
A Theoretical Study of the Oxygen Release Mechanisms of a Cu-Based Oxygen Carrier during Chemical Looping with Oxygen Uncoupling

Abstract
:1. Introduction
2. Results and Discussion
2.1. Calculations of Bulk CuO and O2
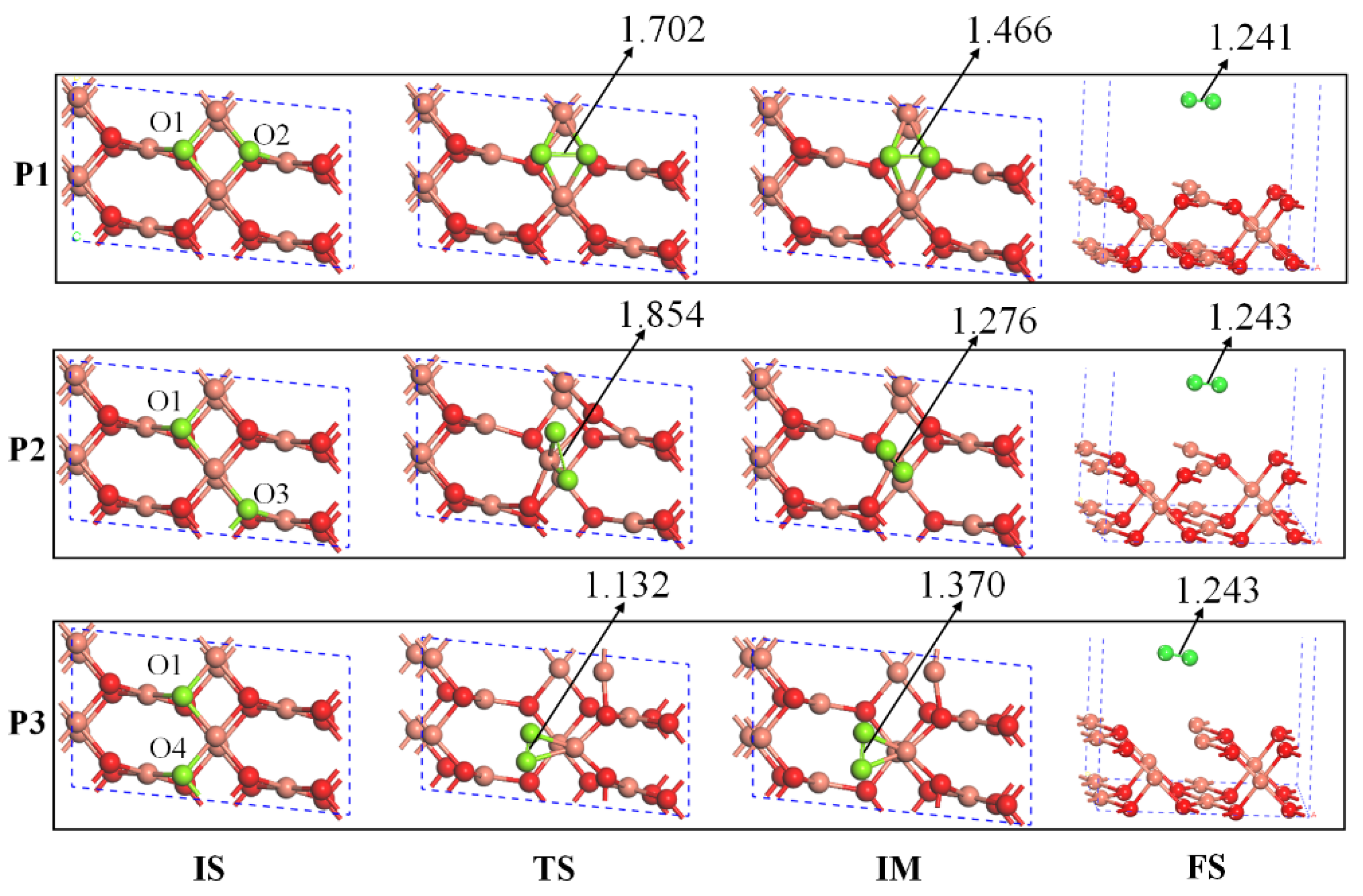
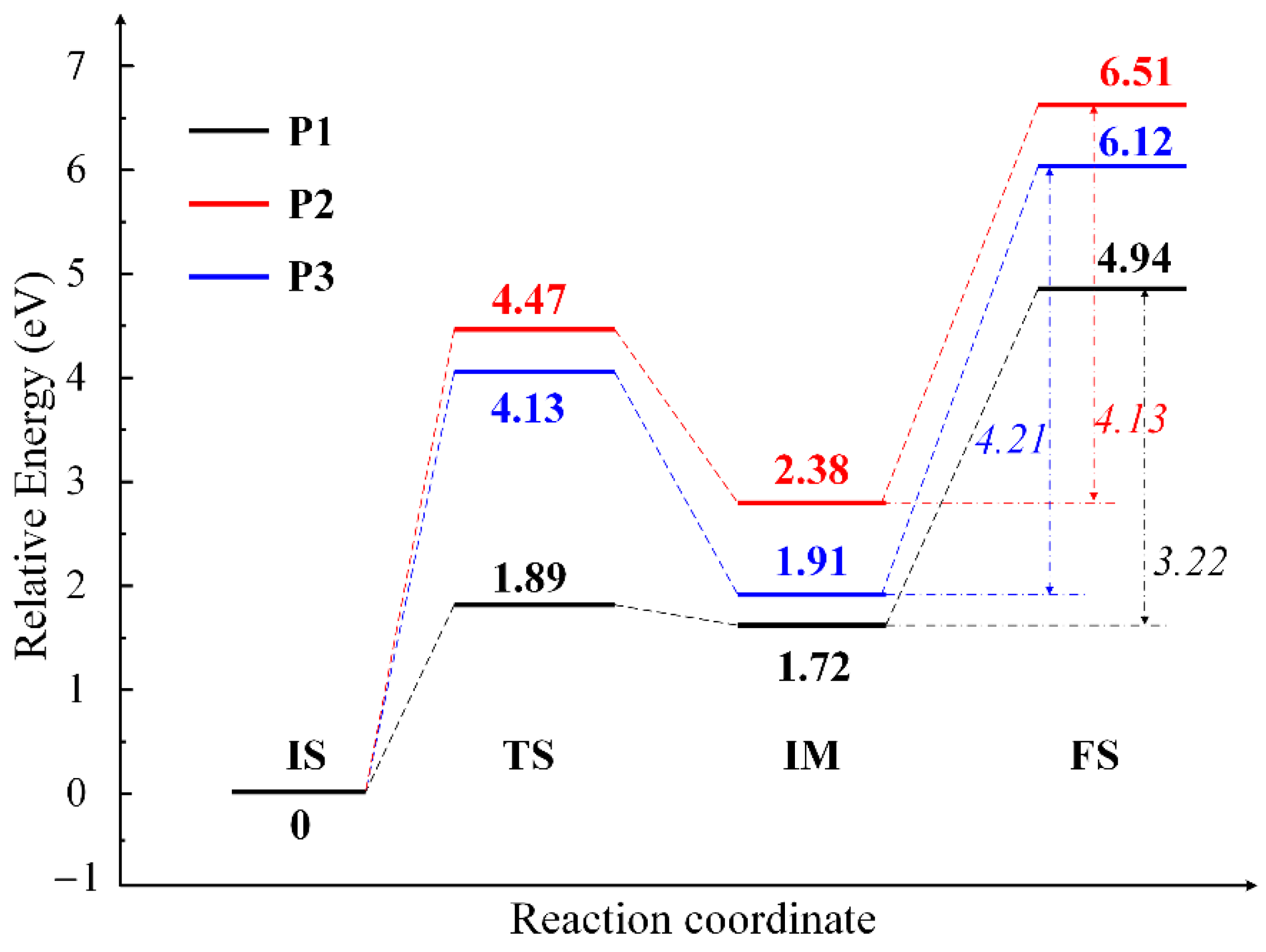
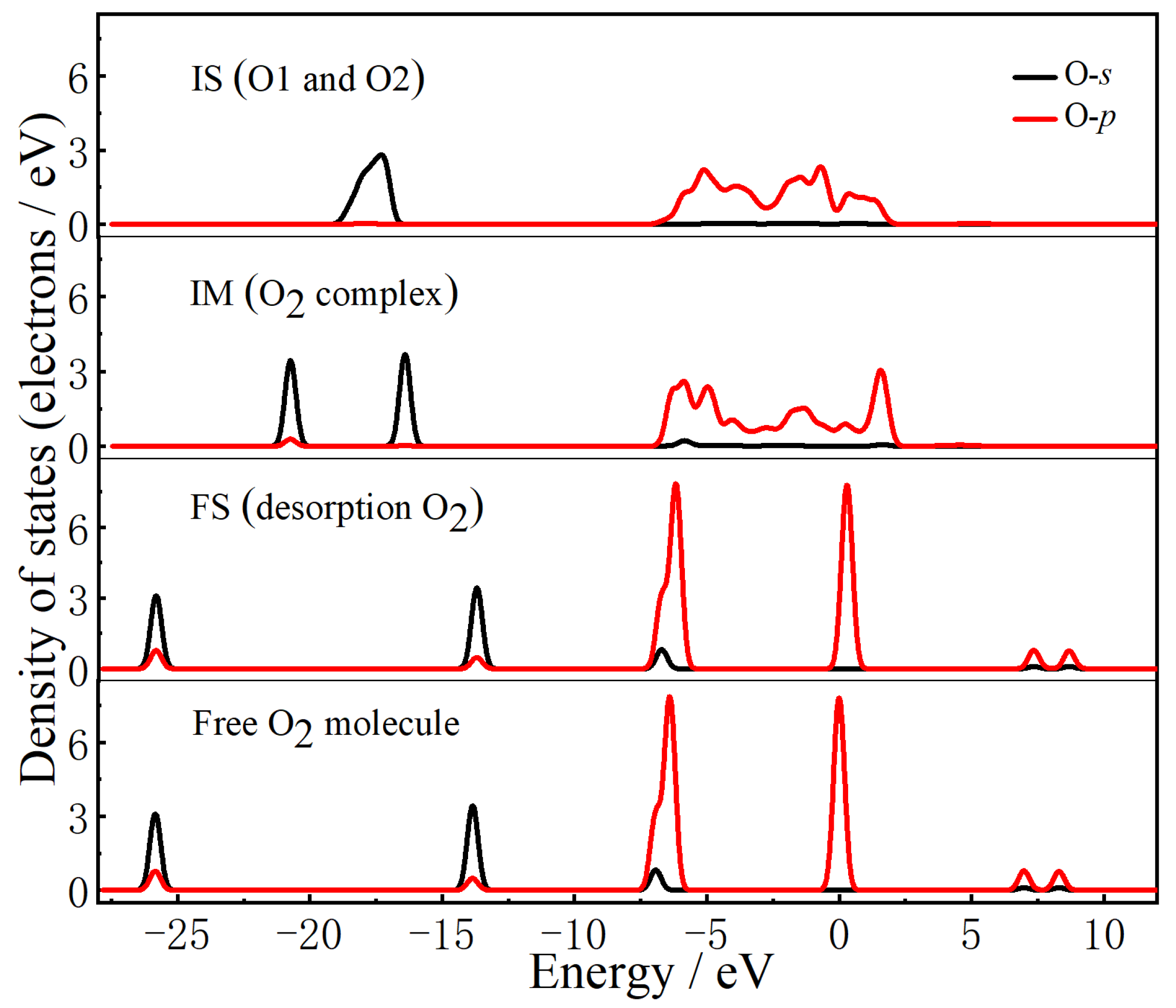
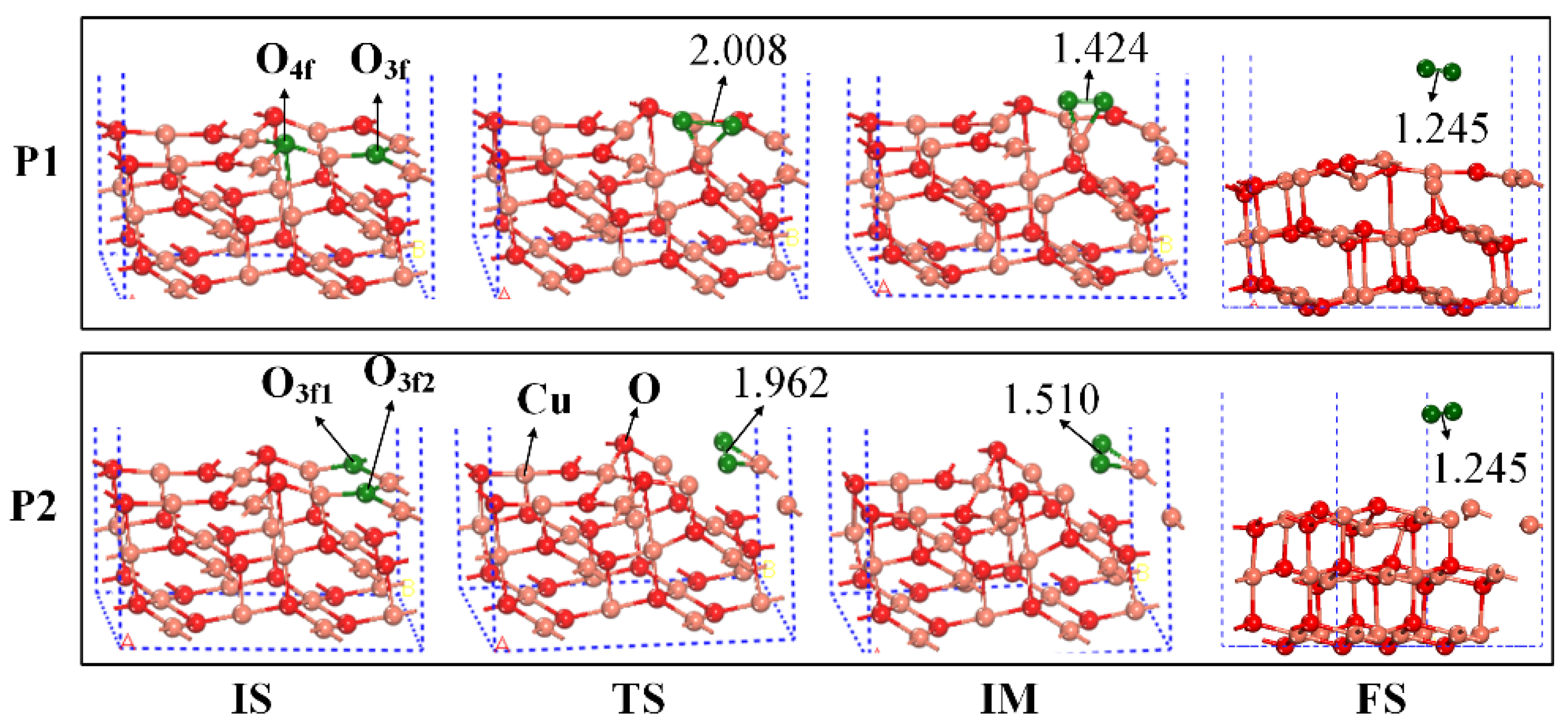
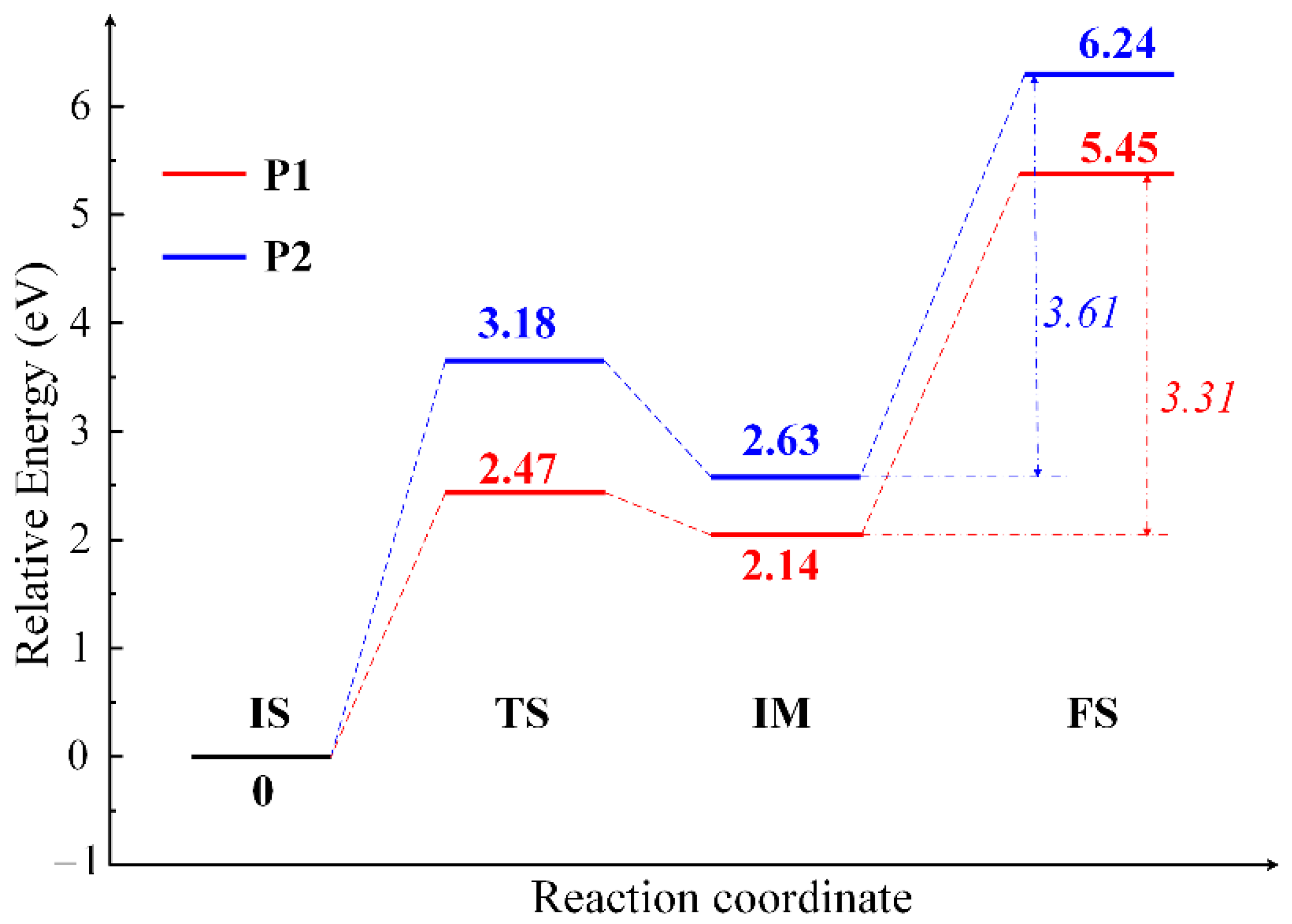
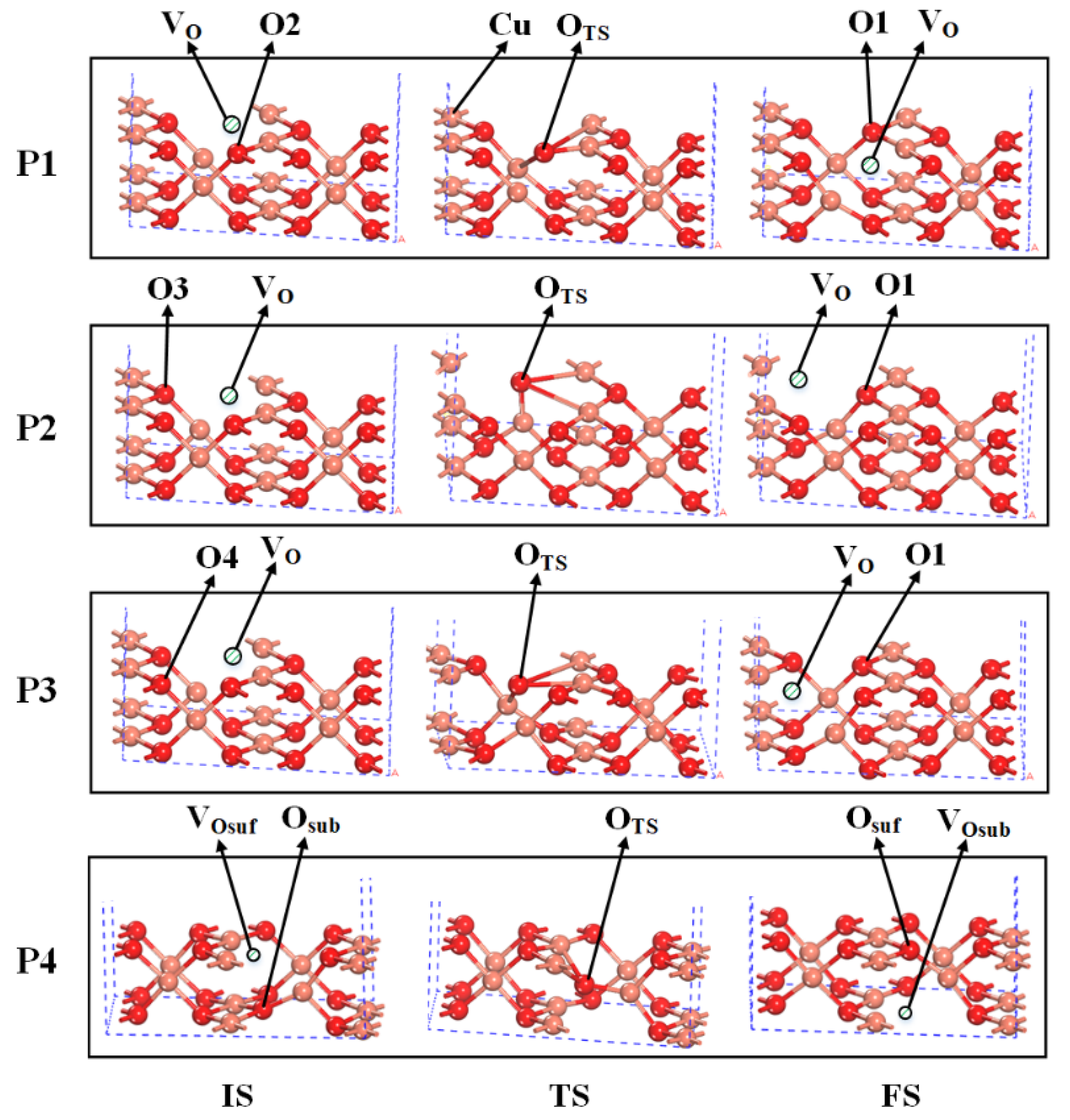
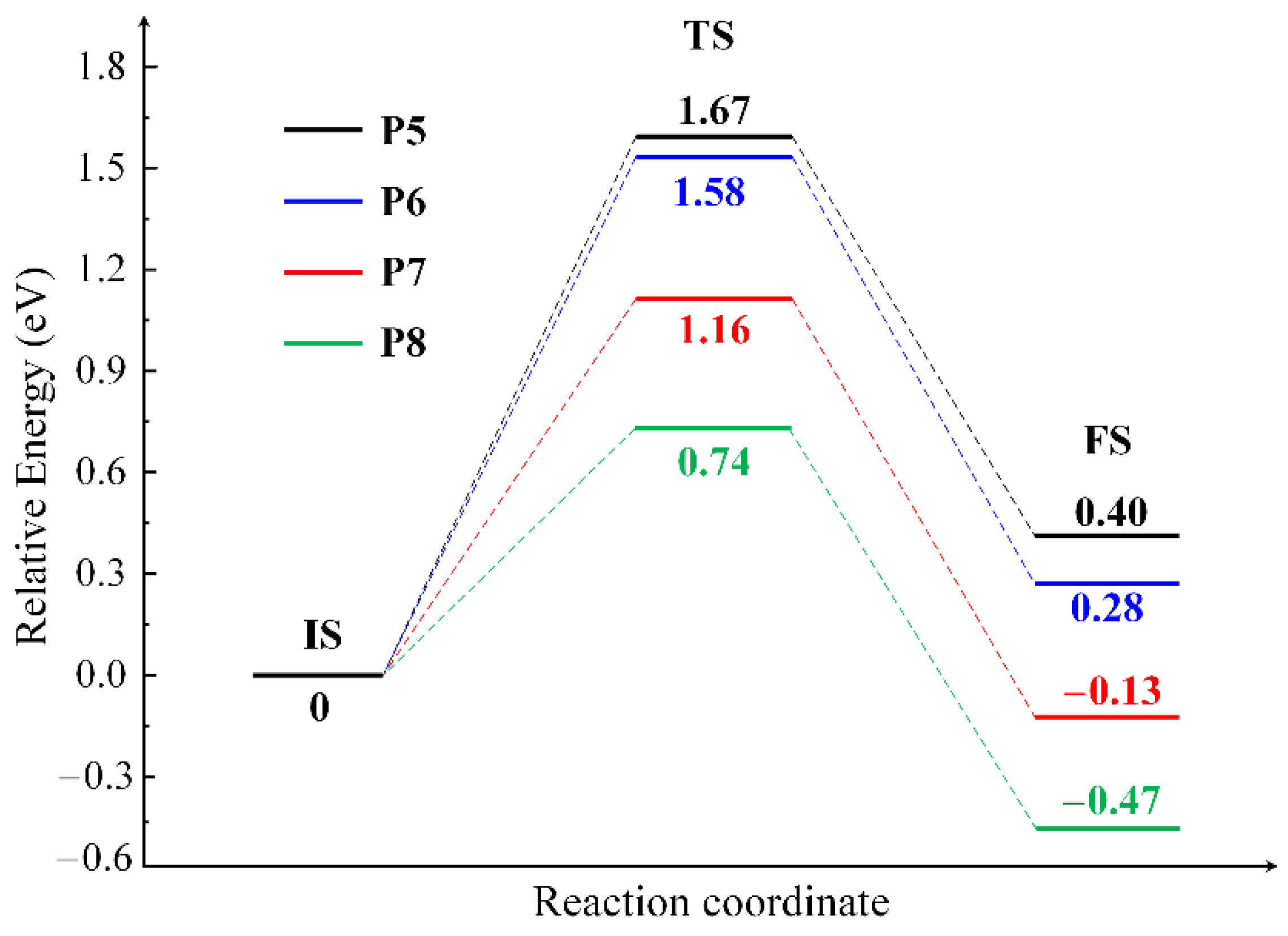
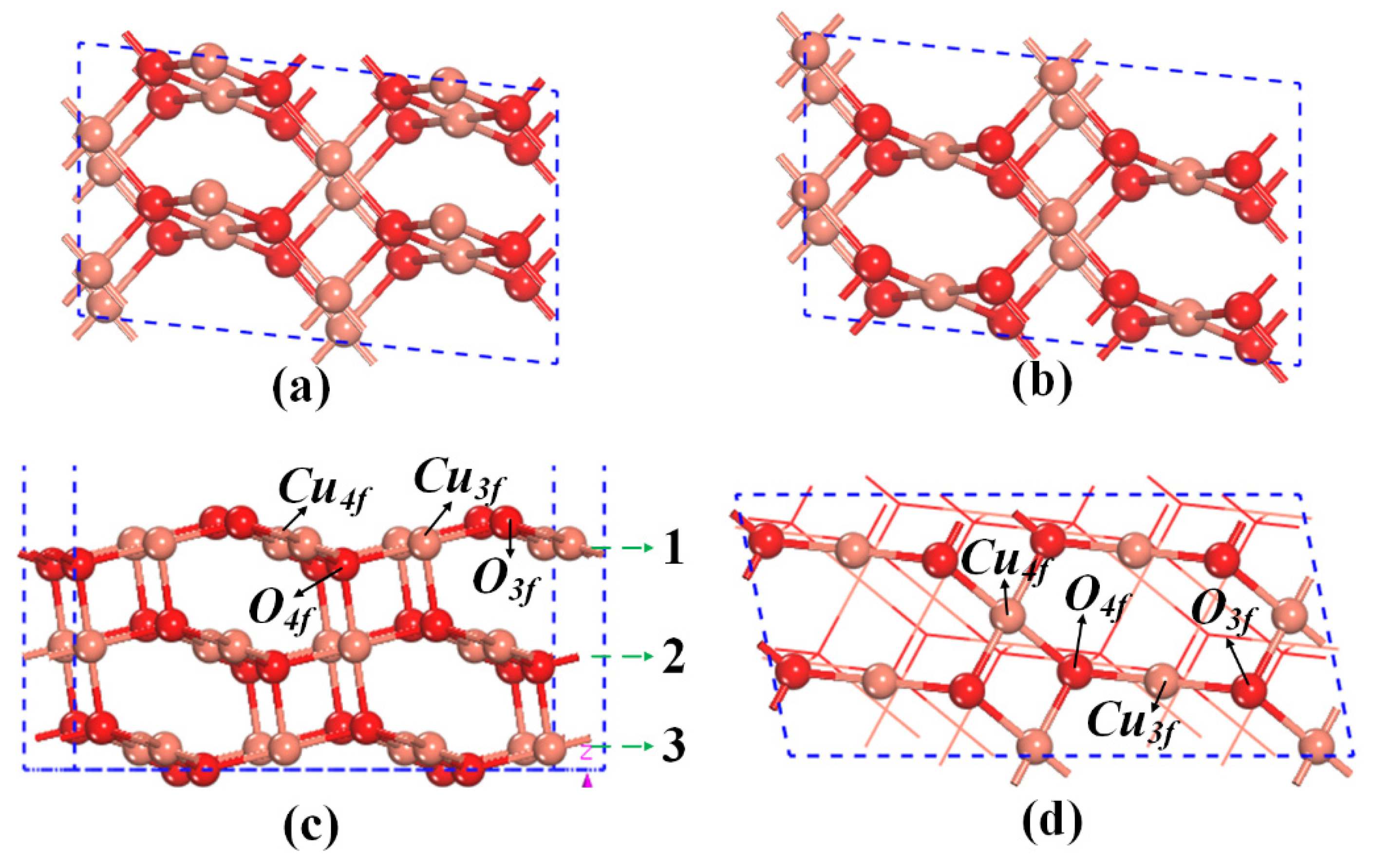
2.2. The Mechanisms of Oxygen Release from the CuO Surface
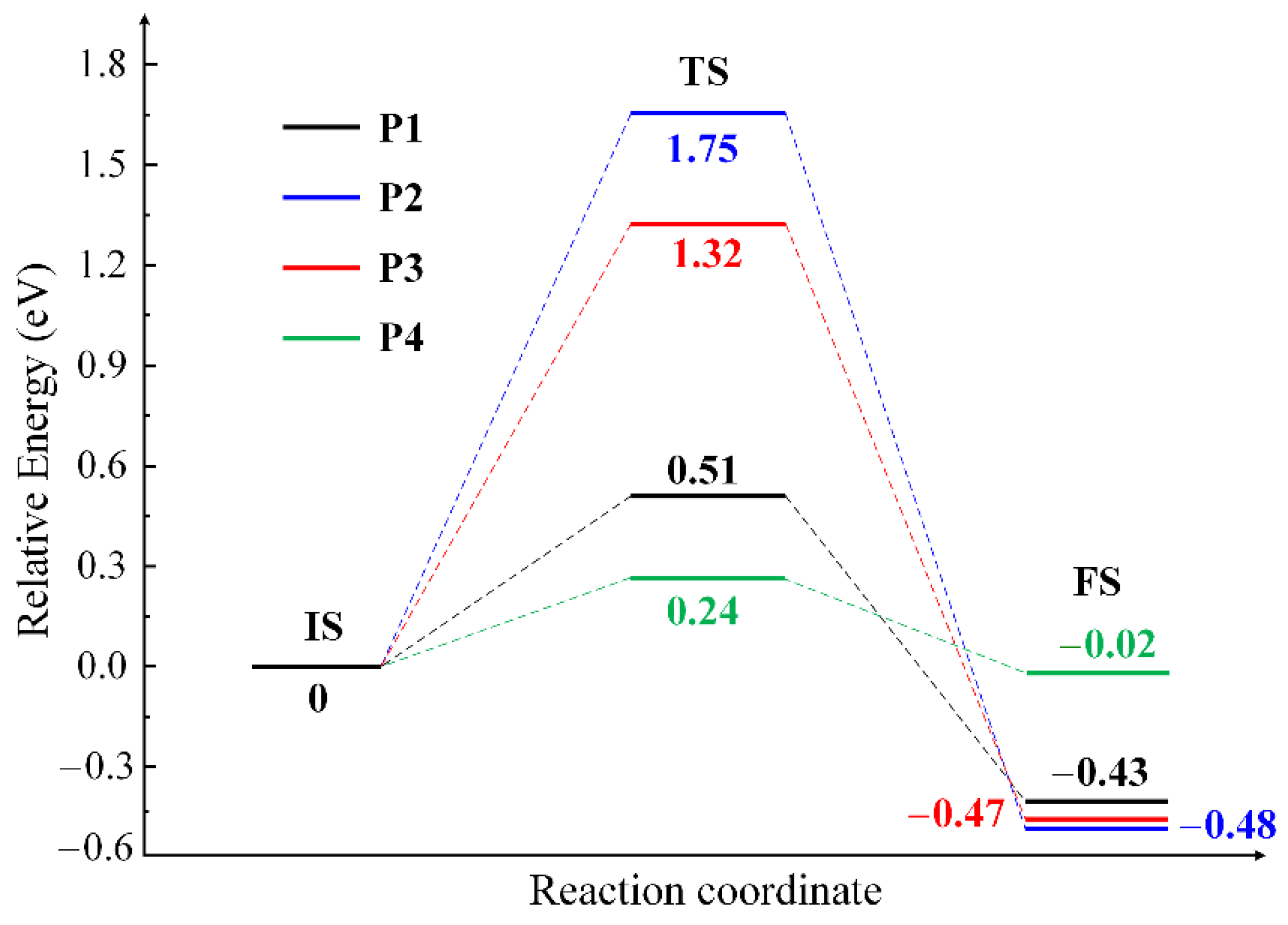
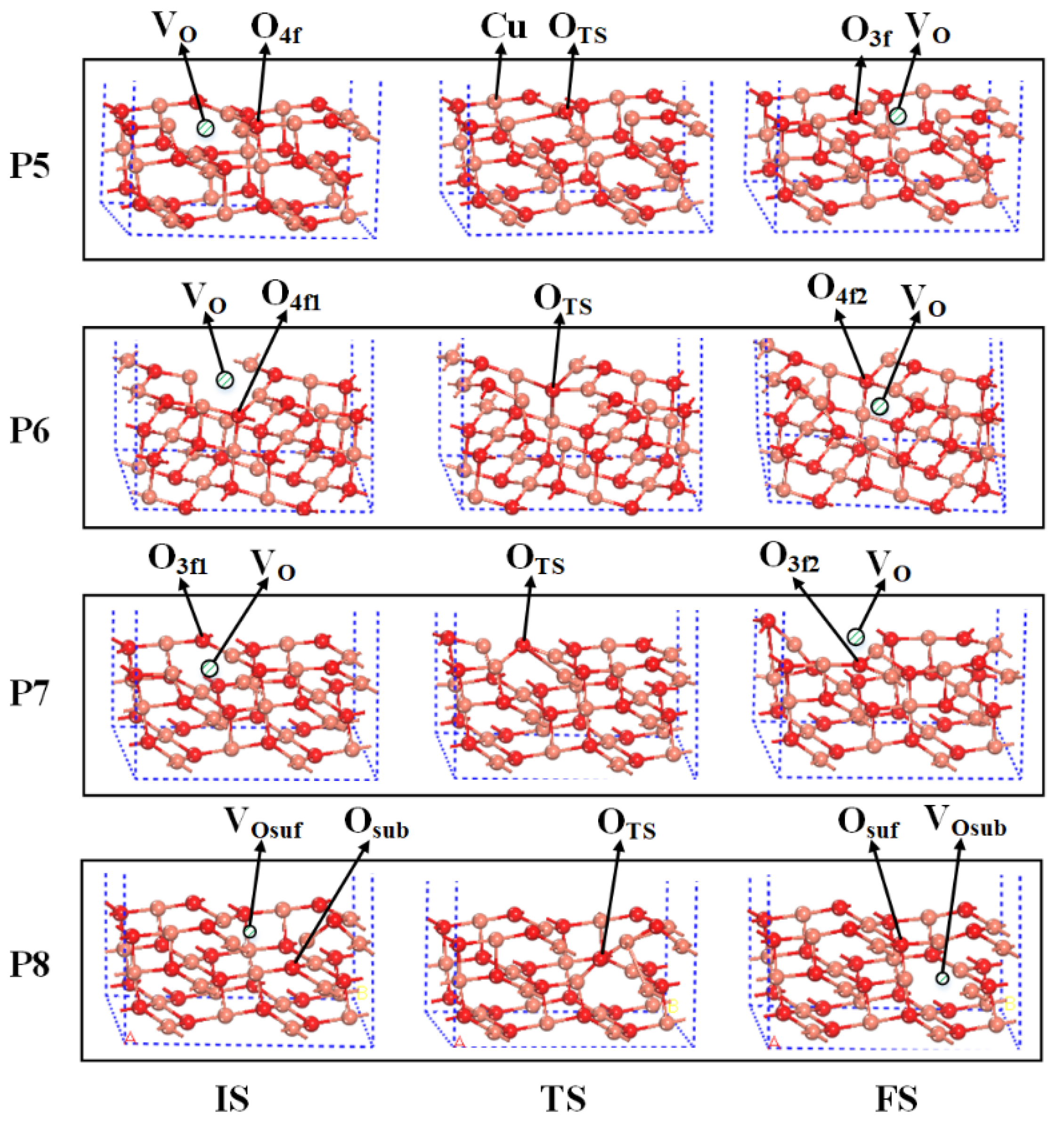
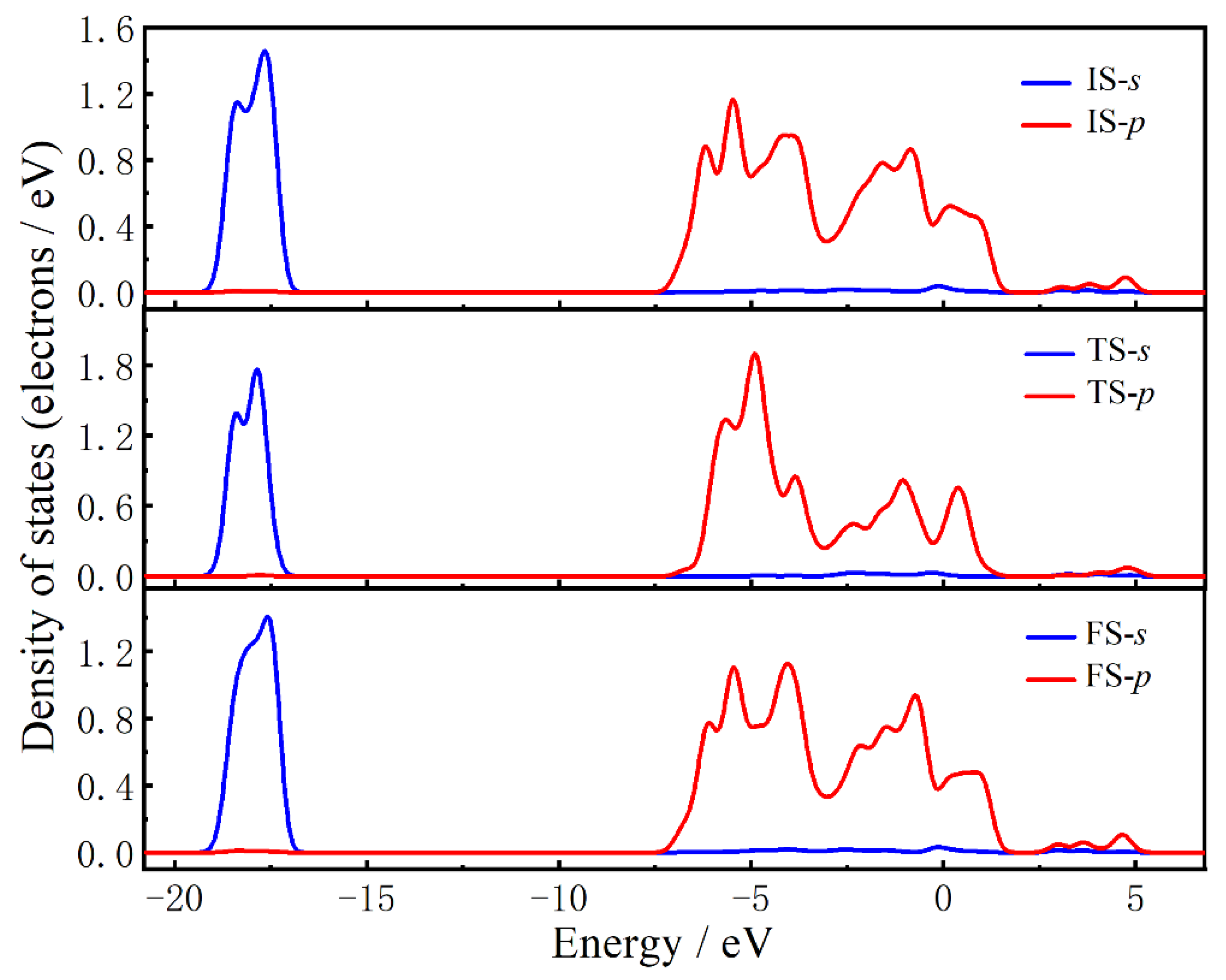
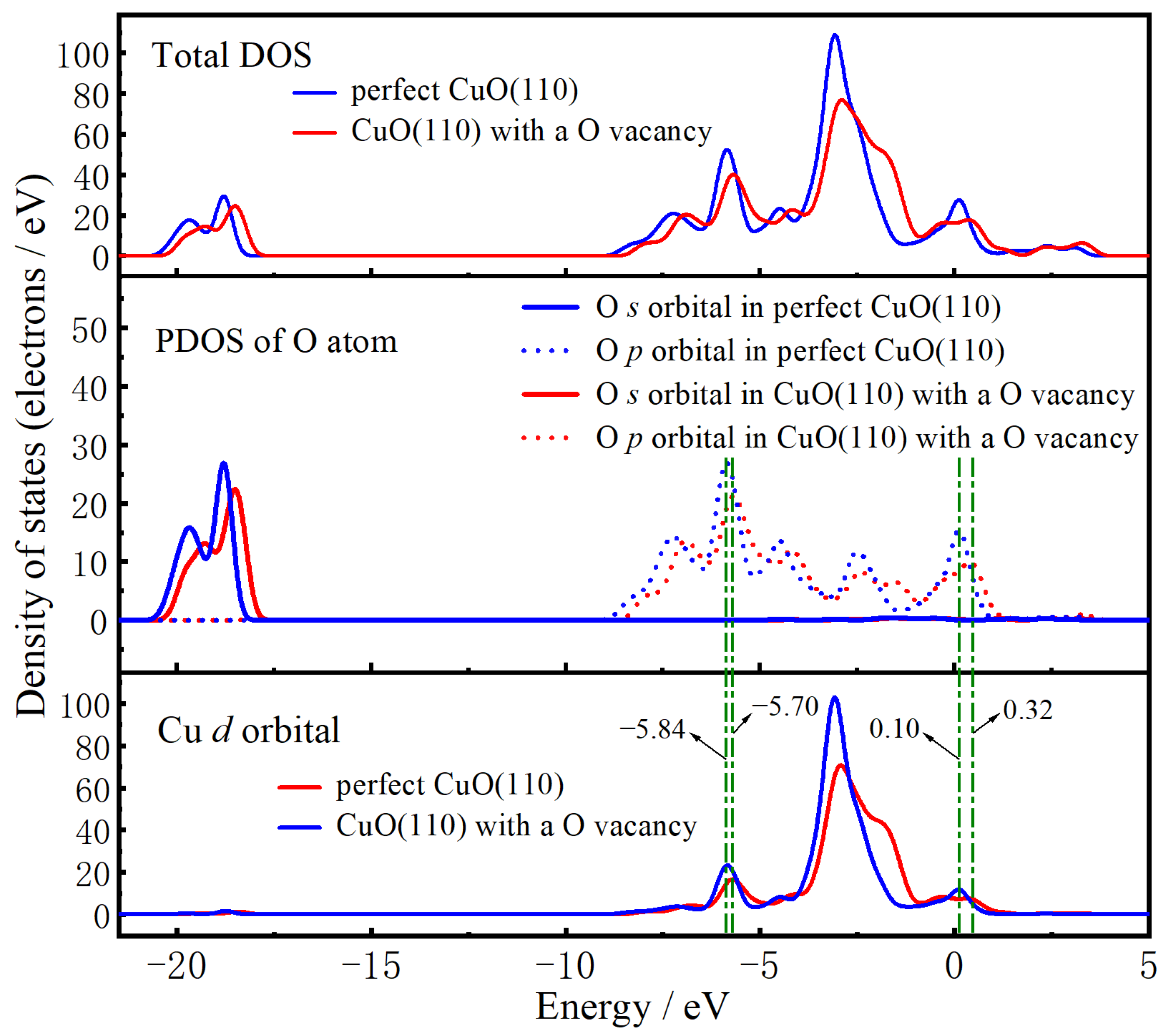
2.3. The O Anion Diffusion Mechanisms in the CuO
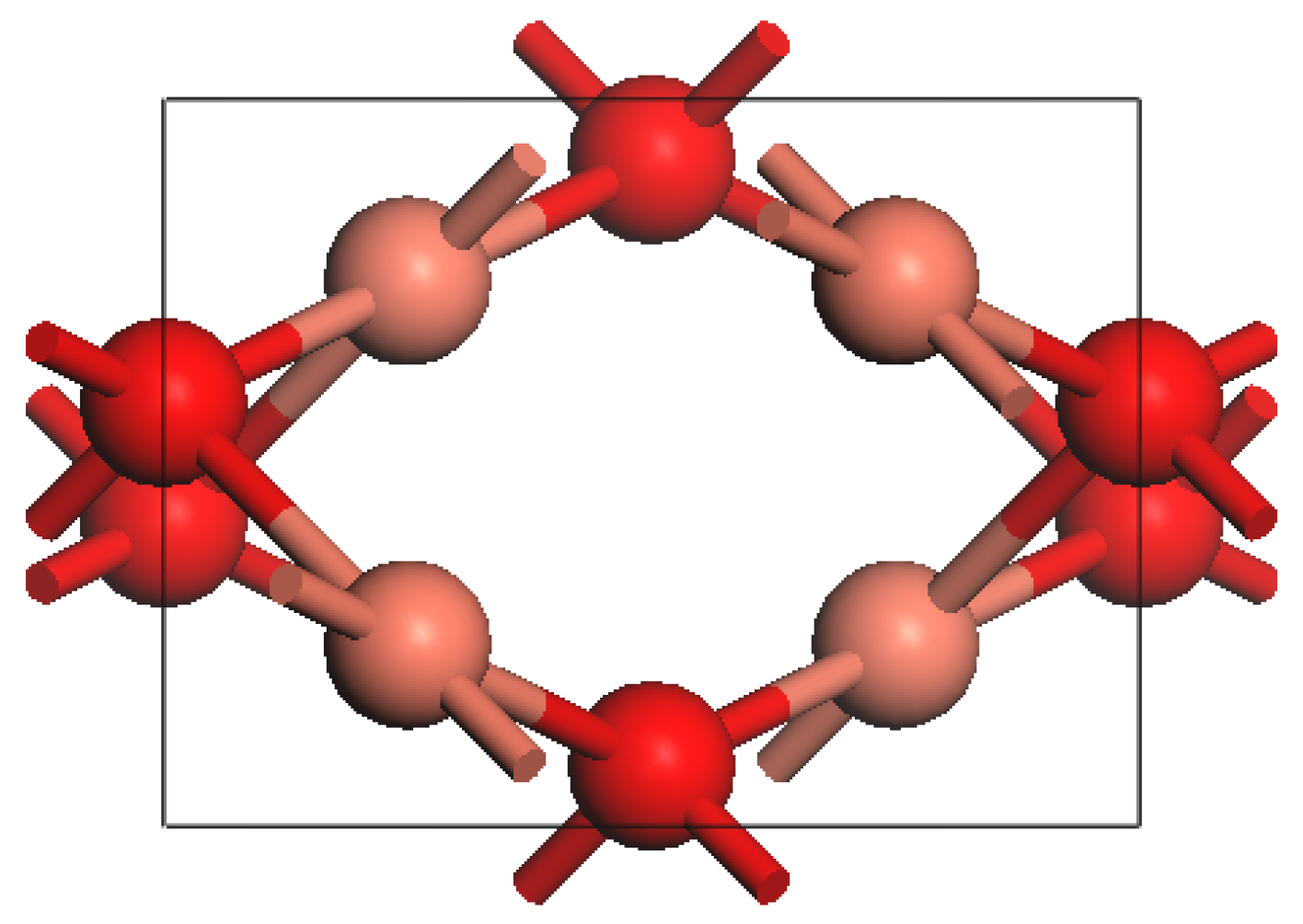
3. Calculation Method and Model
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abuelgasim, S.; Wang, W.; Abdalazeez, A. A brief review for chemical looping combustion as a promising CO2 capture technology: Fundamentals and progress. Sci. Total Environ. 2021, 764, 142892. [Google Scholar] [CrossRef]
- Wang, M.; Liu, J.; Hu, J.; Liu, F. O2–CO2 Mixed Gas Production Using a Zr-Doped Cu-Based Oxygen Carrier. Ind. Eng. Chem. Res. 2015, 54, 9805–9812. [Google Scholar] [CrossRef]
- Li, F.; Luo, S.; Sun, Z.; Bao, X.; Fan, L.-S. Role of metal oxide support in redox reactions of iron oxide for chemical looping applications: Experiments and density functional theory calculations. Energy Environ. Sci. 2011, 4, 3661–3667. [Google Scholar] [CrossRef]
- Hossain, M.M.; Lasa, H. Chemical-looping combustion (CLC) for inherent CO2 separations—A review. Chem. Eng. Sci. 2008, 63, 4433–4451. [Google Scholar] [CrossRef]
- Sarshar, Z.; Sun, Z.; Zhao, D.; Kaliaguine, S. Development of Sinter-Resistant Core-Shell LaMnxFe1-xO3@mSiO(2) Oxygen Carriers for Chemical Looping Combustion. Energy Fuels 2012, 26, 3091–3102. [Google Scholar] [CrossRef]
- Ishida, M.; Jin, H. A Novel Chemical-Looping Combustor without NOx Formation. Ind. Eng. Chem. Res. 1996, 35, 2469–2472. [Google Scholar] [CrossRef]
- Mattisson, T.; Lyngfelt, A.; Leion, H. Chemical-looping with oxygen uncoupling for combustion of solid fuels. Int. J. Greenh. Gas Control 2009, 3, 11–19. [Google Scholar] [CrossRef]
- Ahmad, A.; Al Mamun, M.A.; Al-Mamun, M.; Huque, S.; Ismail, M. LFO Perovskites as Oxygen Carriers for Chemical Looping Oxygen Uncoupling (CLOU). J. Therm. Anal. Calorim. 2021, 1–9. [Google Scholar] [CrossRef]
- Kuang, C.; Wang, S.; Lv, S.; Cai, J.; Luo, M.; Zhao, J. Comparison of metallic oxide, natural ore and synthetic oxygen carrier in chemical looping combustion process. Int. J. Hydrogen Energy 2021, 46, 18032–18041. [Google Scholar] [CrossRef]
- Ksepko, E. Examining SrCuO2 as an oxygen carrier for chemical looping combustion. J. Therm. Anal. Calorim. 2015, 122, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Ksepko, E.; Lysowski, R. Stable Mixed Fe-Mn Oxides Supported on ZrO2 Oxygen Carriers for Practical Utilization in CLC Processes. Catalysts 2021, 11, 1047. [Google Scholar] [CrossRef]
- Ksepko, E.; Lysowski, R. Reactivity Study of Bimetallic Fe-Mn Oxides with Addition of TiO2 for Chemical Looping Combustion Purposes. Catalysts 2021, 11, 1437. [Google Scholar] [CrossRef]
- Siriwardane, R.V.; Ksepko, E.; Tian, H.; Poston, J.; Simonyi, T.; Sciazko, M. Interaction of iron–copper mixed metal oxide oxygen carriers with simulated synthesis gas derived from steam gasification of coal. Appl. Energy 2013, 107, 111–123. [Google Scholar] [CrossRef]
- Adánez, J.; Abad, A. Chemical-looping combustion: Status and research needs. Proc. Combust. Inst. 2018, 37, 4303–4317. [Google Scholar] [CrossRef]
- Alalwan, H.A.; Cwiertny, D.M.; Grassian, V.H. Co3O4 nanoparticles as oxygen carriers for chemical looping combustion: A materials characterization approach to understanding oxygen carrier performance. Chem. Eng. J. 2017, 319, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Vos, Y.D.; Jacobs, M.; Voort, P.; Driessche, I.V.; An, V. Development of Stable Oxygen Carrier Materials for Chemical Looping Processes—A Review. Catalysts 2020, 10, 926. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, H.; Wei, Y.; Zheng, C. Self-assembly template combustion synthesis of a core–shell CuO@TiO2 –Al2O3 hierarchical structure as an oxygen carrier for the chemical-looping processes. Combust. Flame 2015, 162, 3030–3045. [Google Scholar] [CrossRef]
- Imtiaz, Q.; Kierzkowska, A.M.; Broda, M.; Mueller, C.R. Synthesis of Cu-Rich, Al2O3-Stabilized Oxygen Carriers Using a Coprecipitation Technique: Redox and Carbon Formation Characteristics. Environ. Sci. Technol. 2012, 46, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- De Diego, L.F.; García-Labiano, F.; Adánez, J.; Gayán, P.; Abad, A.; Corbella, B.M.; Palacios, J.M. Development of Cu-based oxygen carriers for chemical-looping combustion. Fuel 2004, 83, 1749–1757. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yu, Q.; Qin, Q. Reduction Kinetics of Cu-Based Oxygen Carriers for Chemical Looping Air Separation. Energy Fuels 2013, 27, 5466–5474. [Google Scholar] [CrossRef]
- Gayán, P.; Adánez-Rubio, I.; Abad, A.; Luis, F.; García-Labiano, F.; Adánez, J. Development of Cu-based oxygen carriers for Chemical-Looping with Oxygen Uncoupling (CLOU) process. Fuel 2011, 90, 226–238. [Google Scholar] [CrossRef]
- Li, Z. First-principles-based microkinetic rate equation theory for oxygen carrier reduction in chemical looping. Chem. Eng. Sci. 2021, 247, 117042. [Google Scholar] [CrossRef]
- Adánez-Rubio, I.; Abad, A.; Gayán, P.; De Diego, L.F.; García-Labiano, F.; Adánez, J. Biomass combustion with CO2 capture by chemical looping with oxygen uncoupling (CLOU). Fuel Process. Technol. 2014, 124, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Adánez-Rubio, I.; Abad, A.; Gayán, P.; De Diego, L.F.; García-Labiano, F.; Adánez, J. Performance of CLOU process in the combustion of different types of coal with CO2 capture. Int. J. Greenh. Gas Control 2013, 12, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Abad, A.; Adánez-Rubio, I.; Gayán, P.; García-Labiano, F.; Luis, F.; Adánez, J. Demonstration of chemical-looping with oxygen uncoupling (CLOU) process in a 1.5 kWth continuously operating unit using a Cu-based oxygen-carrier. Int. J. Greenh. Gas Control 2012, 6, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Cheng, Z.; Fan, J.A.; Kopechek, D.; Xu, D.; Deshpande, N.; Fan, L.-S. Nanostructure formation mechanism and ion diffusion in iron–titanium composite materials with chemical looping redox reactions. J. Mater. Chem. A 2015, 3, 11302–11312. [Google Scholar] [CrossRef]
- Su, M.A.; Cao, J.A.; Tian, X.A.; Zhang, Y.B.; Zhao, H.A. Mechanism and kinetics of Cu2O oxidation in chemical looping with oxygen uncoupling. Proc. Combust. Inst. 2019, 37, 4371–4378. [Google Scholar] [CrossRef]
- Li, F.; Sun, Z.; Luo, S.; Fan, L.S. Ionic diffusion in the oxidation of iron—Effect of support and its implications to chemical looping applications. Energy Environ. Sci. 2011, 4, 876–880. [Google Scholar] [CrossRef]
- Shafiefarhood, A.; Galinsky, N.; Huang, A.P.Y.; And, Y.C.; Li, A.P.F. Fe2O3@LaxSr1−xFeO3 Core–Shell Redox Catalyst for Methane Partial Oxidation. ChemCatChem 2014, 6, 790–799. [Google Scholar] [CrossRef]
- Neal, L.M.; Shafiefarhood, A.; Li, F. Dynamic Methane Partial Oxidation Using a Fe2O3@La0.8Sr0.2FeO3-δ Core–Shell Redox Catalyst in the Absence of Gaseous Oxygen. ACS Catal. 2014, 4, 3560–3569. [Google Scholar] [CrossRef]
- Cheng, Z.; Qin, L.; Guo, M.; Xu, M.; Fan, L.S. Oxygen vacancy promoted methane partial oxidation over iron oxide oxygen carriers in the chemical looping process. Phys. Chem. Chem. Phys. 2016, 18, 32418–32428. [Google Scholar] [CrossRef] [PubMed]
- Day, M.; Tachibana, S.; Bell, J.; Lijewski, M.; Beckner, V.; Cheng, R.K. A combined computational and experimental characterization of lean premixed turbulent low swirl laboratory flames I. Methane flames. Combust. Flame 2012, 159, 275–290. [Google Scholar] [CrossRef]
- Meng, Y. Water Gas Shift Reaction Activity on Fe (110): A DFT Study. Catalysts 2021, 12, 27. [Google Scholar]
- Li, H.; Shi, L.; Jin, C.; Ye, R.; Zhang, R. Co and Ni Incorporatedγ-Al2O3(110) Surface: A Density Functional Theory Study. Catalysts 2022, 12, 111. [Google Scholar] [CrossRef]
- Bruix, A.; Margraf, J.T.; Andersen, M.; Reuter, K. First-principles-based multiscale modelling of heterogeneous catalysis. Nat. Catal. 2019, 2, 659–670. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.W.; Bai, M.; Guo, W.P.; Cao, D.B.; Yang, Y.; Li, Y.W.; Wen, X.D. Prediction on morphologies and phase equilibrium diagram of iron oxides nanoparticles. Appl. Surf. Sci. 2019, 480, 478–486. [Google Scholar] [CrossRef]
- Bennett, J.W.; Huang, X.; Fang, Y.; Cwiertny, D.M.; Grassian, V.H.; Mason, S.E. Methane Dissociation on α-Fe2O3(0001) and Fe3O4(111) Surfaces: First-Principles Insights into Chemical Looping Combustion. J. Phys. Chem. C 2019, 123, 6450–6463. [Google Scholar] [CrossRef]
- Dong, C.; Sheng, S.; Qin, W.; Lu, Q.; Zhao, Y.; Wang, X.; Zhang, J. Density functional theory study on activity of α-Fe2O3 in chemical-looping combustion system. Appl. Surf. Sci. 2011, 257, 8647–8652. [Google Scholar] [CrossRef]
- Yuan, Y.; Dong, X.; Ricardez-Sandoval, L. Insights into Syngas Combustion on a Defective NiO Surface for Chemical Looping Combustion: Oxygen Migration and Vacancy Effects. J. Phys. Chem. C 2020, 124, 28359–28370. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Guo, X. Influence mechanism of supports on the reactivity of Ni-based oxygen carriers for chemical looping reforming: A DFT study. Fuel 2018, 229, 88–94. [Google Scholar] [CrossRef]
- Feng, Y.; Guo, X. Study of reaction mechanism of methane conversion over Ni-based oxygen carrier in chemical looping reforming. Fuel 2017, 210, 866–872. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Wei, Y.; Gui, J. Understanding CuO-support interaction in Cu-based oxygen carriers at a microcosmic level. Proc. Combust. Inst. 2017, 36, 4069–4077. [Google Scholar] [CrossRef]
- Wu, L.N.; Tian, Z.Y.; Kasmi, A.E.; Arshad, M.F.; Qin, W. Mechanistic study of the CO oxidation reaction on the CuO(111) surface during chemical looping combustion. Proc. Combust. Inst. 2021, 38, 5289–5297. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Guo, L.; Zheng, C. Decomposition mechanisms of Cu-based oxygen carriers for chemical looping with oxygen uncoupling based on density functional theory calculations. Combust. Flame 2015, 162, 1265–1274. [Google Scholar] [CrossRef]
- Zheng, C.; Cao, J.; Zhang, Y.; Zhao, H. Insight into the Oxidation Mechanism of a Cu-Based Oxygen Carrier (Cu→Cu2O→CuO) in Chemical Looping Combustion. Energy Fuels 2020, 34, 8718–8725. [Google Scholar] [CrossRef]
- Mishra, A.; Li, T.; Li, F.; Santiso, E.E. Oxygen Vacancy Creation Energy in Mn-Containing Perovskites: An Effective Indicator for Chemical Looping with Oxygen Uncoupling. Chem. Mater. 2018, 31, 689–698. [Google Scholar] [CrossRef]
- Xiang, W.; Liu, J.; Chang, M.; Zheng, C. The adsorption mechanism of elemental mercury on CuO(110) surface. Chem. Eng. J. 2012, 200-202, 91–96. [Google Scholar] [CrossRef]
- Moreno, J.L.V.; Padama, A.A.B.; Kasai, H. A density functional theory-based study on the dissociation of NO on a CuO(110) surface. CrystEngComm 2014, 16, 2260–2265. [Google Scholar] [CrossRef]
- Chang, Y.; Zeng, H.C. Controlled Synthesis and Self-Assembly of Single-Crystalline CuO Nanorods and Nanoribbons. Cryst. Growth Des. 2004, 4, 397–402. [Google Scholar] [CrossRef]
- Bari, R.H.; Patil, S.B.; Bari, A.R. Spray-pyrolized nanostructured CuO thin films for H2S gas sensor. Int. Nano Lett. 2013, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, J.; Li, W.; Zhu, C.; Mao, L.-F.; Huang, Q. First-Principles investigation on the behavior of Pt single and triple atoms supported on monolayer CuO (110) in CO oxidation. Appl. Surf. Sci. 2021, 564, 150435. [Google Scholar] [CrossRef]
- Massarotti, V.; Capsoni, D.; Bini, M.; Altomare, A.; Moliterni, A.G.G. X-ray powder diffraction ab initio structure solution of materials from solid state synthesis: The copper oxide case. Z. Kristallogr. 1998, 213, 259–265. [Google Scholar] [CrossRef]
- Åsbrink, S.; Norrby, L.-J. A refinement of the crystal structure of copper(II) oxide with a discussion of some exceptional e.s.d.’s. Acta Crystallogr. B 1970, 26, 8–15. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, H.; Zheng, H.; Ling, L.; Li, Z.; Wang, B. Adsorption and dissociation of O2 on the Cu2O(1 1 1) surface: Thermochemistry, reaction barrier. Appl. Surf. Sci. 2011, 257, 4787–4794. [Google Scholar] [CrossRef]
- Chadda, D.; Ford, J.D.; Fahim, M.A. Chemical energy storage by the reaction cycle cupric oxide/cuprous oxide. Int. J. Energy Res. 1989, 13, 63–73. [Google Scholar] [CrossRef]
- Eyring, E.M.; Konya, G.; Lighty, J.S.; Sahir, A.H.; Sarofim, A.F.; Whitty, K. Chemical Looping with Copper Oxide as Carrier and Coal as Fuel. Oil Gas Sci. Technol. 2011, 66, 209–221. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Wang, M.; Liu, J.; Shen, F.; Cheng, H.; Dai, J.; Long, Y. Theoretical study of stability and reaction mechanism of CuO supported on ZrO2 during chemical looping combustion. Appl. Surf. Sci. 2016, 367, 485–492. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Ef (eV) | Ed (eV) | Et (eV) | Ea (eV) | |
|---|---|---|---|---|---|
| CuO(110) surface | P1 | 1.89 | 3.22 | 4.94 | – |
| P2 | 4.47 | 4.13 | 6.51 | – | |
| P3 | 4.13 | 4.21 | 6.12 | – | |
| CuO(111) surface | P1 | 2.47 | 3.31 | 5.45 | – |
| P2 | 3.18 | 3.61 | 6.24 | – | |
| CuO(111) surface in literature | P1 | 2.3 | 3.5 | 5.2 | |
| P2 | 3.0 | 3.1 | 6.0 | ||
| Experiment values | – | – | – | – | 3.26 |
| – | – | – | – | 3.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhang, S.; Xia, M.; Wang, M. A Theoretical Study of the Oxygen Release Mechanisms of a Cu-Based Oxygen Carrier during Chemical Looping with Oxygen Uncoupling. Catalysts 2022, 12, 332. https://doi.org/10.3390/catal12030332
Wang M, Zhang S, Xia M, Wang M. A Theoretical Study of the Oxygen Release Mechanisms of a Cu-Based Oxygen Carrier during Chemical Looping with Oxygen Uncoupling. Catalysts. 2022; 12(3):332. https://doi.org/10.3390/catal12030332
Chicago/Turabian StyleWang, Minjun, Shixiong Zhang, Ming Xia, and Mengke Wang. 2022. "A Theoretical Study of the Oxygen Release Mechanisms of a Cu-Based Oxygen Carrier during Chemical Looping with Oxygen Uncoupling" Catalysts 12, no. 3: 332. https://doi.org/10.3390/catal12030332