Tandem Hydrogenation/Hydrogenolysis of Furfural to 2-Methylfuran over a Fe/Mg/O Catalyst: Structure–Activity Relationship

 , , , and
, , , and
Abstract
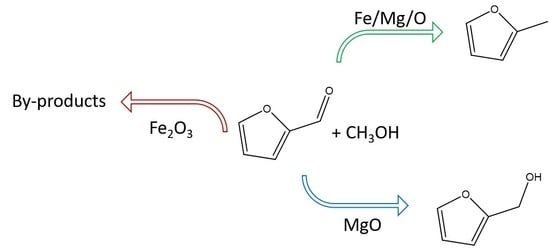
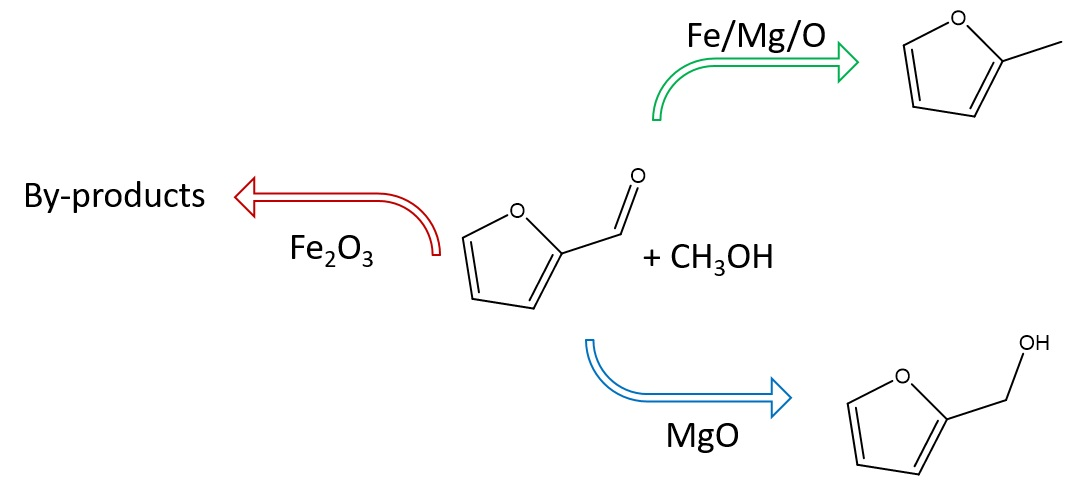
:
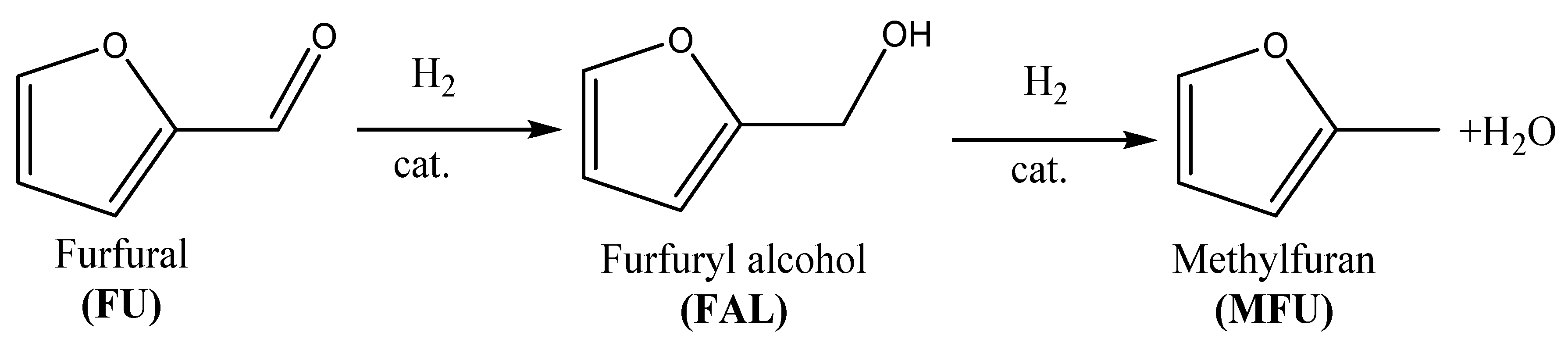
1. Introduction
2. Results and Discussion
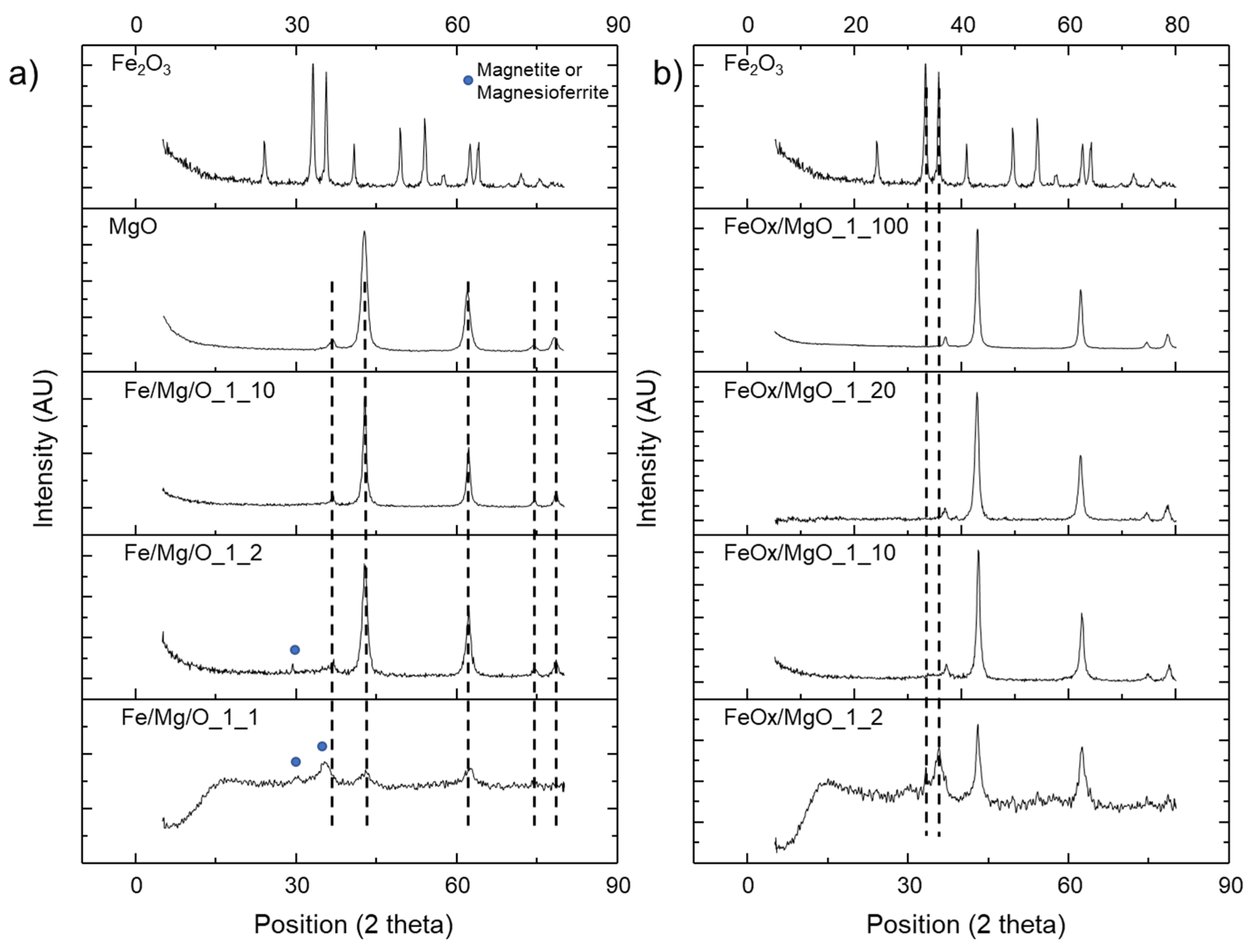
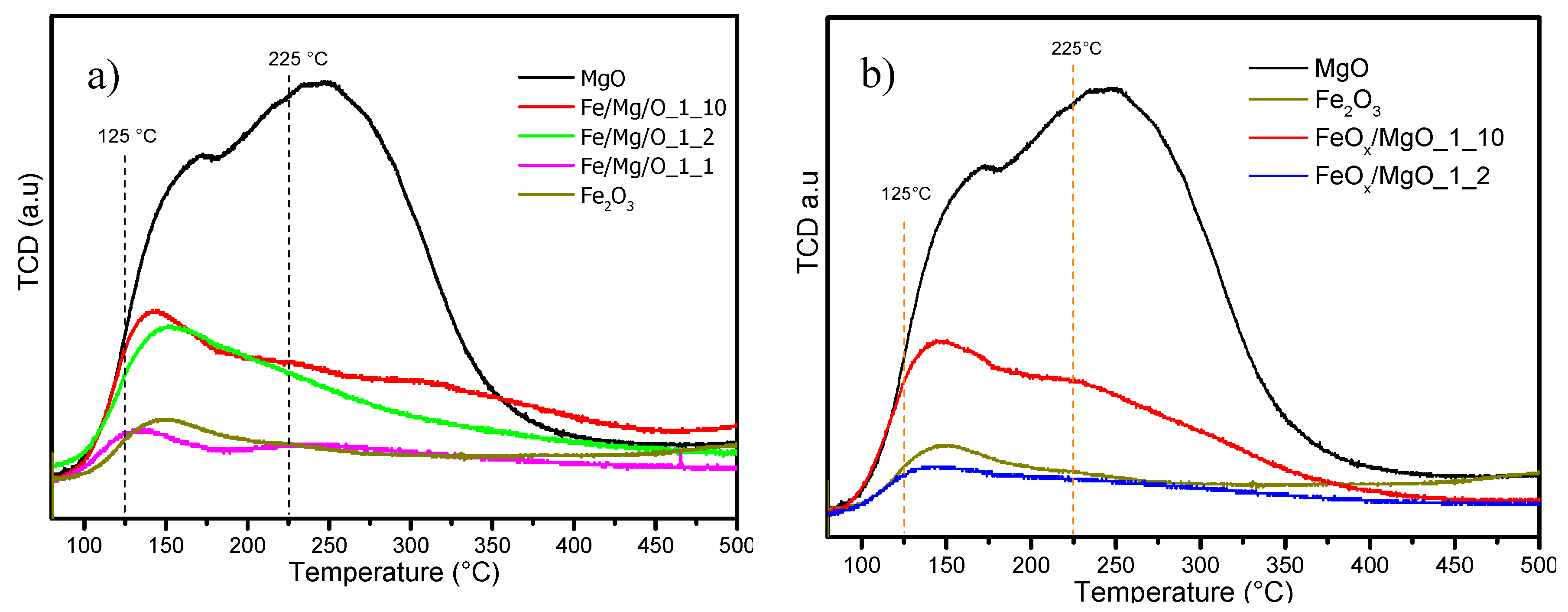
2.1. Physical–Chemical Properties of the Iron-Containing MgO Catalyst
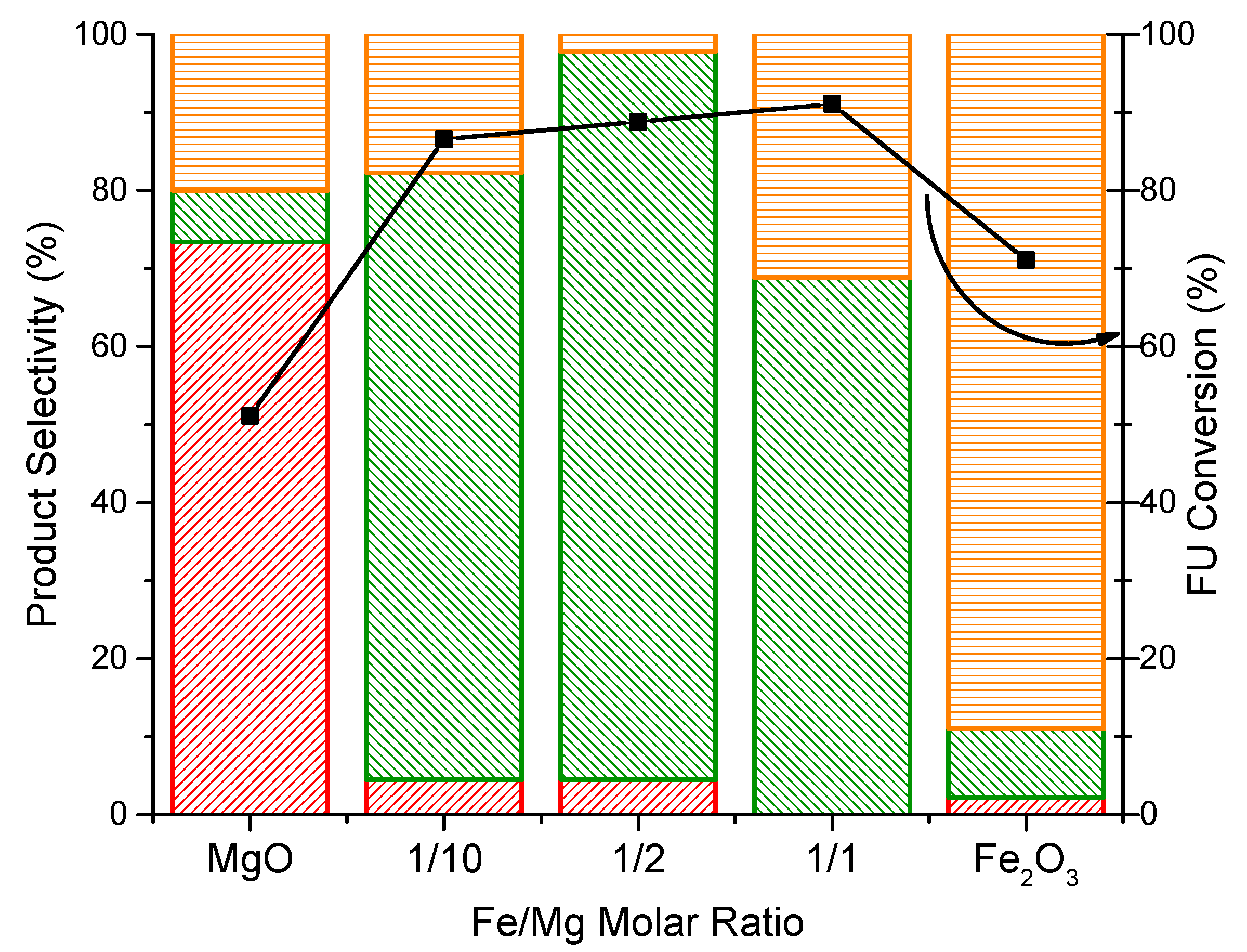
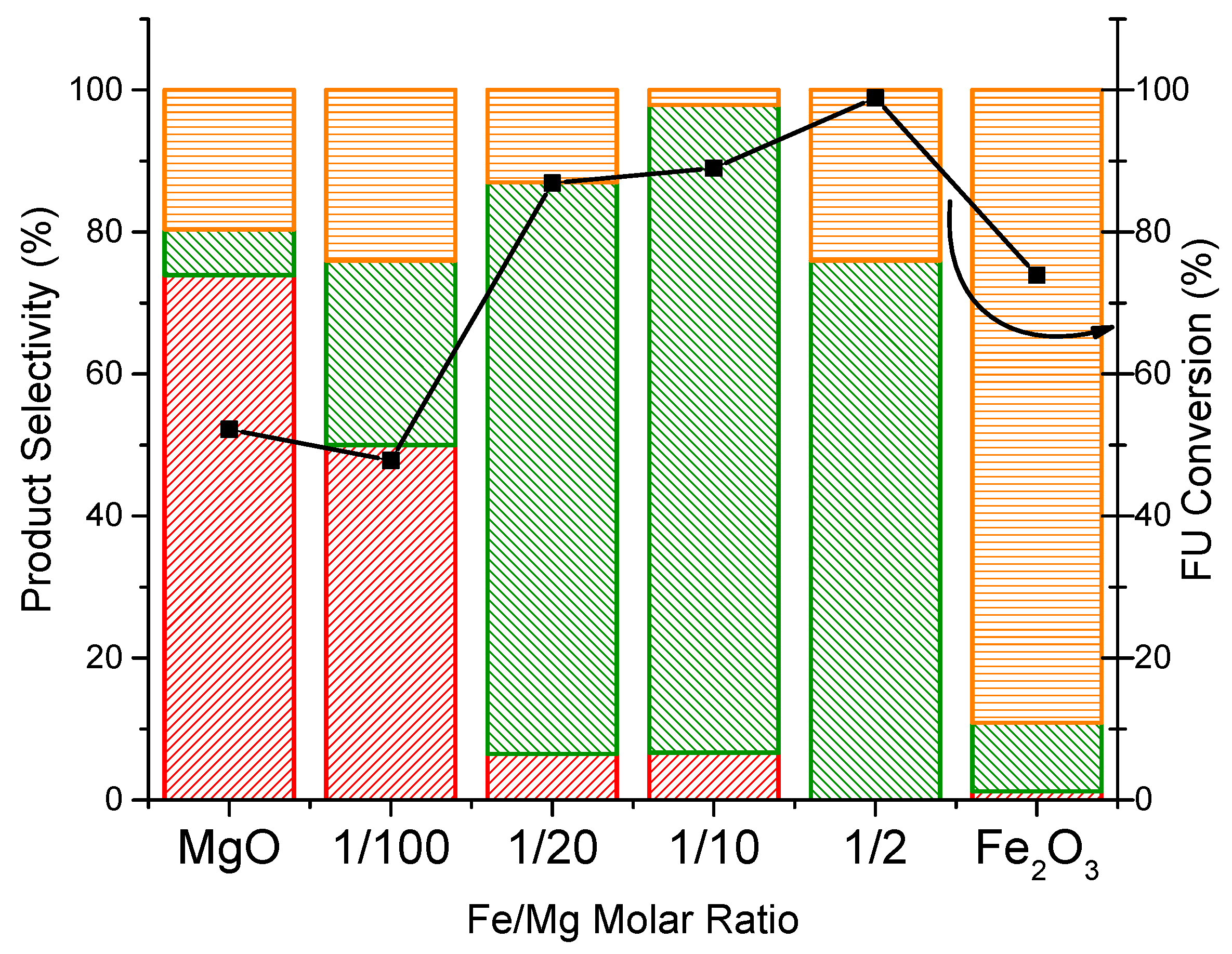
2.2. Reactivity Tests of Iron/Magnesium Oxide Catalysts in the Hydrodeoxygenation of Furfural
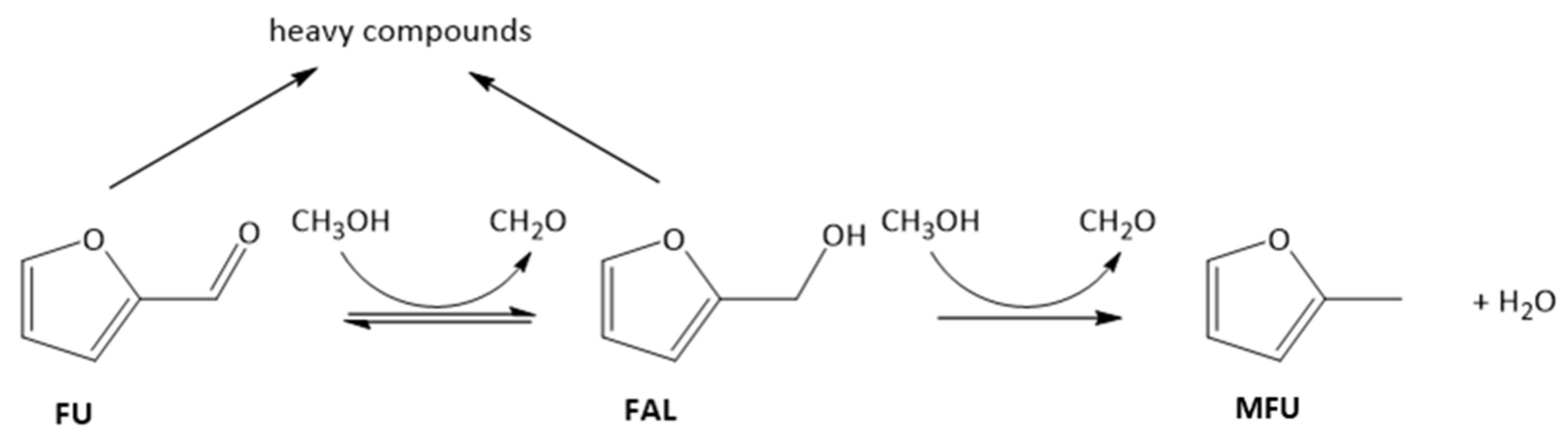
2.3. Effect of the Properties of the Synthesized Catalysts on the Product Distribution of FU Reaction
2.4. Mechanistic Insights
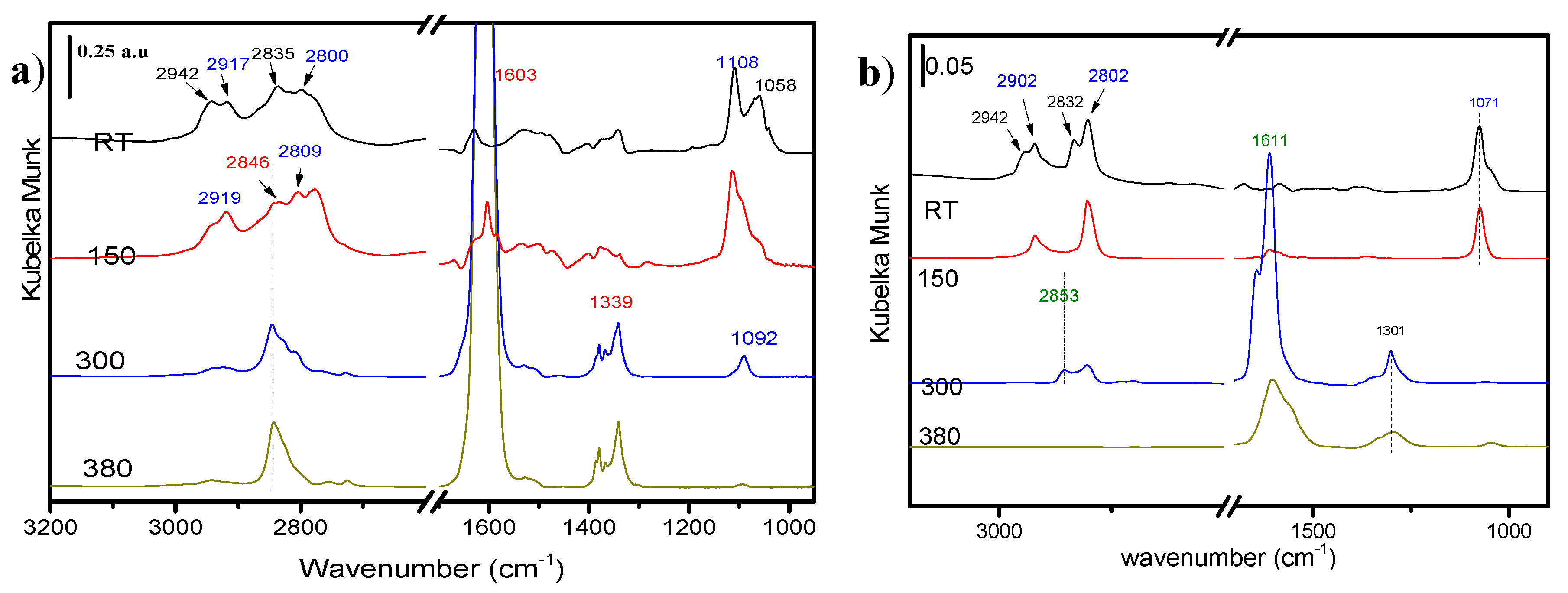
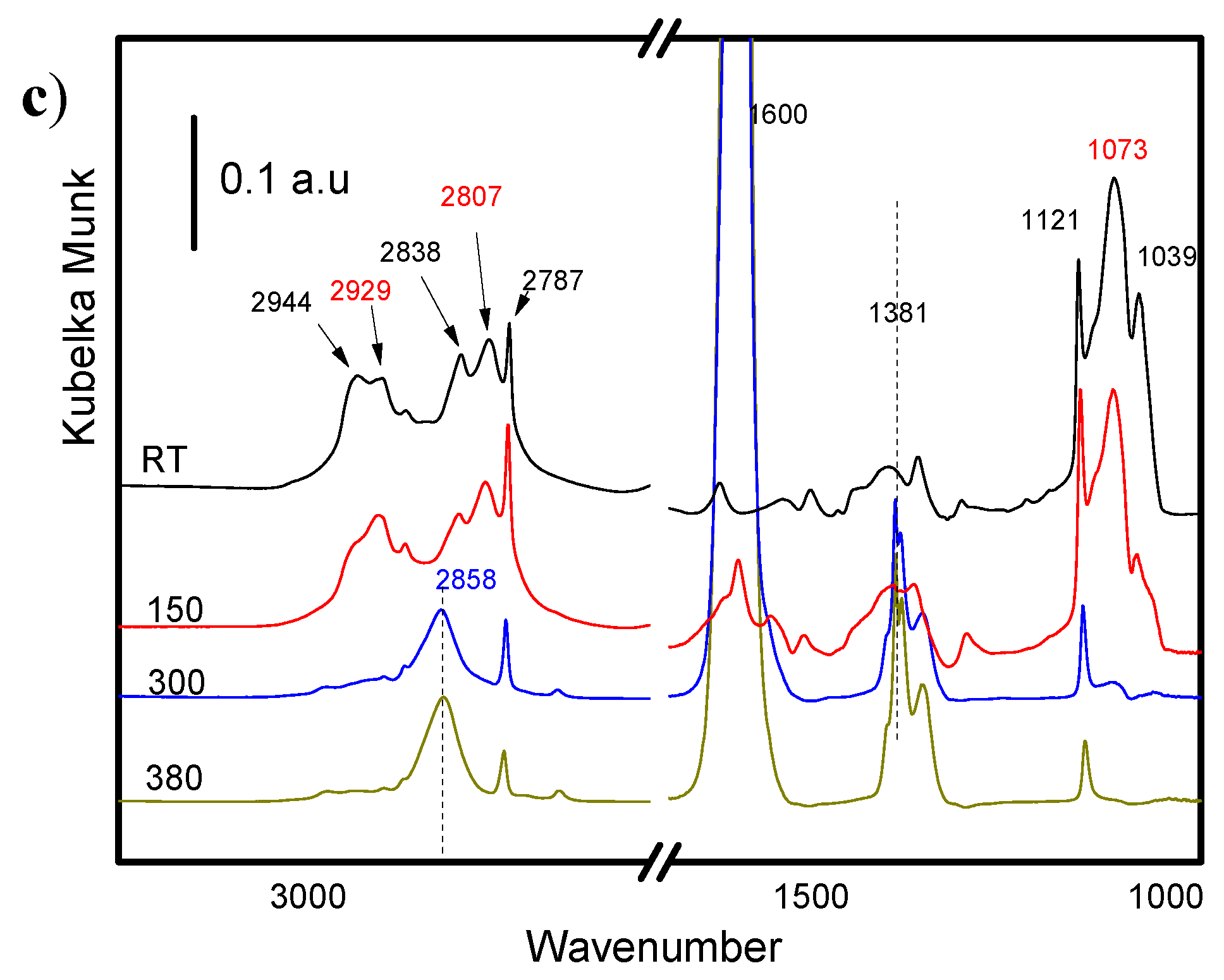
2.4.1. Methanol Adsorption
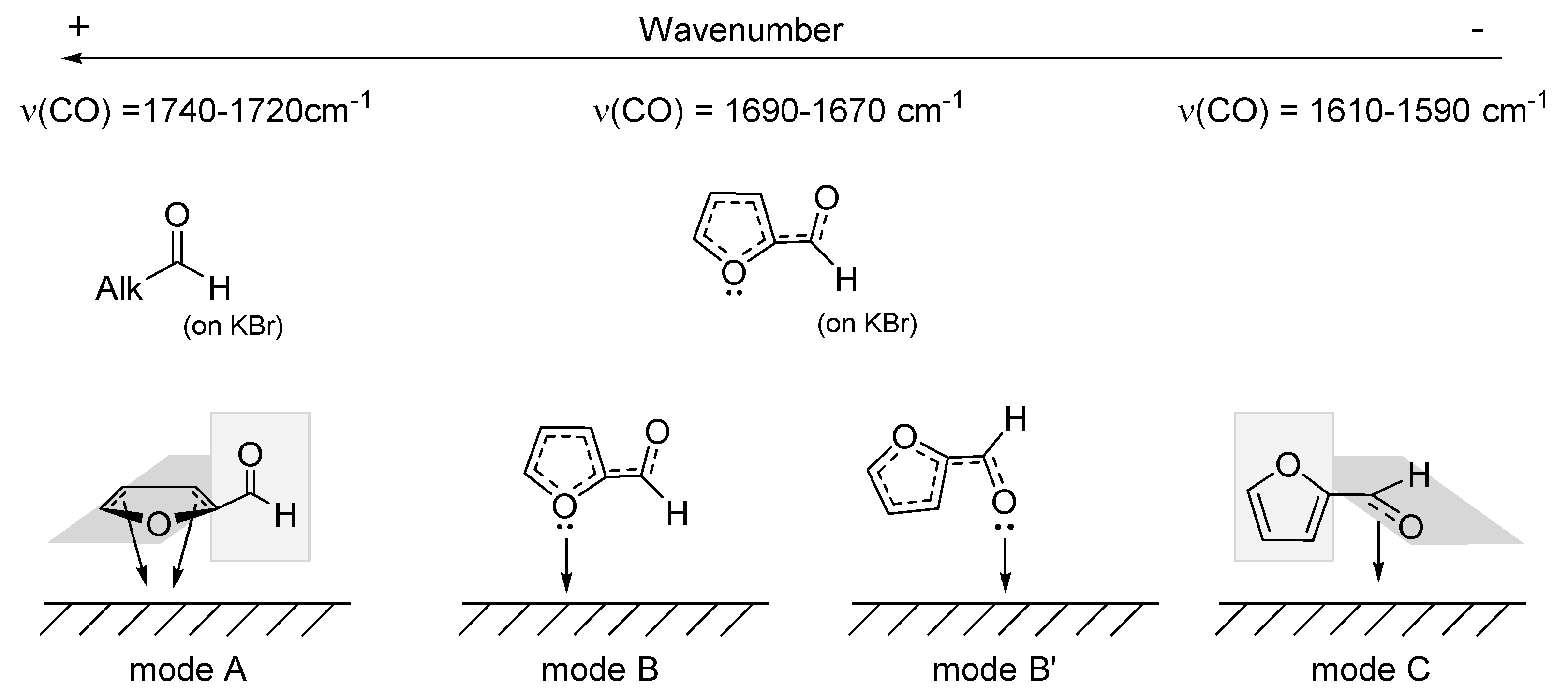
2.4.2. Furfural and Furfuryl Alcohol Adsorption
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Characterization of Catalysts
3.3. Catalytic Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dusselier, M.; Sels, B.F. Selective Catalysis for Cellulose Conversion to Lactic Acid and Other α-Hydroxy Acids. In Selective Catalysis for Renewable Feedstocks and Chemicals; Nicholas, K.M., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 85–125. [Google Scholar]
- Lolli, A.; Zhang, Y.; Basile, F.; Cavani, F.; Albonetti, S. Beyond H2: Exploiting H-Transfer Reaction as a Tool for the Catalytic Reduction of Biomass. In Chemicals and Fuels from Bio-Based Building Blocks; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 349–378. [Google Scholar]
- Gilkey, M.J.; Xu, B. Heterogeneous Catalytic Transfer Hydrogenation as an Effective Pathway in Biomass Upgrading. Acs Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Scholz, D.; Aellig, C.; Hermans, I. Catalytic Transfer Hydrogenation/Hydrogenolysis for Reductive Upgrading of Furfural and 5-(Hydroxymethyl)furfural. ChemSusChem 2014, 7, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Sulmonetti, T.P.; Pang, S.H.; Claure, M.T.; Lee, S.; Cullen, D.A.; Agrawal, P.K.; Jones, C.W. Vapor phase hydrogenation of furfural over nickel mixed metal oxide catalysts derived from layered double hydroxides. Appl. Catal. A Gen. 2016, 517, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Manikandan, M.; Venugopal, A.K.; Nagpure, A.S.; Chilukuri, S.; Raja, T. Promotional effect of Fe on the performance of supported Cu catalyst for ambient pressure hydrogenation of furfural. Rsc Adv. 2016, 6, 3888–3898. [Google Scholar] [CrossRef]
- Lee, J.; Burt, S.P.; Carrero, C.A.; Alba-Rubio, A.C.; Ro, I.; O’Neill, B.J.; Kim, H.J.; Jackson, D.H.K.; Kuech, T.F.; Hermans, I.; et al. Stabilizing cobalt catalysts for aqueous-phase reactions by strong metal-support interaction. J. Catal. 2015, 330, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, H.; Daniel, R.; Ghafourian, A.; Herreros, J.M.; Shuai, S.; Ma, X. Combustion characteristics and emissions of 2-methylfuran compared to 2,5-dimethylfuran, gasoline and ethanol in a DISI engine. Fuel 2013, 103, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Qiu, S.; Long, J.; Wang, C.; Chang, J.; Tan, J.; Liu, Q.; Ma, L.; Wang, T.; Zhang, Q. In situ hydrogenation of furfural with additives over a RANEY Ni catalyst. Rsc Adv. 2015, 5, 91190–91195. [Google Scholar] [CrossRef]
- Villaverde, M.M.; Garetto, T.F.; Marchi, A.J. Liquid-phase transfer hydrogenation of furfural to furfuryl alcohol on Cu–Mg–Al catalysts. Catal. Commun. 2015, 58, 6–10. [Google Scholar] [CrossRef]
- Gong, L.-H.; Cai, Y.-Y.; Li, X.-H.; Zhang, Y.-N.; Su, J.; Chen, J.-S. Room-temperature transfer hydrogenation and fast separation of unsaturated compounds over heterogeneous catalysts in an aqueous solution of formic acid. Green Chem. 2014, 16, 3746–3751. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Martin, N.; Vlachos, D.G. Effect of hydrogen donor on liquid phase catalytic transfer hydrogenation of furfural over a Ru/RuO2/C catalyst. J. Mol. Catal. A: Chem. 2014, 392, 223–228. [Google Scholar] [CrossRef]
- Pasini, T.; Lolli, A.; Albonetti, S.; Cavani, F.; Mella, M. Methanol as a clean and efficient H-transfer reactant for carbonyl reduction: Scope, limitations, and reaction mechanism. J. Catal. 2014, 317, 206–219. [Google Scholar] [CrossRef]
- Mironenko, A.V.; Vlachos, D.G. Conjugation-Driven “Reverse Mars–van Krevelen”-Type Radical Mechanism for Low-Temperature C–O Bond Activation. J. Am. Chem. Soc. 2016, 138, 8104–8113. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Vlachos, D.G. Liquid phase catalytic transfer hydrogenation of furfural over a Ru/C catalyst. Appl. Catal. A: Gen. 2014, 480, 17–24. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Panagiotopoulou, P.; Mironenko, A.V.; Jenness, G.R.; Vlachos, D.G.; Xu, B. Mechanistic Insights into Metal Lewis Acid-Mediated Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran. Acs Catal. 2015, 5, 3988–3994. [Google Scholar] [CrossRef]
- Sitthisa, S.; Sooknoi, T.; Ma, Y.; Balbuena, P.B.; Resasco, D.E. Kinetics and mechanism of hydrogenation of furfural on Cu/SiO2 catalysts. J. Catal. 2011, 277, 1–13. [Google Scholar] [CrossRef]
- Sitthisa, S.; Resasco, D.E. Hydrodeoxygenation of Furfural Over Supported Metal Catalysts: A Comparative Study of Cu, Pd and Ni. Catal. Lett. 2011, 141, 784–791. [Google Scholar] [CrossRef]
- Sitthisa, S.; Pham, T.; Prasomsri, T.; Sooknoi, T.; Mallinson, R.G.; Resasco, D.E. Conversion of furfural and 2-methylpentanal on Pd/SiO2 and Pd–Cu/SiO2 catalysts. J. Catal. 2011, 280, 17–27. [Google Scholar] [CrossRef]
- Sitthisa, S.; An, W.; Resasco, D.E. Selective conversion of furfural to methylfuran over silica-supported NiFe bimetallic catalysts. J. Catal. 2011, 284, 90–101. [Google Scholar] [CrossRef]
- Sheng, H.; Lobo, R.F. Iron-Promotion of Silica-Supported Copper Catalysts for Furfural Hydrodeoxygenation. ChemCatChem 2016, 8, 3402–3408. [Google Scholar] [CrossRef]
- Grazia, L.; Lolli, A.; Folco, F.; Zhang, Y.; Albonetti, S.; Cavani, F. Gas-phase cascade upgrading of furfural to 2-methylfuran using methanol as a H-transfer reactant and MgO based catalysts. Catal. Sci. Technol. 2016, 6, 4418–4427. [Google Scholar] [CrossRef]
- Crocella, V.; Cerrato, G.; Magnacca, G.; Morterra, C.; Cavani, F.; Maselli, L.; Passeri, S. Gas-phase phenol methylation over Mg/Me/O (Me = Al, Cr, Fe) catalysts: mechanistic implications due to different acid-base and dehydrogenating properties. Dalton Trans. 2010, 39, 8527–8537. [Google Scholar] [CrossRef] [PubMed]
- Valente, J.S.; Figueras, F.; Gravelle, M.; Kumbhar, P.; Lopez, J.; Besse, J.P. Basic Properties of the Mixed Oxides Obtained by Thermal Decomposition of Hydrotalcites Containing Different Metallic Compositions. J. Catal. 2000, 189, 370–381. [Google Scholar] [CrossRef]
- Sato, T.; Wakabayashi, T.; Shimada, M. Adsorption of various anions by magnesium aluminum oxide of (Mg0.7Al0.3O1.15). Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 89–92. [Google Scholar] [CrossRef]
- Tichit, D.; Lhouty, M.H.; Guida, A.; Chiche, B.H.; Figueras, F.; Auroux, A.; Bartalini, D.; Garrone, E. Textural Properties and Catalytic Activity of Hydrotalcites. J. Catal. 1995, 151, 50–59. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Ren, J.; Liu, X.; Li, X.; Xia, Y.; Lu, G.; Wang, Y. Mesoporous niobium phosphate: an excellent solid acid for the dehydration of fructose to 5-hydroxymethylfurfural in water. Catal. Sci. Technol. 2012, 2, 2485–2491. [Google Scholar] [CrossRef]
- Wang, F.; Ta, N.; Shen, W. MgO nanosheets, nanodisks, and nanofibers for the Meerwein–Ponndorf–Verley reaction. Appl. Catal. A Gen. 2014, 475, 76–81. [Google Scholar] [CrossRef]
- Ballarini, N.; Cavani, F.; Maselli, L.; Montaletti, A.; Passeri, S.; Scagliarini, D.; Flego, C.; Perego, C. The transformations involving methanol in the acid- and base-catalyzed gas-phase methylation of phenol. J. Catal. 2007, 251, 423–436. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Liu, X.; Lu, G.; Wang, Y. Pd/Nb2O5/SiO2 Catalyst for the Direct Hydrodeoxygenation of Biomass-Related Compounds to Liquid Alkanes under Mild Conditions. ChemSusChem 2015, 8, 1761–1767. [Google Scholar] [CrossRef]
- Hong, D.-Y.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Hydrodeoxygenation and coupling of aqueous phenolics over bifunctional zeolite-supported metal catalysts. Chem. Commun. 2010, 46, 1038–1040. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Díez, V.K.; Xu, M.; Iglesia, E.; Apesteguía, C.R. Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Routray, K.; Zhou, W.; Kiely, C.J.; Wachs, I.E. Catalysis Science of Methanol Oxidation over Iron Vanadate Catalysts: Nature of the Catalytic Active Sites. Acs Catal. 2011, 1, 54–66. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. Use of methanol as an IR molecular probe to study the surface of polycrystalline ceria. J. Chem. Soc. Faraday Trans. 1997, 93, 1159–1168. [Google Scholar] [CrossRef]
- Tabanelli, T.; Passeri, S.; Guidetti, S.; Cavani, F.; Lucarelli, C.; Cargnoni, F.; Mella, M. A cascade mechanism for a simple reaction: The gas-phase methylation of phenol with methanol. J. Catal. 2019, 370, 447–460. [Google Scholar] [CrossRef]
- Tabanelli, T.; Cocchi, S.; Gumina, B.; Izzo, L.; Mella, M.; Passeri, S.; Cavani, F.; Lucarelli, C.; Schütz, J.; Bonrath, W.; et al. Mg/Ga mixed-oxide catalysts for phenol methylation: Outstanding performance in 2,4,6-trimethylphenol synthesis with co-feeding of water. Appl. Catal. A Gen. 2018, 552, 86–97. [Google Scholar] [CrossRef]
- Lucarelli, C.; Galli, S.; Maspero, A.; Cimino, A.; Bandinelli, C.; Lolli, A.; Velasquez Ochoa, J.; Vaccari, A.; Cavani, F.; Albonetti, S. Adsorbent–Adsorbate Interactions in the Oxidation of HMF Catalyzed by Ni-Based MOFs: A DRIFT and FT-IR Insight. J. Phys. Chem. C 2016, 120, 15310–15321. [Google Scholar] [CrossRef]
- Chandramohan, P.; Srinivasan, M.P.; Velmurugan, S.; Narasimhan, S.V. Cation distribution and particle size effect on Raman spectrum of CoFe2O4. J. Solid State Chem. 2011, 184, 89–96. [Google Scholar] [CrossRef]
- Nowicka, E.; Hofmann, J.P.; Parker, S.F.; Sankar, M.; Lari, G.M.; Kondrat, S.A.; Knight, D.W.; Bethell, D.; Weckhuysen, B.M.; Hutchings, G.J. In situ spectroscopic investigation of oxidative dehydrogenation and disproportionation of benzyl alcohol. Phys. Chem. Chem. Phys. 2013, 15, 12147–12155. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.; Ferri, D.; Campisi, S.; Chan-Thaw, C.E.; Lu, Y.; Kröcher, O.; Prati, L. Operando Attenuated Total Reflectance FTIR Spectroscopy: Studies on the Different Selectivity Observed in Benzyl Alcohol Oxidation. ChemCatChem 2015, 7, 2534–2541. [Google Scholar] [CrossRef]
- Shekhar, R.; Barteau, M.A.; Plank, R.V.; Vohs, J.M. Adsorption and Reaction of Aldehydes on Pd Surfaces. J. Phys. Chem. B 1997, 101, 7939–7951. [Google Scholar] [CrossRef]
- Davis, J.L.; Barteau, M.A. Spectroscopic identification of alkoxide, aldehyde, and acyl intermediates in alcohol decomposition on Pd(111). Surf. Sci. 1990, 235, 235–248. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Dimas-Rivera, G.L.; De la Rosa, J.R.; Lucio-Ortiz, C.J.; De los Reyes Heredia, J.A.; González, V.G.; Hernández, T. Desorption of Furfural from Bimetallic Pt-Fe Oxides/Alumina Catalysts. Materials 2014, 7, 527–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 27 October 2019).
- Rogojerov, M.; Keresztury, G.; Jordanov, B. Vibrational spectra of partially oriented molecules having two conformers in nematic and isotropic solutions: furfural and 2-chlorobenzaldehyde. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, M.; Wu, Y.-n.; Li, F. Preparation of mesoporous carbon nanofibers from the electrospun poly(furfuryl alcohol)/poly(vinyl acetate)/silica composites. Rsc Adv. 2014, 4, 21089–21092. [Google Scholar] [CrossRef]
- Cesari, C.; Mazzoni, R.; Matteucci, E.; Baschieri, A.; Sambri, L.; Mella, M.; Tagliabue, A.; Basile, F.L.; Lucarelli, C. Hydrogen Transfer Activation via Stabilization of Coordinatively Vacant Sites: Tuning Long-Range π-System Electronic Interaction between Ru(0) and NHC Pendants. Organometallics 2019, 38, 1041–1051. [Google Scholar] [CrossRef]
- Albonetti, S.; Boanini, E.; Jiménez-Morales, I.; Lucarelli, C.; Mella, M.; Molinari, C.; Vaccari, A. Novel thiotolerant catalysts for the on-board partial dehydrogenation of jet fuels. Rsc Adv. 2016, 6, 48962–48972. [Google Scholar] [CrossRef]
- Lucarelli, C.; Giugni, A.; Moroso, G.; Vaccari, A. FT-IR Investigation of Methoxy Substituted Benzenes Adsorbed on Solid Acid Catalysts. J. Phys. Chem. C 2012, 116, 21308–21317. [Google Scholar] [CrossRef] [Green Version]
- Grazia, L.; Bonincontro, D.; Lolli, A.; Tabanelli, T.; Lucarelli, C.; Albonetti, S.; Cavani, F. Exploiting H-transfer as a tool for the catalytic reduction of bio-based building blocks: the gas-phase production of 2-methylfurfural using a FeVO4 catalyst. Green Chem. 2017, 19, 4412–4422. [Google Scholar] [CrossRef]




 MFU
MFU  Carbon loss
Carbon loss  FU conversion
FU conversion  .
MFU Carbon loss FU conversion .
.
MFU Carbon loss FU conversion .
 MFU
MFU  Carbon loss
Carbon loss  FU conversion
FU conversion  .
MFU Carbon loss FU conversion .
.
MFU Carbon loss FU conversion .




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Fe:Mg | Surface Area (m2·g−1) | Crystalline Phase (XRD) | Lewis Acidity (mmol·g−1) (a) | Brønsted Acidity (mmol·g−1) (a) | Basicity (mmol·g−1) (b) |
|---|---|---|---|---|---|---|
| MgO | 172 | Periclase MgO | 0 | 0 | 7.51 | |
| Fe2O3 | 51 | Hematite | - (d) | - (d) | 1.38 | |
| Fe/Mg/O_1_10 | 1:10 | 102 | Periclase MgO | 0 | 0 | 2.62 |
| Fe/Mg/O_1_2 | 1:2 | 140 | Periclase MgO and Magnetite/Magnesioferrite | 0.43 [0.20 + 0.07 + 0.16] (c) | 0 | 2.34 |
| Fe/Mg/O_1_1 | 1:1 | 74 | Periclase MgO and Magnetite/Magnesioferrite | 0.62 [0.25 + 0.24 + 0.13] (c) | 0 | 1.09 |
| FeOx/MgO_1_100 | 1:100 | 150 | Periclase MgO | 0 | 0 | 2.62 |
| FeOx/MgO_1_20 | 1:20 | 129 | Periclase MgO and Hematite | 0 | 0 | 2.34 |
| FeOx/MgO_1_10 | 1:10 | 94 | Periclase MgO and Hematite | 0.15 [0.05 + 0.06 + 0.04] (c) | 0 | 2.67 |
| FeOx/MgO_1_2 | 1:2 | 33 | Periclase MgO and Hematite | 0.29 [0.04 + 0.17 + 0.08] (c) | 0 | 0.96 |
| Catalyst | Surface Area (m2·g−1) | Crystalline Phase (XRD) | Lewis Acidity (mmol·g−1) (a) | Brønsted Acidity (mmol·g−1) (a) | Basicity (mmol·g−1) (b) |
|---|---|---|---|---|---|
| Al/Mg/O_1_2 | 132 | Periclase MgO | 0.80 [059 + 0.03 + 0.17] (c) | 0 | 4.48 |
| AlOx/MgO_1_10 | 28 | Periclase MgO | 0.59 [0.40 + 0.13 + 0.06] (c) | 0 | 2.54 |
| Catalyst | M/Mg Molar Ratio | FU Conv (%) | FAL Sel (%) | MFU Sel. (%) | C-Loss (%) |
|---|---|---|---|---|---|
| MgO | - | 52 | 75 | 5 | 20 |
| Al/Mg/O_1_2 | 1:2 | 63 | 41 | 22 | 37 |
| Fe/Mg/O_1_2 | 1:2 | 93 | 5 | 92 | 3 |
| AlOx/MgO_1_10 | 1:10 | 40 | 76 | 5 | 19 |
| FeOx/MgO_1_10 | 1:10 | 89 | 5 | 88 | 7 |
| Entry | Catalyst | Fe/Si Molar Ratio | FU Conv (%) | FAL Sel (%) | MFU Sel. (%) | C-Loss (%) |
|---|---|---|---|---|---|---|
| 1 | MgO | - | 52 | 75 | 5 | 20 |
| 2 | Fe2O3 | - | 73 | 2 | 10 | 88 |
| 3 | SiO2 | - | 0 | 0 | 0 | 0 |
| 4 | FeOx/SiO2_1_10 | 1:10 | 19 | 1 | 25 | 74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucarelli, C.; Bonincontro, D.; Zhang, Y.; Grazia, L.; Renom-Carrasco, M.; Thieuleux, C.; Quadrelli, E.A.; Dimitratos, N.; Cavani, F.; Albonetti, S. Tandem Hydrogenation/Hydrogenolysis of Furfural to 2-Methylfuran over a Fe/Mg/O Catalyst: Structure–Activity Relationship. Catalysts 2019, 9, 895. https://doi.org/10.3390/catal9110895
Lucarelli C, Bonincontro D, Zhang Y, Grazia L, Renom-Carrasco M, Thieuleux C, Quadrelli EA, Dimitratos N, Cavani F, Albonetti S. Tandem Hydrogenation/Hydrogenolysis of Furfural to 2-Methylfuran over a Fe/Mg/O Catalyst: Structure–Activity Relationship. Catalysts. 2019; 9(11):895. https://doi.org/10.3390/catal9110895
Chicago/Turabian StyleLucarelli, Carlo, Danilo Bonincontro, Yu Zhang, Lorenzo Grazia, Marc Renom-Carrasco, Chloé Thieuleux, Elsje Alessandra Quadrelli, Nikolaos Dimitratos, Fabrizio Cavani, and Stefania Albonetti. 2019. "Tandem Hydrogenation/Hydrogenolysis of Furfural to 2-Methylfuran over a Fe/Mg/O Catalyst: Structure–Activity Relationship" Catalysts 9, no. 11: 895. https://doi.org/10.3390/catal9110895