
Uncovering the Mechanism of the Hydrogen Poisoning on Ru Nanoparticles via Density Functional Theory Calculations

 ,
,
Abstract
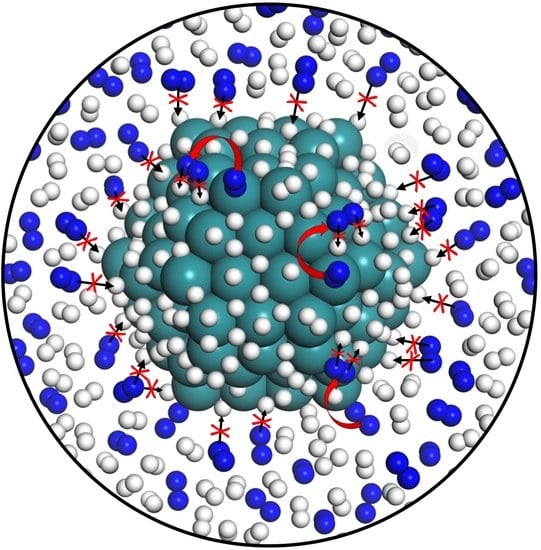
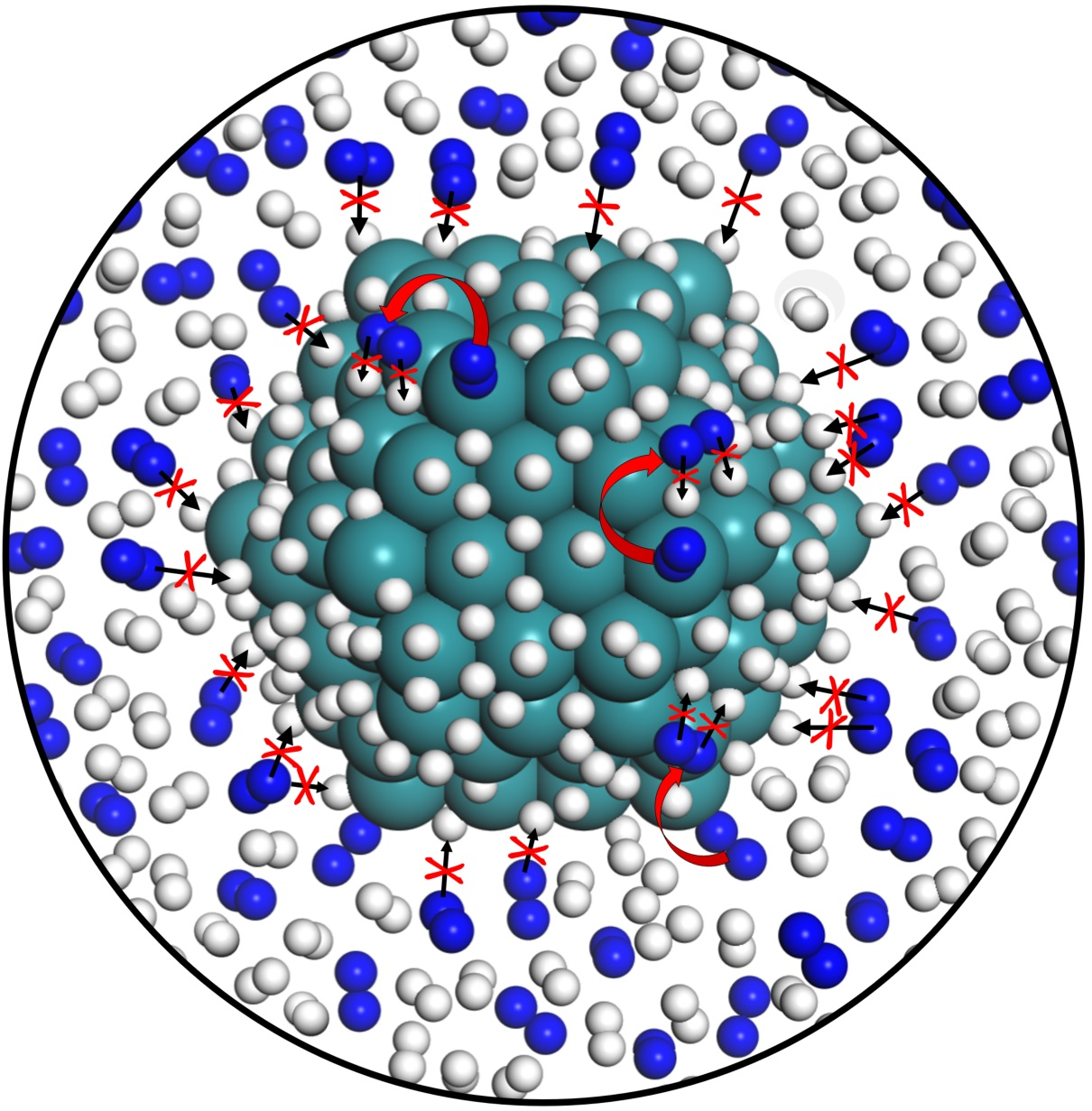
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
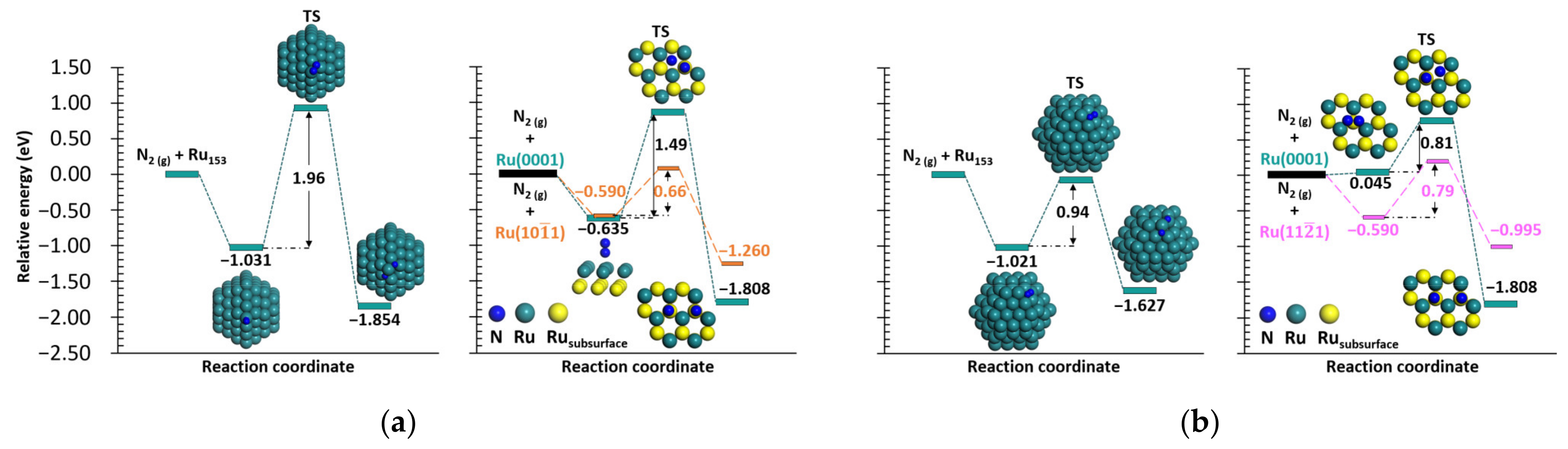
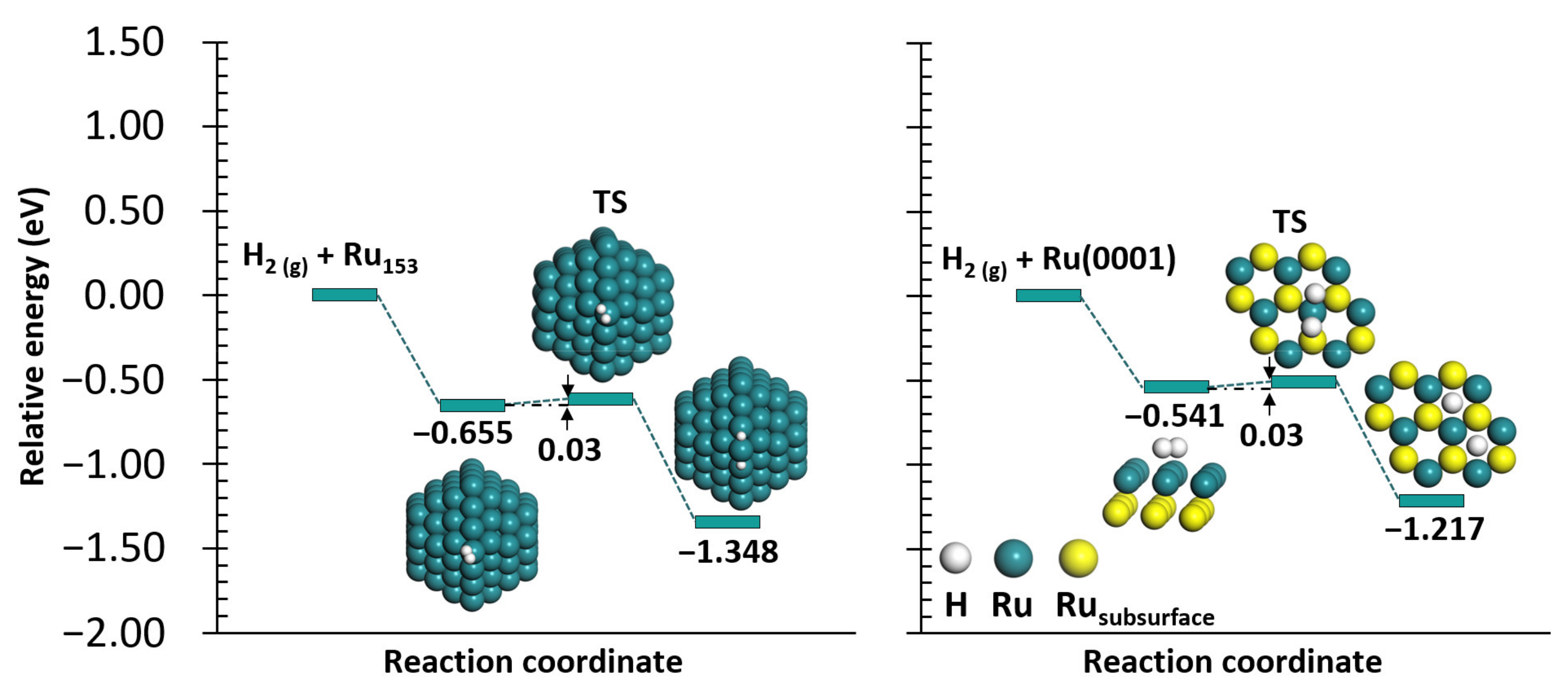
2.1. N2 and H2 Dissociation
2.1.1. N2 Dissociation
2.1.2. H2 Dissociation
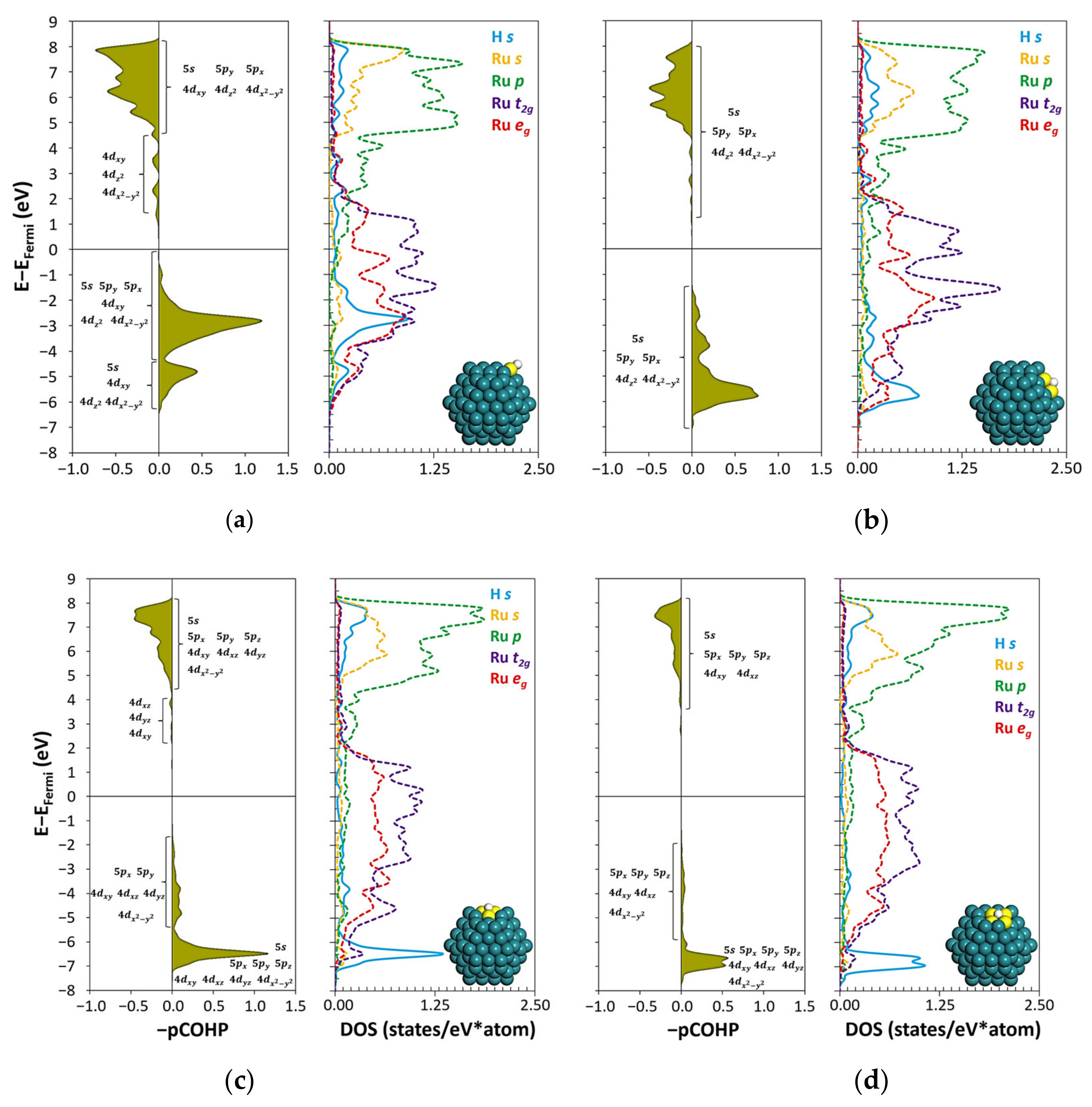
2.2. H Atom Binding
3. Discussion
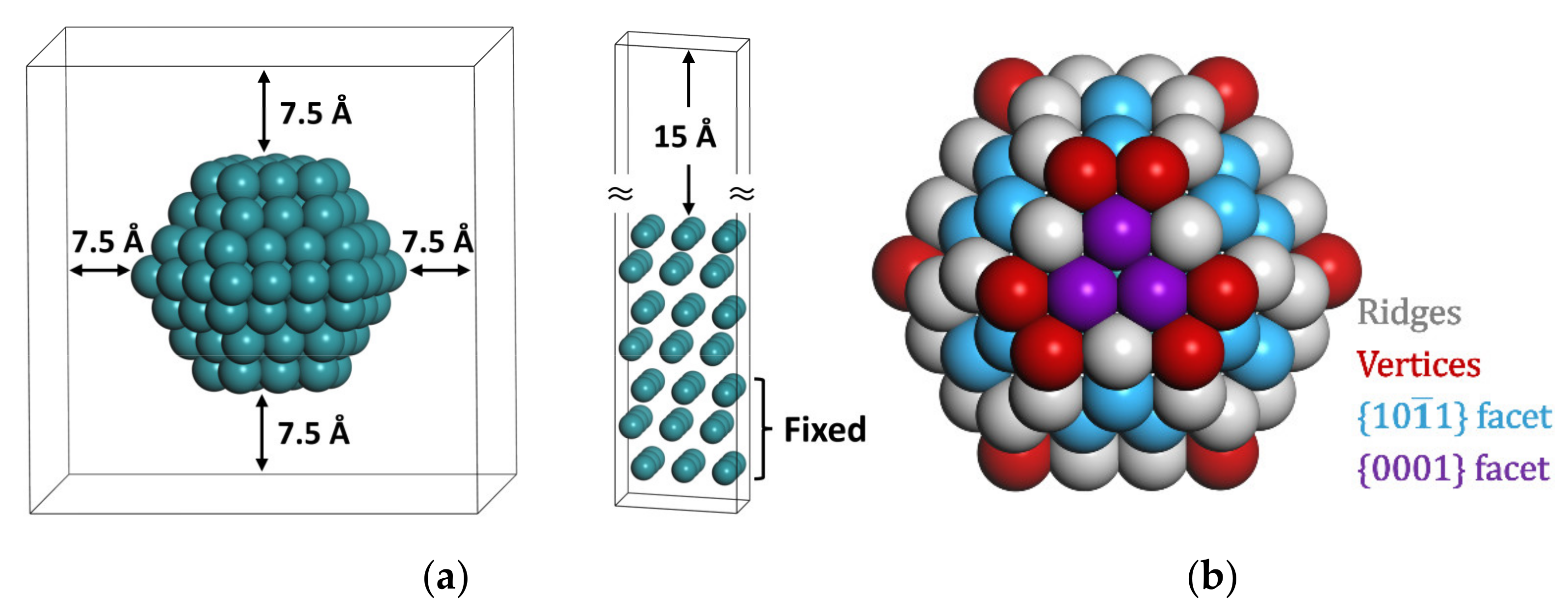
4. Computational Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ogura, Y.; Sato, K.; Miyahara, S.; Kawano, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Hosokawa, S.; Nagaoka, K. Efficient ammonia synthesis over a Ru/La0.5Ce0.5O1.75 catalyst pre-reduced at high temperature. Chem. Sci. 2018, 9, 2230–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valera-Medina, A.; Amer-Hatem, F.; Azad, A.K.; Dedoussi, I.C.; de Joannon, M.; Fernandes, R.X.; Glarborg, P.; Hashemi, H.; He, X.; Mashruk, S.; et al. Review on Ammonia as a Potential Fuel: From Synthesis to Economics. Energy Fuels 2021, 35, 6964–7029. [Google Scholar] [CrossRef]
- Sato, K.; Imamura, K.; Kawano, Y.; Miyahara, S.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. A low-crystalline ruthenium nano-layer supported on praseodymium oxide as an active catalyst for ammonia synthesis. Chem. Sci. 2017, 8, 674–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Miyahara, S.; Ogura, Y.; Tsujimaru, K.; Wada, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. Surface Dynamics for Creating Highly Active Ru Sites for Ammonia Synthesis: Accumulation of a Low-Crystalline, Oxygen-Deficient Nanofraction. ACS Sustain. Chem. Eng. 2020, 8, 2726–2734. [Google Scholar] [CrossRef]
- Siporin, S.; Davis, R. Use of kinetic models to explore the role of base promoters on Ru/MgO ammonia synthesis catalysts. J. Catal. 2004, 225, 359–368. [Google Scholar] [CrossRef]
- Murata, S. Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis: 2. A lanthanide oxide-promoted Ru/Al2O3 catalyst. J. Catal. 1992, 136, 118–125. [Google Scholar] [CrossRef]
- Aika, K.; Kumasaka, M.; Oma, T.; Kato, O.; Matsuda, H.; Watanabe, N.; Yamazaki, K.; Ozaki, A.; Onishi, T. Support and promoter effect of ruthenium catalyst. III. Kinetics of ammonia synthesis over various Ru catalysts. Appl. Catal. 1986, 28, 57–68. [Google Scholar] [CrossRef]
- Aika, K. Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis: 3. A magnesia-supported ruthenium catalyst. J. Catal. 1992, 136, 126–140. [Google Scholar] [CrossRef]
- Szmigiel, D.; Bielawa, H.; Kurtz, M.; Hinrichsen, O.; Muhler, M.; Raróg, W.; Jodzis, S.; Kowalczyk, Z.; Znak, L.; Zieliński, J. The Kinetics of Ammonia Synthesis over Ruthenium-Based Catalysts: The Role of Barium and Cesium. J. Catal. 2002, 205, 205–212. [Google Scholar] [CrossRef]
- Raróg, W.; Kowalczyk, Z.; Sentek, J.; Składanowski, D.; Zieliński, J. Effect of K, Cs and Ba on the kinetics of NH3 synthesis over carbon-based ruthenium catalysts. Catal. Lett. 2000, 68, 163–168. [Google Scholar] [CrossRef]
- Kitano, M.; Inoue, Y.; Ishikawa, H.; Yamagata, K.; Nakao, T.; Tada, T.; Matsuishi, S.; Yokoyama, T.; Hara, M.; Hosono, H. Essential role of hydride ion in ruthenium-based ammonia synthesis catalysts. Chem. Sci. 2016, 7, 4036–4043. [Google Scholar] [CrossRef] [Green Version]
- Üner, D.; Aslan, M.Y. Using spilled over hydrogen in NH3 synthesis over supported Ru catalysts. Catal. Today 2016, 272, 49–57. [Google Scholar] [CrossRef]
- Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M.; Ertl, G. Ruthenium catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics. Appl. Catal. A Gen. 1997, 151, 443–460. [Google Scholar] [CrossRef]
- Hara, M.; Kitano, M.; Hosono, H. Ru-Loaded C12A7:e− Electride as a Catalyst for Ammonia Synthesis. ACS Catal. 2017, 7, 2313–2324. [Google Scholar] [CrossRef]
- Ghuman, K.K.; Tozaki, K.; Sadakiyo, M.; Kitano, S.; Oyabe, T.; Yamauchi, M. Tailoring widely used ammonia synthesis catalysts for H and N poisoning resistance. Phys. Chem. Chem. Phys. 2019, 21, 5117–5122. [Google Scholar] [CrossRef]
- Zhang, C.J.; Lynch, M.; Hu, P. A density functional theory study of stepwise addition reactions in ammonia synthesis on Ru(0001). Surf. Sci. 2002, 496, 221–230. [Google Scholar] [CrossRef]
- Truflandier, L.A.; Del Rosal, I.; Chaudret, B.; Poteau, R.; Gerber, I.C. Where does Hydrogen Adsorb on Ru Nanoparticles? A Powerful Joint 2H MAS-NMR/DFT Approach. ChemPhysChem 2009, 10, 2939–2942. [Google Scholar] [CrossRef]
- Mak, C.H.; George, S.M. Surface diffusion of hydrogen on Ru(001): Transition state theory calculations. Chem. Phys. Lett. 1987, 135, 381–386. [Google Scholar] [CrossRef]
- Kristinsdóttir, L.; Skúlason, E. A systematic DFT study of hydrogen diffusion on transition metal surfaces. Surf. Sci. 2012, 606, 1400–1404. [Google Scholar] [CrossRef]
- Del Rosal, I.; Mercy, M.; Gerber, I.C.; Poteau, R. Ligand-Field Theory-Based Analysis of the Adsorption Properties of Ruthenium Nanoparticles. ACS Nano 2013, 7, 9823–9835. [Google Scholar] [CrossRef]
- Ge, G.-X.; Yan, H.-X.; Jing, Q.; Luo, Y.-H. Theoretical Study of Hydrogen Adsorption on Ruthenium Clusters. J. Clust. Sci. 2011, 22, 473–489. [Google Scholar] [CrossRef] [Green Version]
- Cusinato, L.; Martínez-Prieto, L.M.; Chaudret, B.; del Rosal, I.; Poteau, R. Theoretical characterization of the surface composition of ruthenium nanoparticles in equilibrium with syngas. Nanoscale 2016, 8, 10974–10992. [Google Scholar] [CrossRef] [PubMed]
- Rivera Rocabado, D.S.; Noguchi, T.G.; Hayashi, S.; Maeda, N.; Yamauchi, M.; Ishimoto, T. Adsorption States of N2/H2 Activated on Ru Nanoparticles Uncovered by Modulation–Excitation Infrared Spectroscopy and Density Functional Theory Calculations. ACS Nano 2021, 15, 20079–20086. [Google Scholar] [CrossRef]
- Dean, J.; Taylor, M.G.; Mpourmpakis, G. Unfolding Adsorption on Metal Nanoparticles: Connecting Stability with Catalysis. Sci. Adv. 2019, 5, eaax5101. [Google Scholar] [CrossRef] [Green Version]
- Dahl, S.; Tornqvist, E.; Chorkendorff, I. Dissociative Adsorption of N2 on Ru: A Surface Reaction Totally Dominated by Steps. J. Catal. 2000, 192, 381–390. [Google Scholar] [CrossRef]
- Dahl, S.; Logadottir, A.; Egeberg, R.C.; Larsen, J.H.; Chorkendorff, I.; Törnqvist, E.; Nørskov, J.K. Role of Steps in N2 Activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814–1817. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Jansen, A.P.J.; van Santen, R.A. Active Sites for N2 Dissociation on Ruthenium. J. Phys. Chem. C 2008, 112, 17768–17771. [Google Scholar] [CrossRef]
- Herron, J.A.; Tonelli, S.; Mavrikakis, M. Atomic and Molecular Adsorption on Ru(0001). Surf. Sci. 2013, 614, 64–74. [Google Scholar] [CrossRef]
- Mortensen, J.J.; Morikawa, Y.; Hammer, B.; Nørskov, J.K. Density Functional Calculations of N2 Adsorption and Dissociation on a Ru(0001) Surface. J. Catal. 1997, 169, 85–92. [Google Scholar] [CrossRef]
- De Paola, R.A.; Hoffmann, F.M.; Heskett, D.; Plummer, E.W. Adsorption of Molecular Nitrogen on Clean and Modified Ru(001) Surfaces: The Role of σ Bonding. Phys. Rev. B Condens. Matter 1987, 35, 4236–4249. [Google Scholar] [CrossRef]
- Anton, A.B.; Avery, N.R.; Toby, B.H.; Weinberg, W.H. The Chemisorption of Nitrogen on the (001) Surface of Ruthenium. J. Electron Spectros. Relat. Phenomena 1983, 29, 181–186. [Google Scholar] [CrossRef]
- Ishikawa, A.; Doi, T.; Nakai, H. Catalytic Performance of Ru, Os, and Rh Nanoparticles for Ammonia Synthesis: A Density Functional Theory Analysis. J. Catal. 2018, 357, 213–222. [Google Scholar] [CrossRef]
- Kuganathan, N.; Hosono, H.; Shluger, A.L.; Sushko, P.V. Enhanced N2 Dissociation on Ru-Loaded Inorganic Electride. J. Am. Chem. Soc. 2014, 136, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Seets, D.C.; Wheeler, M.C.; Mullins, C.B. Thermal desorption of hydrogen from atomic nitrogen precovered Ru(001). J. Chem. Phys. 1995, 103, 10399–10400. [Google Scholar] [CrossRef]
- Casey-Stevens, C.A.; Lambie, S.G.; Ruffman, C.; Skúlason, E.; Garden, A.L. Geometric and Electronic Effects Contributing to N2 Dissociation Barriers on a Range of Active Sites on Ru Nanoparticles. J. Phys. Chem. C 2019, 123, 30458–30466. [Google Scholar] [CrossRef]
- Pozzo, M.; Alfè, D. Hydrogen dissociation and diffusion on transition metal (=Ti, Zr, V, Fe, Ru, Co, Rh, Ni, Pd, Cu, Ag)-doped Mg(0001) surfaces. Int. J. Hydrogen Energy 2009, 34, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Onwudinanti, C.; Tranca, I.; Morgan, T.; Tao, S. Tin, The Enabler—Hydrogen Diffusion into Ruthenium. Nanomaterials 2019, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Thiel, P.A.; Weinberg, W.H. Adsite symmetry and vibrational structure of NO and H2 co-adsorbed on the Ru(001) surface. J. Chem. Phys. 1980, 73, 4081–4085. [Google Scholar] [CrossRef]
- Urabe, K. Activation of nitrogen by alkali metal-promoted transition metal VI. Hydrogen effect on isotopic equilibration of nitrogen and rate-determining step of ammonia synthesis on potassium-promoted ruthenium catalysts. J. Catal. 1976, 42, 197–204. [Google Scholar] [CrossRef]
- Aika, K. Role of alkali promoter in ammonia synthesis over ruthenium catalysts—Effect on reaction mechanism. Catal. Today 2017, 286, 14–20. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B—Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Manz, T.A. Introducing DDEC6 atomic population analysis: Part 3. Comprehensive method to compute bond orders. RSC Adv. 2017, 7, 45552–45581. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Dronskowski, R.; Bloechl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera Rocabado, D.S.; Aizawa, M.; Noguchi, T.G.; Yamauchi, M.; Ishimoto, T. Uncovering the Mechanism of the Hydrogen Poisoning on Ru Nanoparticles via Density Functional Theory Calculations. Catalysts 2022, 12, 331. https://doi.org/10.3390/catal12030331
Rivera Rocabado DS, Aizawa M, Noguchi TG, Yamauchi M, Ishimoto T. Uncovering the Mechanism of the Hydrogen Poisoning on Ru Nanoparticles via Density Functional Theory Calculations. Catalysts. 2022; 12(3):331. https://doi.org/10.3390/catal12030331
Chicago/Turabian StyleRivera Rocabado, David S., Mika Aizawa, Tomohiro G. Noguchi, Miho Yamauchi, and Takayoshi Ishimoto. 2022. "Uncovering the Mechanism of the Hydrogen Poisoning on Ru Nanoparticles via Density Functional Theory Calculations" Catalysts 12, no. 3: 331. https://doi.org/10.3390/catal12030331