomponent reactions. Nanocatalysis 2019, 15, 7–12. [Google Scholar] [CrossRef]
- Park, S.; Biligetu, T.; Wang, Y.; Nishitoba, T.; Kondo, J.N.; Yokoi, T. Acidic and catalytic properties of ZSM-5 zeolites with different Al distributions. Catal. Today 2018, 303, 64–70. [Google Scholar] [CrossRef]
- Arya, K.; Rajesh, U.R.; Rawat, D.S. Proline confined FAU zeolite: Heterogeneous hybrid catalyst for the synthesis of spiroheterocycles via a Mannich type reaction. Green Chem. 2012, 14, 3344–3351. [Google Scholar] [CrossRef]
- Narasimharao, K.; Hartmann, M.; Thiel, H.H.; Ernst, S. Novel solid basic catalysts by nitridation of zeolite beta at low temperature. Microporous Mesoporous Mater. 2006, 90, 377–383. [Google Scholar] [CrossRef]
- Saravanamurugan, S.; Riisager, A. Zeolite catalized transformation of carbohydrates to alkyl levulinates. ChemCatChem 2013, 5, 1754–1757. [Google Scholar] [CrossRef]
- Adinolfi, M.; Barone, G.; Iadonisi, A.; Schiattarella, M. Activation of Glycoyl Trihaloacetimidates with Acid-washed Molecular Sieves in the Glycosidation Reaction. Org. Lett. 2003, 5, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, M.; Iadonisi, A.; Pezzella, A.; Ravidà, A. Regioselective Phenol or Carbinol Glycosidation of 17p-Estradiol and Derivatives Thereof. Synlett 2005, 12, 1848–1852. [Google Scholar] [CrossRef]
- Adinolfi, M.; Iadonisi, A.; Ravidà, A.; Schiattarella, M. Moisture Stable Promoters for Selective α-Fucosylation Reactions: Synthesis of Antigen Fragments. Synlett 2004, 2, 0275–0278. [Google Scholar] [CrossRef]
- Adinolfi, M.; Iadonisi, A.; Ravidà, A.; Schiattarella, M. Versatile Use of Ytterbium(III) Triflate and Acid Washed Molecular Sieves in the Activation of Glycosyl Trifluoroacetamide Donors. Assemblage of a Biologically Relevant Tetrasaccharide Sequence of Globo H. J. Org. Chem. 2005, 70, 5316–5319. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, M.; Barone, G.; Iadonisi, A.; Schiattarella, M. Mild benzhydrylation and tritylation of saccharidic hydroxyls promoted by acid washed molecular sieves. Tetrahedron Lett. 2003, 44, 3733–3735. [Google Scholar] [CrossRef]
- Adinolfi, M.; Barone, G.; Iadonisi, A.; Schiattarella, M. An easy approach for the acetylation of saccharidic alcohols. Applicability for regioselective protections. Tetrahedron Lett. 2003, 44, 4661–4663. [Google Scholar] [CrossRef]
- Kartha, R.K.P.; Mukhopadhyay, B.; Field, R.A. Practical de-O-acylation reactions promoted by molecular sieves. Carbohydr. Res. 2004, 339, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Monfregola, L.; De Luca, S. Synthetic strategy for side chain mono-N-alkylation of Fmoc-amino acids promoted by molecular sieves. Amino Acids 2011, 41, 981–990. [Google Scholar] [CrossRef]
- Monfregola, L.; Leone, M.; Calce, E.; De Luca, S. Postsynthetic Modification of Peptide via Chemoselective N-Alkylation of Their Side Chains. Org. Lett. 2012, 14, 1664–1667. [Google Scholar] [CrossRef]
- Calce, E.; De Luca, S. The Cysteine S-Alkylation Reaction as a Synthetic Method to Covalently Modify Peptide Sequences. Chem. Eur. J. 2016, 22, 1–11. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Chemical Modifications of Peptide Sequences via S-Alkylation Reaction. Org. Lett. 2013, 15, 5354–5357. [Google Scholar] [CrossRef] [PubMed]
- Calce, E.; De Luca, S. Microwave heating in peptide side chain modification via cysteine alkylation. Amino Acids 2016, 48, 2267–2271. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Lipidated peptides via post-synthetic thioalkylation promoted by molecular sieves. Amino Acids 2014, 46, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Calce, E.; Leone, M.; Mercurio, F.A.; Monfregola, L.; De Luca, S. Solid-Phase S-Alkylaion Promoted by Molecular Sieves. Org. Lett. 2015, 17, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Calce, E.; Digilio, G.; Menchise, V.; Saviano, M.; De Luca, S. Chemoselective Glycosylation of Peptides through S-Alkylation Reaction. Chem. Eur. J. 2018, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Luca, S.; Digilio, G.; Verdoliva, V.; Saviano, M.; Menchise, V.; Tovillas, P.; Jiménez-Osés, G.; Peregrina, J.M. A Late-Stage synthetic approach to lanthionine-containing peptides via S-alkylation on cyclic sulfamidates promoted by molecular sieves. Org. Lett. 2018, 20, 7478–7482. [Google Scholar] [CrossRef] [PubMed]
- Azarifar, D.; Soleimanei, F.; Aliani, F. Benzylation of arylcyanamides catalyzed by acidic zeolites. J. Mol. Catal. A Chem. 2013, 377, 7–15. [Google Scholar] [CrossRef]
- Costarrosa, L.; Ruiz-Martínez, J.; Rios, R.V.R.A.; Silvestre-Albero, J.; Rojas-Cervantes, M.L.; Sepúlveda-Escribano, A. Basic zeolites as catalysts in the N-alkylation of imidazole: Activation by microwave irradiation. Microporous Mesoporous Mater. 2009, 120, 115–121. [Google Scholar] [CrossRef]
- Bahari, S.; Sajadi, S.M. Natrolite zeolite: A natural and reusable catalyst for one-pot synthesis of α-aminophosphonates under solvent-free conditions. Arabian J. Chem. 2017, 10, S700–S704. [Google Scholar] [CrossRef]
- Hasegawa, M.; Ono, F.; Kanemasa, S. Molecular sieves 4Å work to mediate the catalytic metal enolization of nucleophile precursors: Application to catalyzed enantioselective Michael addition reactions. Tetrahedron Lett. 2008, 49, 5220–5223. [Google Scholar] [CrossRef]
- Martins, L.; Hӧlderich, W.; Cardoso, D. Methylammonium-FAU zeolite: Investigation of the basic sites in base catalyzed reactions and its performance. J. Catal. 2008, 258, 14–24. [Google Scholar] [CrossRef]
- Emana, A.N.; Chand, S. Alkylation of benzene with ethanol over modified HZSM-5 zeolite catalysts. Appl. Petrochem. Res. 2015, 5, 121–134. [Google Scholar] [CrossRef]
- Sarmah, B.; Satpati, B.; Srivastava, R. Highly efficient and recyclable basic mesoporous zeolite catalyzed condensation, hydroxylation, and cycloaddition reactions. J. Colloid Interface Sci. 2017, 493, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Rama, V.; Kanagaraj, K.; Subramanian, T.; Suresh, P.; Pitchumani, K. Pyridinium ylide-assisted KY zeolite catalyzed tandem synthesis of polysubstituted cyclopropanes. Catal. Commun. 2012, 26, 39–43. [Google Scholar] [CrossRef]
- Jiao, J.; Kanellopoulos, J.; Wang, W.; Ray, S.S.; Foerster, H.; Freude, D.; Hunger, M. Characterization of framework and extra-framework aluminum species in non-hydrated zeolites Y by 27Al spin-echo, high-speed MAS, and MQMAS NMR spectroscopy at B0 = 9.4 to 17.6 T. Phys. Chem. Chem. Phys. 2005, 7, 3221–3226. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Lee, S.; Shin, A.; Hong, S.B.; Prins, R.; van Bokhoven, J.A. Influence of framework silicon to aluminium ratio on aluminium coordination and distribution in zeolite Beta investigated by 27Al MAS and 27Al MQ MAS NMR. Phys. Chem. Chem. Phys. 2004, 6, 3031–3036. [Google Scholar] [CrossRef]
- Kim, H.; Cho, H.S.; Kim, C.; Choi, M. Gradual Disordering of LTA Zeolite for Continuous Tuning of the Molecular Sieving Effect. J. Phys. Chem. C 2017, 121, 6807–6812. [Google Scholar] [CrossRef]
- Rimer, J.D. Rational design of zeolite catalysts. Nat. Catal. 2018, 1, 488–489. [Google Scholar] [CrossRef]































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdoliva, V.; Saviano, M.; De Luca, S. Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments. Catalysts 2019, 9, 248. https://doi.org/10.3390/catal9030248
Verdoliva V, Saviano M, De Luca S. Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments. Catalysts. 2019; 9(3):248. https://doi.org/10.3390/catal9030248
Chicago/Turabian StyleVerdoliva, Valentina, Michele Saviano, and Stefania De Luca. 2019. "Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments" Catalysts 9, no. 3: 248. https://doi.org/10.3390/catal9030248