
Influence of the Support on Propene Oxidation over Gold Catalysts

Abstract
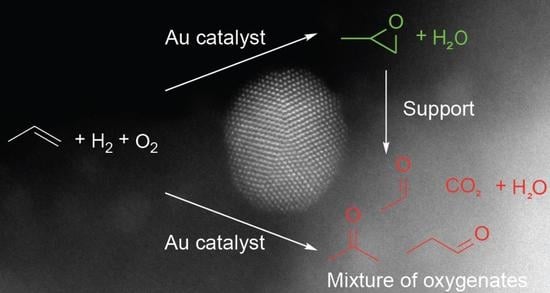
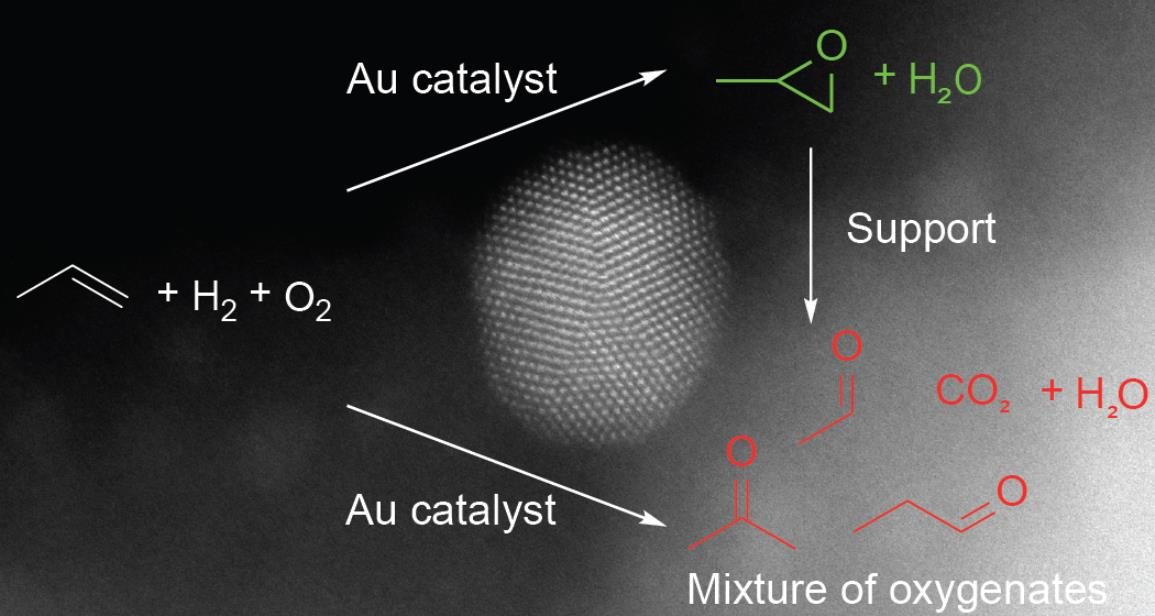
:
1. Introduction
2. Results and Discussion
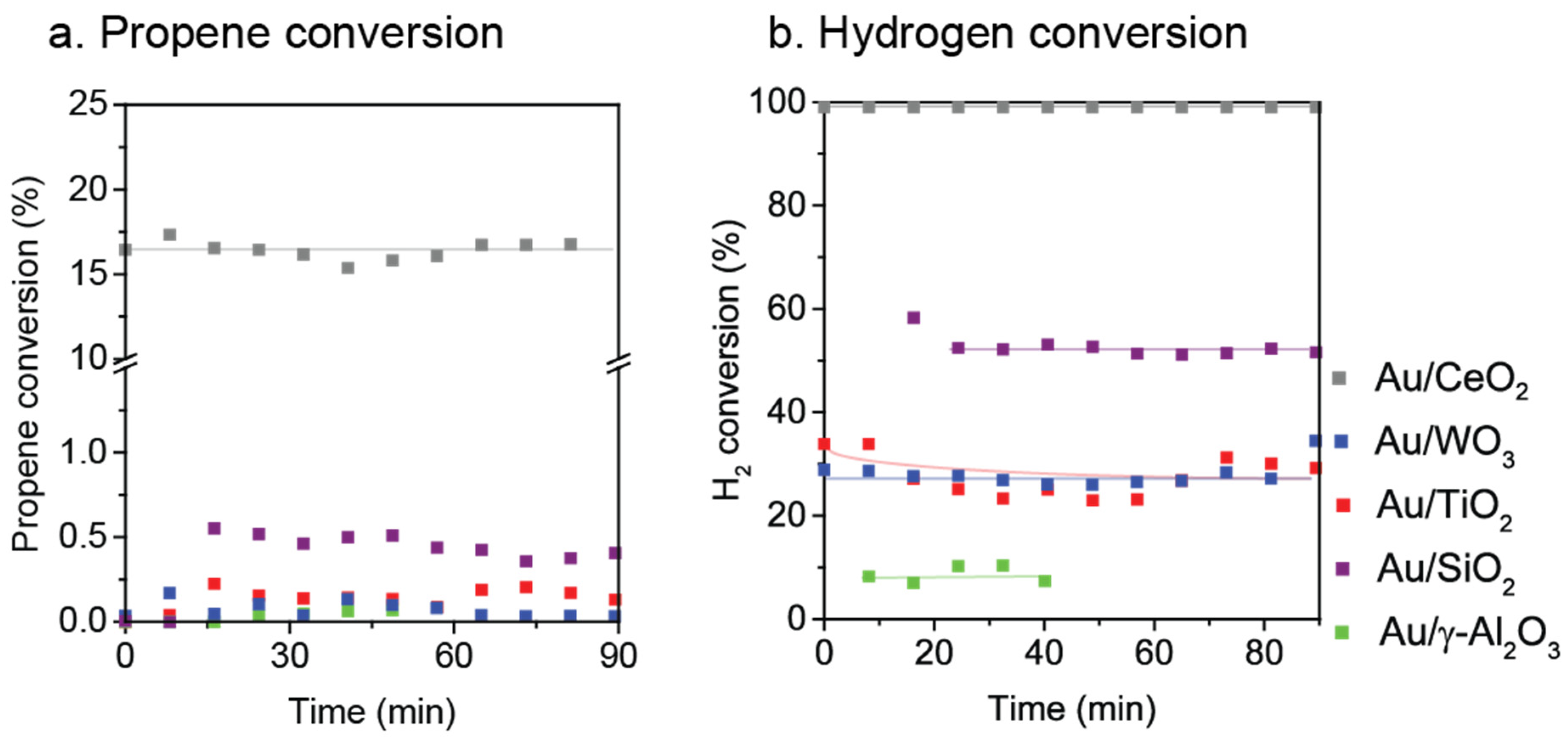
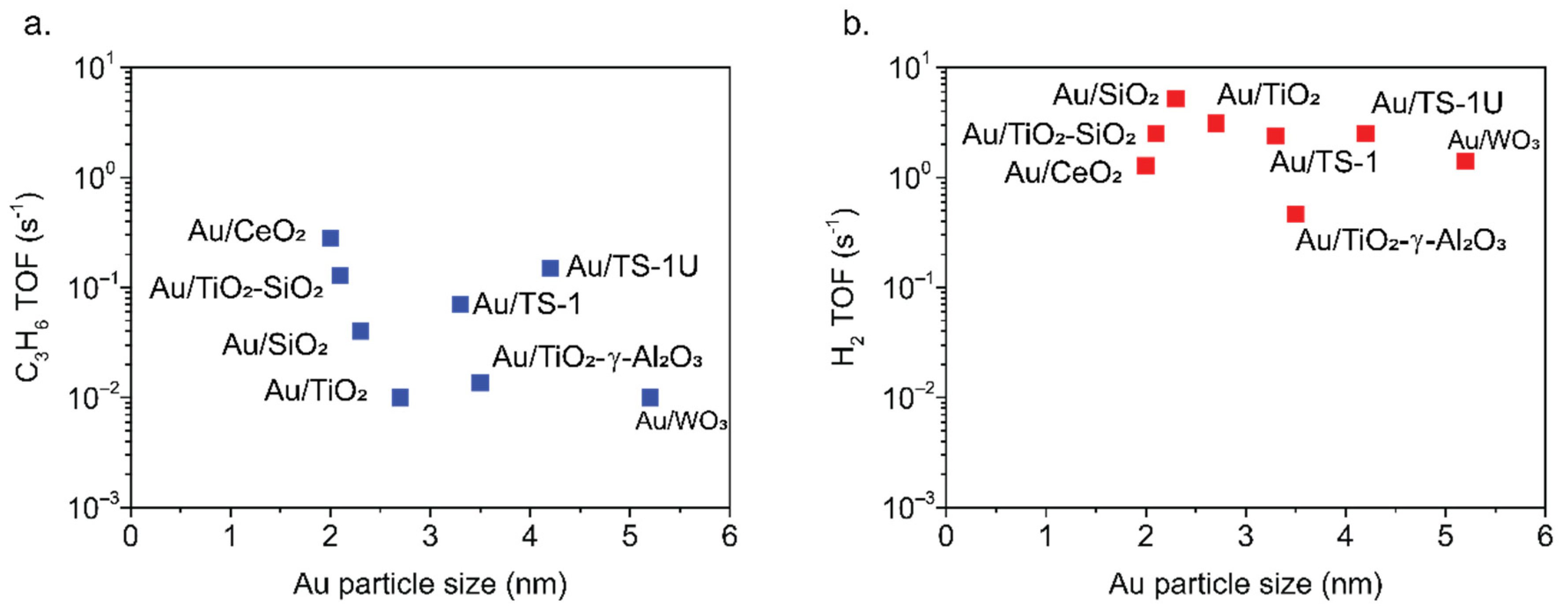
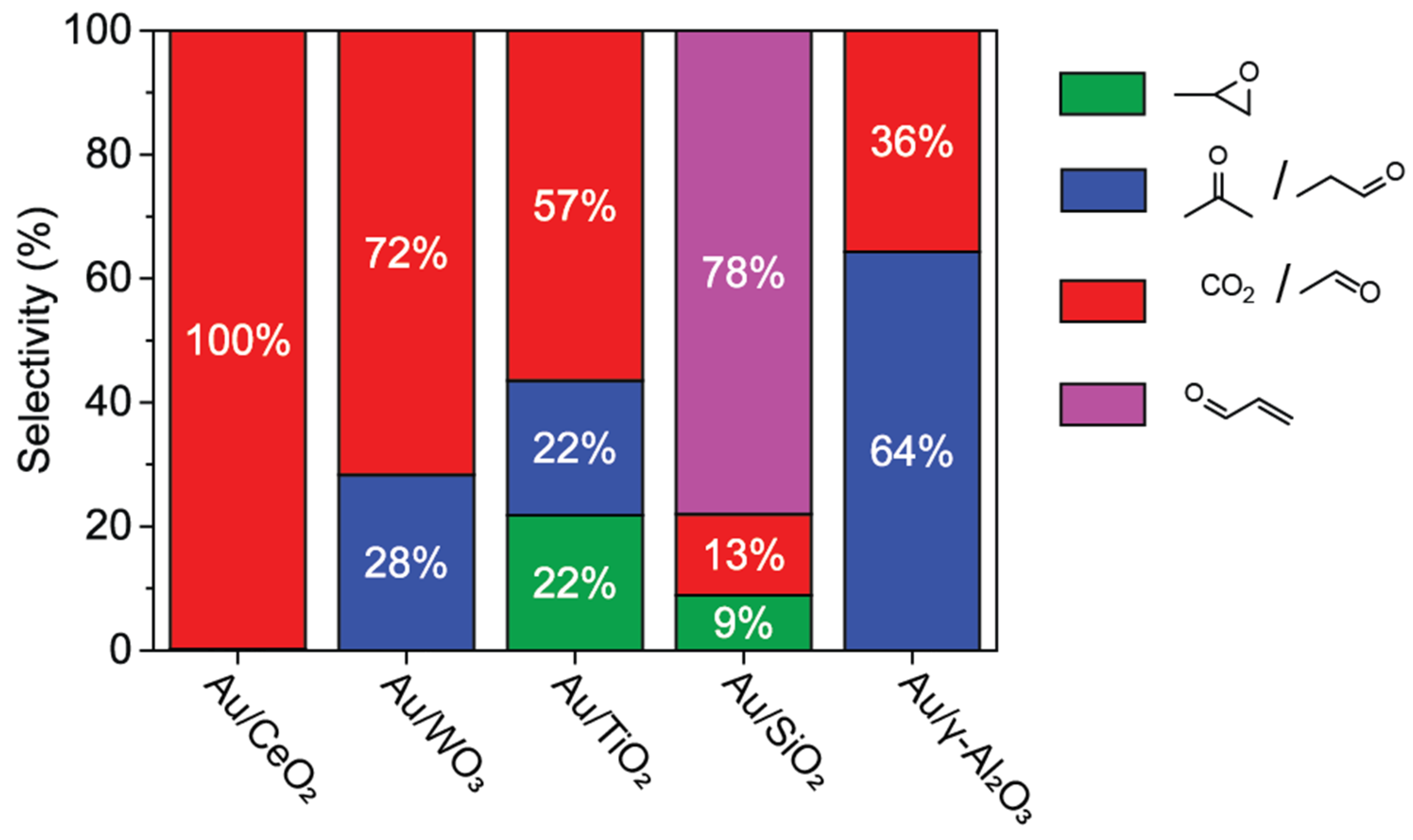
2.1. Metal Oxide Supported Gold Catalysts
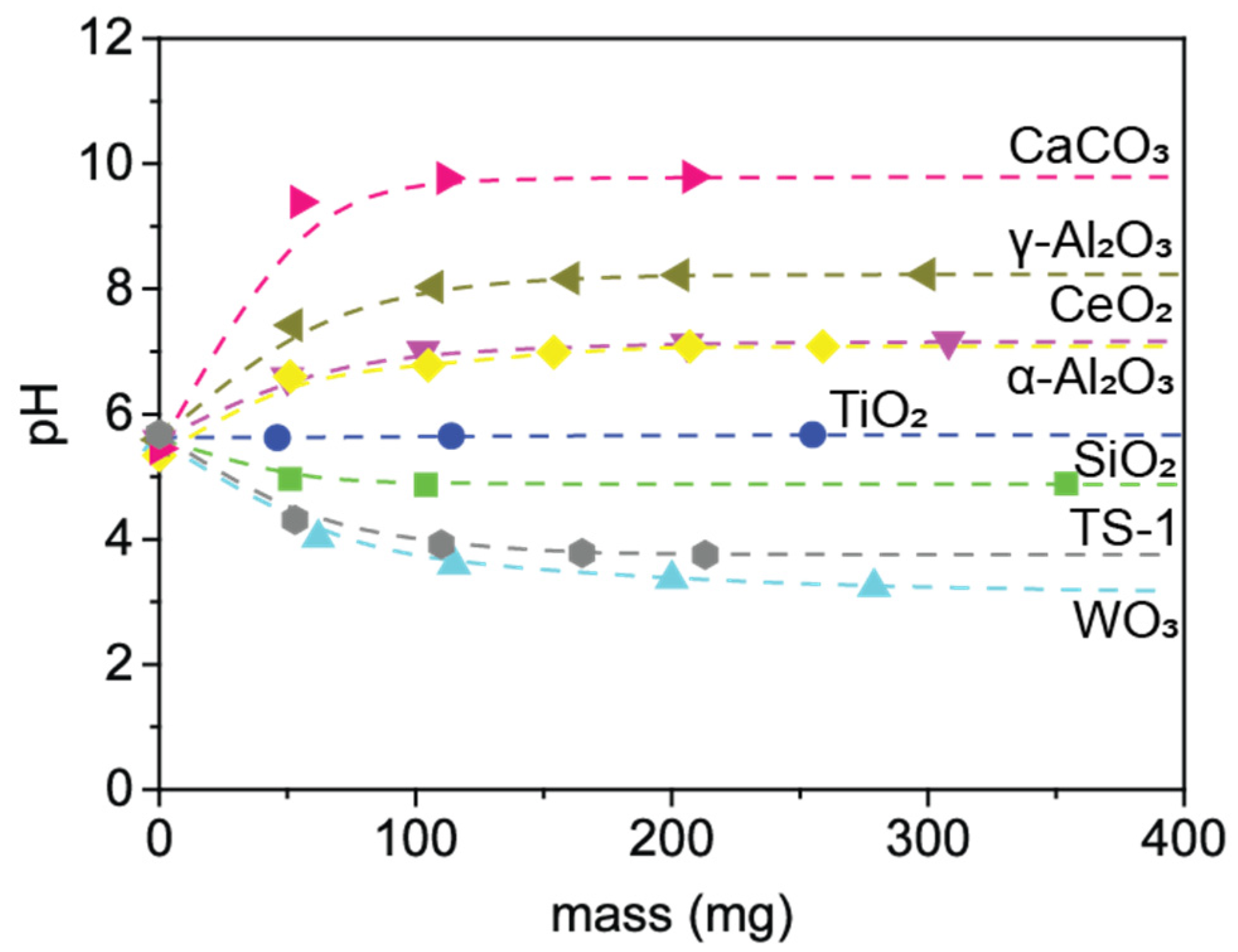
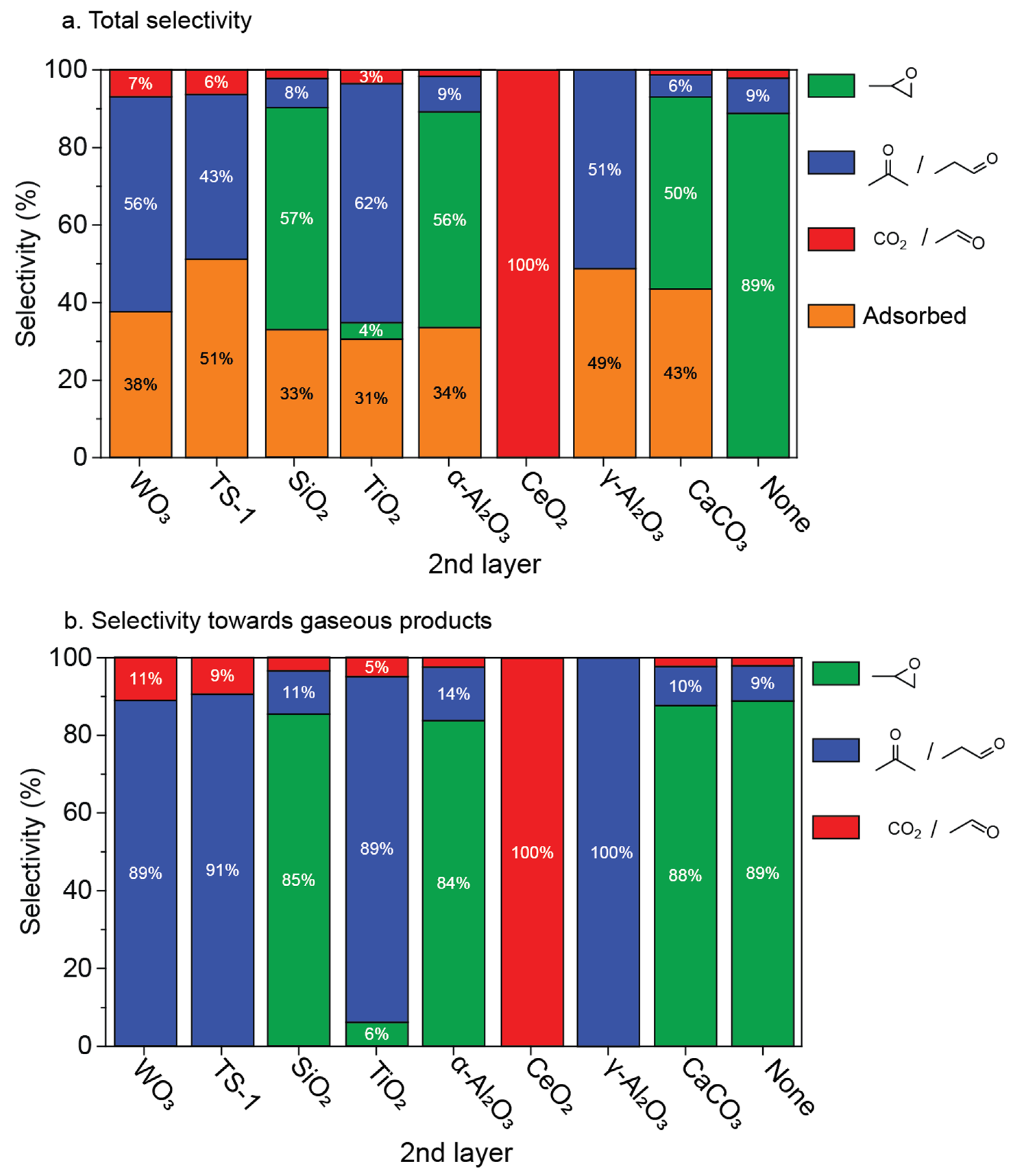
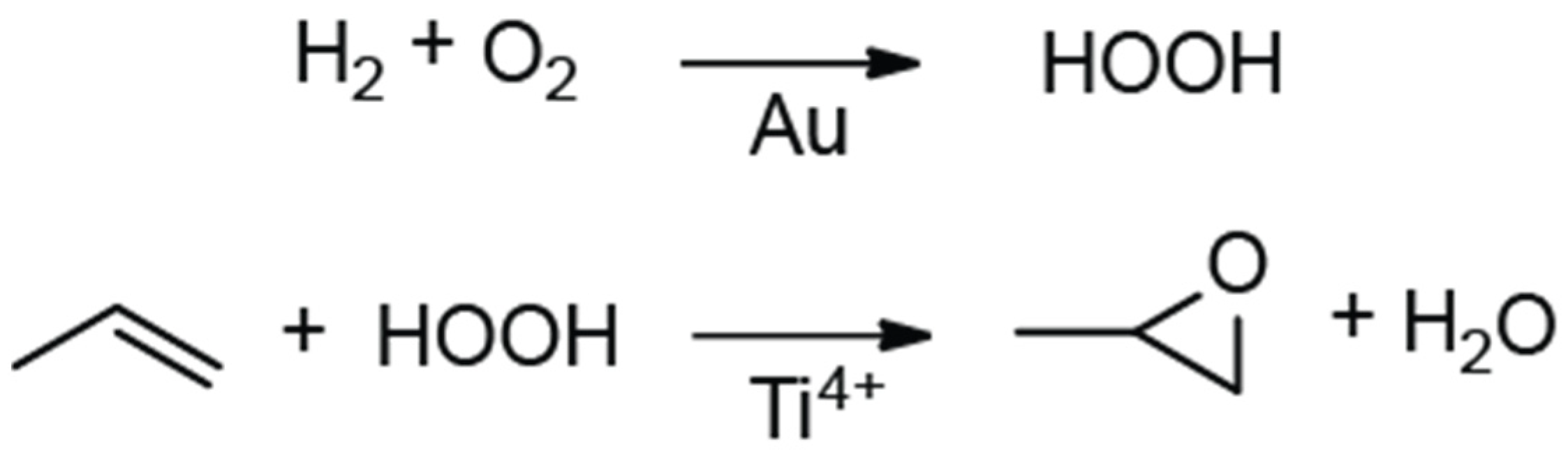
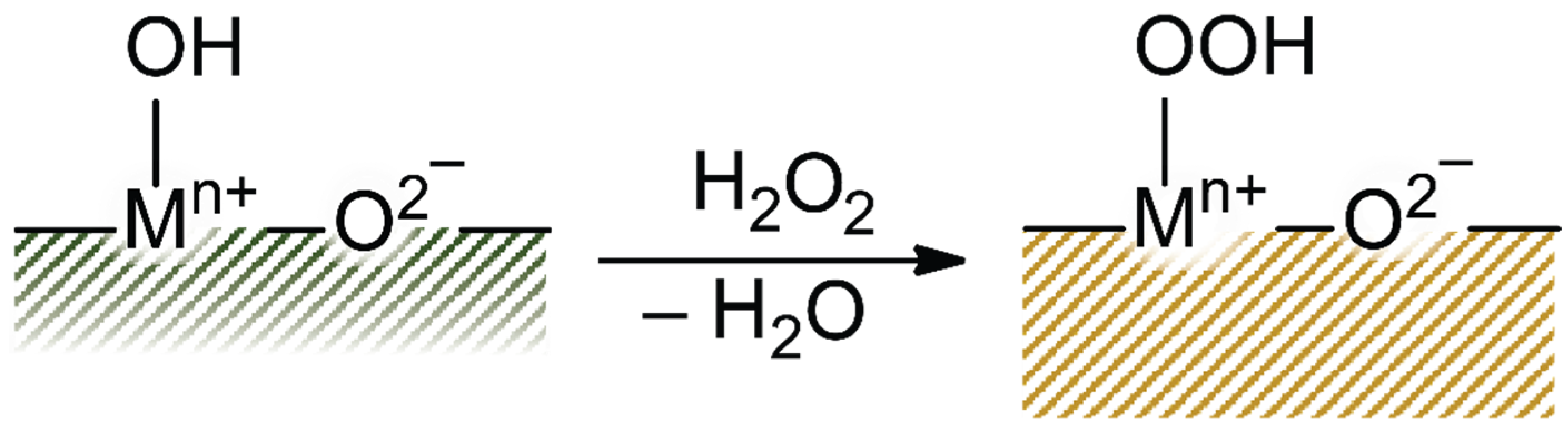
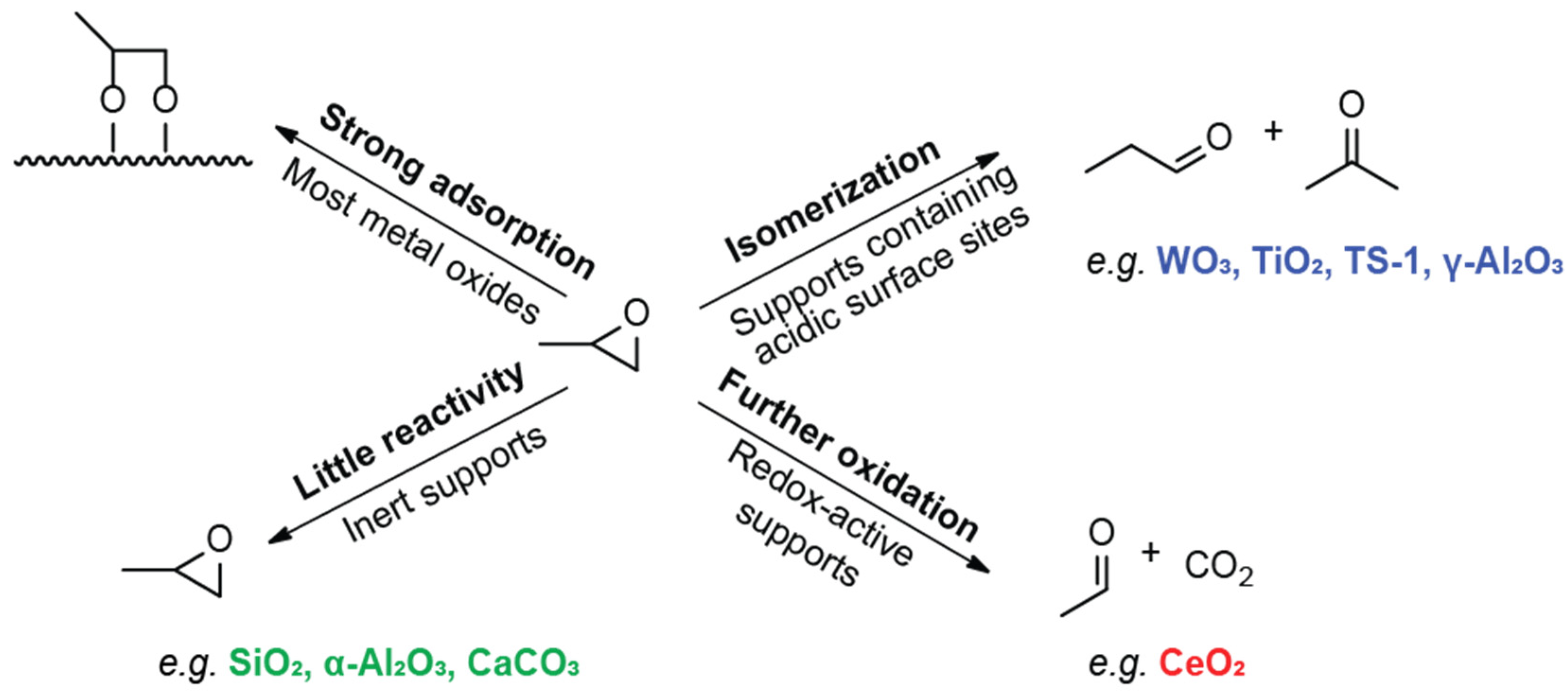
2.2. Reactivity of Propene Oxide with Metal Oxide Supports

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | BET S.A. (m2/g) | PZC (pH) | -OH Coverage (OH/nm2) | Acid Concentration (μmol/gcat) |
|---|---|---|---|---|
| CeO2 | 66 | 7.2 | 3.8 [69] | 17.7 a LA; 0 a BA |
| WO3 | 8 | 3.1 | n.d. | 22.3 b |
| TiO2 | 52 | 5.7 | 3.3 [70] | 239.3 b 111.3 a LA; 0.4 a BA |
| TS-1 | 549 | 3.8 | n.d. | 82.3 b |
| SiO2 | 499 | 4.9 | 5 [71] | 38.2 b 0 a LA; 0 a BA |
| γ-Al2O3 | 249 | 8.2 | 8 [72,73] | 385.3 b |
| α -Al2O3 | 8 | 7.1 | 3.5 [74] | - |
| CaCO3 | 1 | 9.8 | n.d. | - |
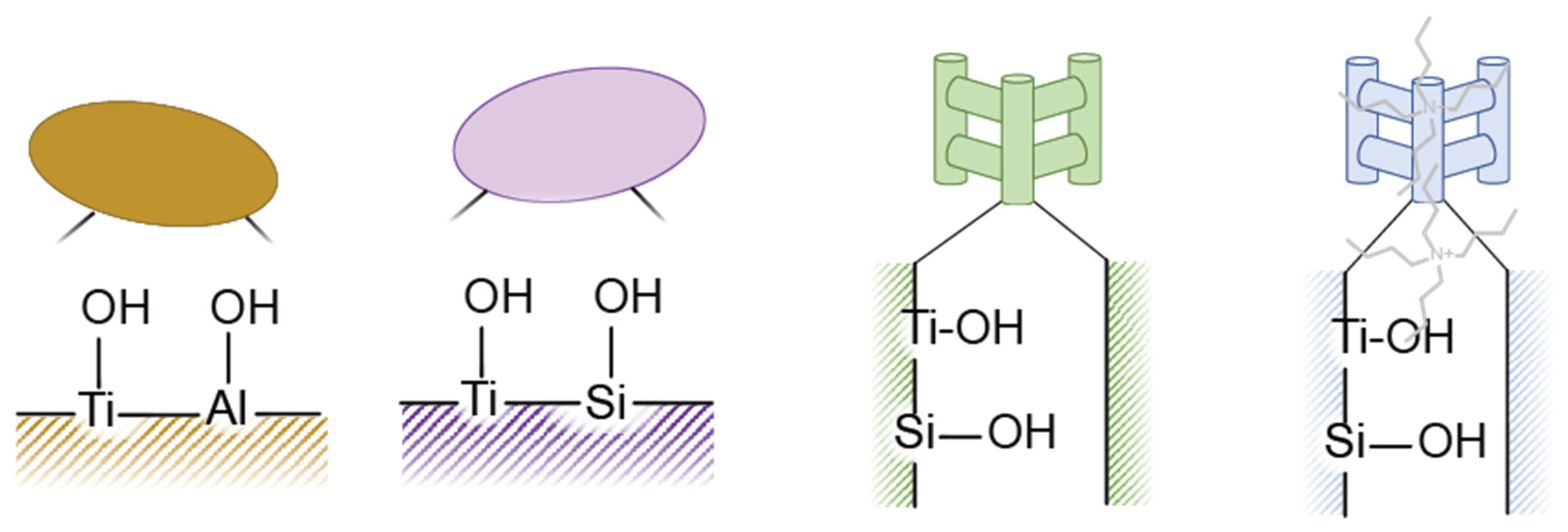
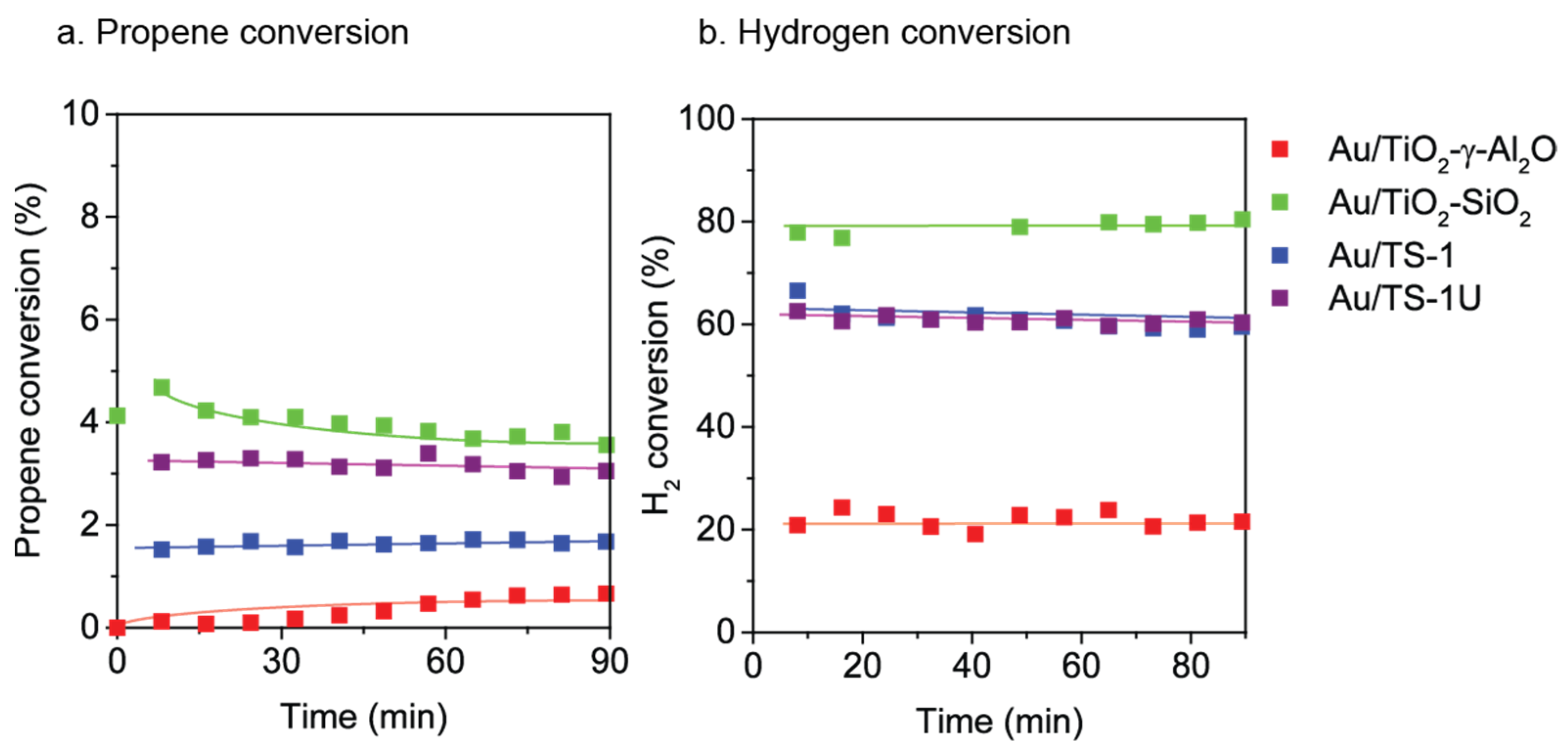
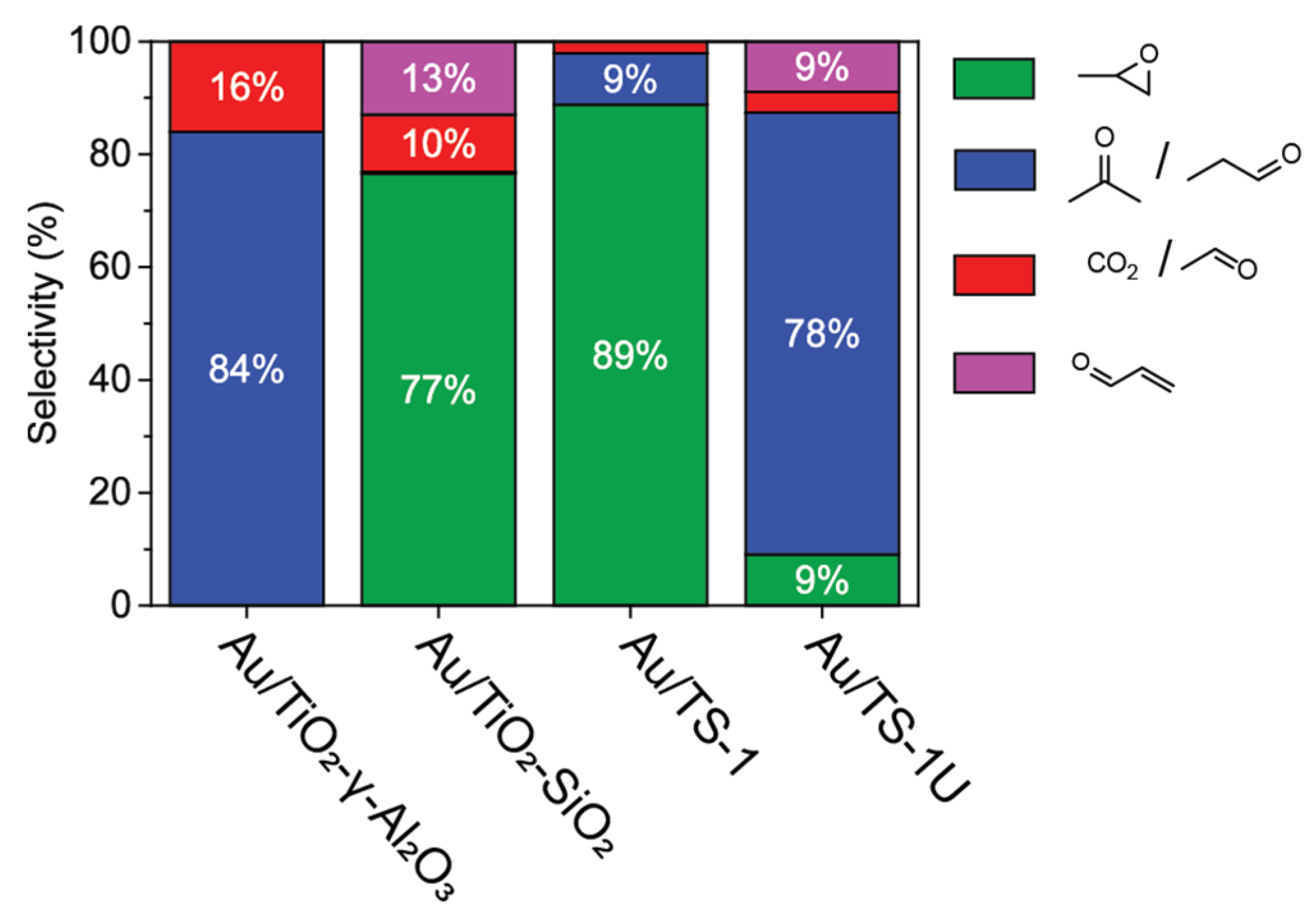
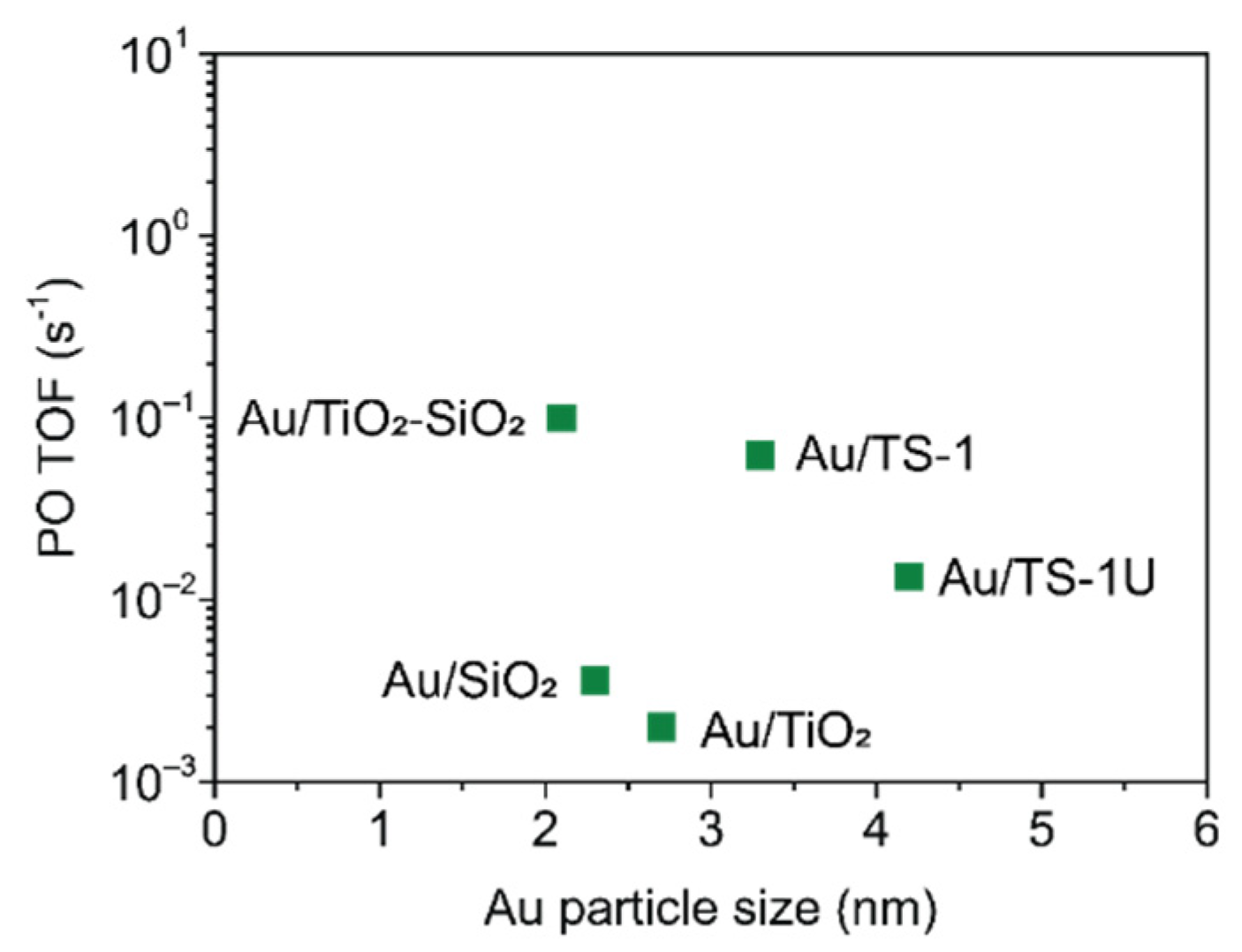
2.3. Au Supported on Ti4+ Modified Oxides
| Catalyst. | Au Loading (wt%) a | Gold Particle Size (nm) b | PZC TiO2-Support (pH) | PZC Catalyst (pH) |
|---|---|---|---|---|
| Au/TiO2-γ-Al2O3 | 0.08 | 3.5 ± 0.5 | 6.0 | 7.7 |
| Au/TiO2-SiO2 | 0.16 | 2.1 ± 0.7 | 2.9 | 6.8 |
| Au/TS-1 | 0.19 | 3.3 ± 1.0 | 3.8 | 6.7 |
| Au/TS-1U | 0.22 | 4.2 ± 1.1 | 3.8 c | 3.8 |
3. Materials and Methods
3.1. Materials
3.2. General Considerations
3.2.1. Support Preparation: Titanosilicate-1
3.2.2. Support Preparation: TiO2-SiO2 and TiO2-γ-Al2O3
3.2.3. Bis(ethylenediamine) Gold (III) Chloride
3.2.4. Catalyst Preparation: Deposition Precipitation Urea
3.2.5. Catalyst Preparation: Cation Adsorption of Au(en)2Cl3
3.2.6. Post Synthesis Treatment of Au/TS-1U
3.3. Catalyst Characterization
3.3.1. Catalytic Testing
3.3.2. Stacked Bed Experiments
3.3.3. Turnover Frequency Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trent, D.L. Propylene Oxide. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: New York, NY, USA, 2001; ISBN 9780471238966. [Google Scholar]
- Nijhuis, T.A.; Makkee, M.; Moulijn, J.A.; Weckhuysen, B.M. The production of propene oxide: Catalytic processes and recent developments. Ind. Eng. Chem. Res. 2006, 45, 3447–3459. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Tanaka, K.; Haruta, M. Selective Vapor-Phase Epoxidation of Propylene over Au/TiO2Catalysts in the Presence of Oxygen and Hydrogen. J. Catal. 1998, 178, 566–575. [Google Scholar] [CrossRef]
- Chen, J.; Halin, S.J.A.; Schouten, J.C.; Nijhuis, T.A. Kinetic study of propylene epoxidation with H2 and O2 over Au/Ti-SiO2 in the explosive regime. Faraday Discuss. 2011, 152, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Partial oxidation of propylene to propylene oxide over a neutral gold trimer in the gas phase: A density functional theory study. J. Phys. Chem. B 2006, 110, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Uphade, B.S.; Akita, T.; Nakamura, T.; Haruta, M. Vapor-phase epoxidation of propene using H2 and O2 over Au/Ti-MCM-48. J. Catal. 2002, 209, 331–340. [Google Scholar] [CrossRef]
- Chowdhury, B.; Bravo-Suárez, J.J.; Mimura, N.; Jiqing, L.; Bando, K.K.; Tsubota, S.; Haruta, M. In situ UV-vis and EPR study on the formation of hydroperoxide species during direct gas phase propylene epoxidation over Au/Ti-SiO2 catalyst. J. Phys. Chem. B 2006, 110, 22995–22999. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, T.A.; Visser, T.; Weckhuysen, B.M. Mechanistic Study into the Direct Epoxidation of Propene over Gold/Titania Catalysts. J. Phys. Chem. B 2005, 109, 19309–19319. [Google Scholar] [CrossRef]
- Sinha, A.K.; Seelan, S.; Tsubota, S.; Haruta, M. Catalysis by Gold Nanoparticles: Epoxidation of Propene. Top. Catal. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Lee, W.S.; Cem Akatay, M.; Stach, E.A.; Ribeiro, F.H.; Nicholas Delgass, W. Reproducible preparation of Au/TS-1 with high reaction rate for gas phase epoxidation of propylene. J. Catal. 2012, 287, 178–189. [Google Scholar] [CrossRef]
- Gaudet, J.; Bando, K.K.; Song, Z.; Fujitani, T.; Zhang, W.; Su, D.S.; Oyama, S.T. Effect of gold oxidation state on the epoxidation and hydrogenation of propylene on Au/TS-1. J. Catal. 2011, 280, 40–49. [Google Scholar] [CrossRef]
- Kropp, T.; Mavrikakis, M. BrØnsted–Evans–Polanyi relation for CO oxidation on metal oxides following the Mars–van Krevelen mechanism. J. Catal. 2019, 377, 577–581. [Google Scholar] [CrossRef]
- Doornkamp, C.; Ponec, V. The universal character of the Mars and Van Krevelen mechanism. J. Mol. Catal. A Chem. 2000, 162, 19–32. [Google Scholar] [CrossRef]
- Gaálová, J.; Topka, P. Gold and ceria as catalysts for voc abatement: A review. Catalysts 2021, 11, 789. [Google Scholar] [CrossRef]
- Delannoy, L.; Fajerwerg, K.; Lakshmanan, P.; Potvin, C.; Méthivier, C.; Louis, C. Supported gold catalysts for the decomposition of VOC: Total oxidation of propene in low concentration as model reaction. Appl. Catal. B Environ. 2010, 94, 117–124. [Google Scholar] [CrossRef]
- Chen, S.; Luo, L.; Jiang, Z.; Huang, W. Size-dependent reaction pathways of low-temperature CO oxidation on Au/CeO2 catalysts. ACS Catal. 2015, 5, 1653–1662. [Google Scholar] [CrossRef]
- Helali, Z.; Jedidi, A.; Syzgantseva, O.A.; Calatayud, M.; Minot, C. Scaling reducibility of metal oxides. Theor. Chem. Acc. 2017, 136, 1–16. [Google Scholar] [CrossRef]
- Pinto, F.M.; Suzuki, V.Y.; Silva, R.C.; La Porta, F.A. Oxygen Defects and Surface Chemistry of Reducible Oxides. Front. Mater. 2019, 6, 260. [Google Scholar] [CrossRef]
- Hernández-Ramírez, E.; Wang, J.A.; Chen, L.F.; Valenzuela, M.A.; Dalai, A.K. Partial oxidation of methanol catalyzed with Au/TiO2, Au/ZrO2 and Au/ZrO2-TiO2 catalysts. Appl. Surf. Sci. 2017, 399, 77–85. [Google Scholar] [CrossRef]
- Aguado, J.; Escola, J.M.; Castro, M.C.; Paredes, B. Metathesis of 1-hexene over rhenium oxide supported on ordered mesoporous aluminas: Comparison with Re2O7/ γ-Al2O3. Appl. Catal. A Gen. 2005, 284, 47–57. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Rooney, D.W.; Halawy, S.A.; Mohamed, M.A.; Abdelkader, A. Effect of precursor on the performance of alumina for the dehydration of methanol to dimethyl ether. Appl. Catal. B Environ. 2012, 127, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Gong, J.; Wang, S.; Gao, N.; Wang, D.; Yang, X.; He, F. Reactivity and surface properties of silica supported molybdenum oxide catalysts for the transesterification of dimethyl oxalate with phenol. Catal. Commun. 2004, 5, 101–106. [Google Scholar] [CrossRef]
- Nakajima, K.; Baba, Y.; Noma, R.; Kitano, M.; Kondo, J.N.; Hayashi, S.; Hara, M. Nb2O5·nH2O as a heterogeneous catalyst with water-tolerant lewis acid sites. J. Am. Chem. Soc. 2011, 133, 4224–4227. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.G.; Marquez, D.T.; Crites, C.O.L.; Netto-Ferreira, J.C.; Scaiano, J.C. Plasmon heating mediated Friedel-Crafts alkylation of anisole using supported AuNP@Nb2O5 catalysts. Tetrahedron Lett. 2017, 58, 427–431. [Google Scholar] [CrossRef]
- Tao, Y.; De Luca, O.; Singh, B.; Kamphuis, A.J.; Chen, J.; Rudolf, P.; Pescarmona, P.P. WO3–SiO2 nanomaterials synthesized using a novel template-free method in supercritical CO2 as heterogeneous catalysts for epoxidation with H2O2. Mater. Today Chem. 2020, 18, 100373. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Ẑalac, S.; Kallay, N. Application of mass titration to the point of zero charge determination. J. Colloid Interface Sci. 1992, 149, 233–240. [Google Scholar] [CrossRef]
- Bravo-Suárez, J.J.; Bando, K.K.; Lu, J.; Haruta, M.; Fujitani, T.; Oyama, S.T. Transient technique for identification of true reaction intermediates: Hydroperoxide species in propylene epoxidation on gold/titanosilicate catalysts by X-ray absorption fine structure spectroscopy. J. Phys. Chem. C 2008, 112, 1115–1123. [Google Scholar] [CrossRef]
- Xiong, G.; Cao, Y.; Guo, Z.; Jia, Q.; Tian, F.; Liu, L. The roles of different titanium species in TS-1 zeolite in propylene epoxidation studied by in situ UV Raman spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 190–196. [Google Scholar] [CrossRef]
- Spanó, E.; Tabacchi, G.; Gamba, A.; Fois, E. On the role of Ti(IV) as a lewis acid in the chemistry of titanium zeolites: Formation, structure, reactivity, and aging of ti-peroxo oxidizing intermediates. A first principles study. J. Phys. Chem. B 2006, 110, 21651–21661. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous Catalysts for Liquid-Phase Oxidations: Philosophers’ Stones or Trojan Horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Ghosh, S.; Acharyya, S.S.; Tiwari, R.; Sarkar, B.; Singha, R.K.; Pendem, C.; Sasaki, T.; Bal, R. Selective oxidation of propylene to propylene oxide over silver-supported tungsten oxide nanostructure with molecular oxygen. ACS Catal. 2014, 4, 2169–2174. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Patil, N.S.; Bhargava, S.K. Epoxidation of styrene by anhydrous H2O2 over TS-1 and γ-Al2O3 catalysts: Effect of reaction water, poisoning of acid sites and presence of base in the reaction mixture. Catal. Lett. 2003, 89, 55–62. [Google Scholar] [CrossRef]
- Solsona, B.E.; Edwards, J.K.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Al2O3 supported Au-Pd catalysts. Chem. Mater. 2006, 18, 2689–2695. [Google Scholar] [CrossRef]
- Jildeh, Z.B.; Oberländer, J.; Kirchner, P.; Wagner, P.H.; Schöning, M.J. Thermocatalytic behavior of manganese (IV) oxide as nanoporous material on the dissociation of a gas mixture containing hydrogen peroxide. Nanomaterials 2018, 8, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, R.A. Synthetic and mechanistic aspects of metal-catalysed epoxidations with hydroperoxides. J. Mol. Catal. 1980, 7, 107–126. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Huizinga, B.J.; Makkee, M.; Moulijn, J.A. Direct Epoxidation of Propene Using Gold Dispersed on TS-1 and Other Titanium-Containing Supports. Ind. Eng. Chem. Res. 1999, 38, 884–891. [Google Scholar] [CrossRef]
- van den Reijen, J.E.; Kanungo, S.; Welling, T.A.J.; Versluijs-Helder, M.; Nijhuis, T.A.; de Jong, K.P.; de Jongh, P.E. Preparation and particle size effects of Ag/α-Al2O3 catalysts for ethylene epoxidation. J. Catal. 2017, 356, 65–74. [Google Scholar] [CrossRef]
- Mao, C.-F.; Albert Vannice, M. High surface area α-aluminas III. Oxidation of ethylene, ethylene oxide, and acetaldehyde over silver dispersed on high surface area α-alumina. Appl. Catal. A Gen. 1995, 122, 61–76. [Google Scholar] [CrossRef]
- Lu, J.; Bravo-Suárez, J.J.; Haruta, M.; Oyama, S.T. Direct propylene epoxidation over modified Ag/CaCO3 catalysts. Appl. Catal. A Gen. 2006, 302, 283–295. [Google Scholar] [CrossRef]
- Zemichael, F.W.; Palermo, A.; Tikhov, M.S.; Lambert, R.M. Propene epoxidation over K-promoted Ag/CaCO3 catalysts: The effect of metal particle size. Catal. Lett. 2002, 80, 93–98. [Google Scholar] [CrossRef]
- Teržan, J.; Djinović, P.; Zavašnik, J.; Arčon, I.; Žerjav, G.; Spreitzer, M.; Pintar, A. Alkali and earth alkali modified CuOx/SiO2 catalysts for propylene partial oxidation: What determines the selectivity? Appl. Catal. B Environ. 2018, 237, 214–227. [Google Scholar] [CrossRef]
- He, J.; Zhai, Q.; Zhang, Q.; Deng, W.; Wang, Y. Active site and reaction mechanism for the epoxidation of propylene by oxygen over CuOx/SiO2 catalysts with and without Cs + modification. J. Catal. 2013, 299, 53–66. [Google Scholar] [CrossRef]
- Diekmann, M.; Koch, G.; König, M.; Ressler, T. Correlation between Copper Oxide Particle Size and Selectivity towards Propylene Oxide in Selective Oxidation of Propene. ChemCatChem 2018, 10, 5459–5467. [Google Scholar] [CrossRef]
- Mul, G.; Zwijnenburg, A.; van der Linden, B.; Makkee, M.; Moulijn, J.A. Stability and Selectivity of Au/TiO2 and Au/TiO2/SiO2 Catalysts in Propene Epoxidation: An in Situ FT-IR Study. J. Catal. 2001, 201, 128–137. [Google Scholar] [CrossRef]
- Ruiz, A.; van der Linden, B.; Makkee, M.; Mul, G. Acrylate and propoxy-groups: Contributors to deactivation of Au/TiO2 in the epoxidation of propene. J. Catal. 2009, 266, 286–290. [Google Scholar] [CrossRef]
- Sinha, A.K.; Seelan, S.; Akita, T.; Tsubota, S.; Haruta, M. Vapor phase propylene epoxidation over Au/Ti-MCM-41 catalysts prepared by different Ti incorporation modes. Appl. Catal. A Gen. 2003, 240, 243–252. [Google Scholar] [CrossRef]
- Stangland, E.E.; Taylor, B.; Andres, R.P.; Delgass, W.N. Direct Vapor Phase Propylene Epoxidation over Deposition-Precipitation Gold-Titania Catalysts in the Presence of H2/O2: Effects of Support, Neutralizing Agent, and Pretreatment. J. Phys. Chem. B 2005, 109, 2321–2330. [Google Scholar] [CrossRef]
- Harris, J.W.; Arvay, J.; Mitchell, G.; Delgass, W.N.; Ribeiro, F.H. Propylene oxide inhibits propylene epoxidation over Au/TS-1. J. Catal. 2018, 365, 105–114. [Google Scholar] [CrossRef]
- de Boed, E.J.J.; de Rijk, J.W.; de Jongh, P.E.; Donoeva, B. Steering the Selectivity in Gold–Titanium-Catalyzed Propene Oxidation by Controlling the Surface Acidity. J. Phys. Chem. C 2021, 125, 16557–16568. [Google Scholar] [CrossRef]
- Zanella, R.; Delannoy, L.; Louis, C. Mechanism of deposition of gold precursors onto TiO2 during the preparation by cation adsorption and deposition-precipitation with NaOH and urea. Appl. Catal. A Gen. 2005, 291, 62–72. [Google Scholar] [CrossRef]
- Zhu, H.; Liang, C.; Yan, W.; Overbury, S.H.; Dai, S. Preparation of highly active silica-supported Au catalysts for Co oxidation by a solution-based technique. J. Phys. Chem. B 2006, 110, 10842–10848. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Ma, Z.; Overbury, S.H.; Dai, S. Promotion of Au(en)2Cl3-Derived Au/Fumed SiO2 by Treatment with KMnO4. Society 2008, 2, 8349–8358. [Google Scholar] [CrossRef]
- Feng, X.; Duan, X.; Qian, G.; Zhou, X.; Chen, D.; Yuan, W. Insights into size-dependent activity and active sites of Au nanoparticles supported on TS-1 for propene epoxidation with H2 and O2. J. Catal. 2014, 317, 99–104. [Google Scholar] [CrossRef]
- Stangland, E.E.; Stavens, K.B.; Andres, R.P.; Delgass, W.N. Characterization of gold-titania catalysts via oxidation of propylene to propylene oxide. J. Catal. 2000, 191, 332–347. [Google Scholar] [CrossRef]
- Kanungo, S.; Perez Ferrandez, D.M.; Neira D’Angelo, F.; Schouten, J.C.; Nijhuis, T.A. Kinetic study of propene oxide and water formation in hydro-epoxidation of propene on Au/Ti-SiO2 catalyst. J. Catal. 2016, 338, 284–294. [Google Scholar] [CrossRef]
- Barton, D.G.; Podkolzin, S.G. Kinetic study of a direct water synthesis over silica-supported gold nanoparticles. J. Phys. Chem. B 2005, 109, 2262–2274. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Sacaliuc, E.; Beale, A.M.; van der Eerden, A.M.J.; Schouten, J.C.; Weckhuysen, B.M. Spectroscopic evidence for the adsorption of propene on gold nanoparticles during the hydro-epoxidation of propene. J. Catal. 2008, 258, 256–264. [Google Scholar] [CrossRef]
- Aneggi, E.; Boaro, M.; De Leitenburg, C.; Dolcetti, G.; Trovarelli, A. Insights into the redox properties of ceria-based oxides and their implications in catalysis. J. Alloys Compd. 2006, 408–412, 1096–1102. [Google Scholar] [CrossRef]
- Haruta, M.; Uphade, B.S.; Tsubota, S.; Miyamoto, A. Selective oxidation of propylene over gold deposited on titanium-based oxides. Res. Chem. Intermed. 1998, 24, 329–336. [Google Scholar] [CrossRef]
- Huang, J.; Takei, T.; Ohashi, H.; Haruta, M. Propene epoxidation with oxygen over gold clusters: Role of basic salts and hydroxides of alkalis. Appl. Catal. A Gen. 2012, 435–436, 115–122. [Google Scholar] [CrossRef]
- Cant, N.W.; Hall, W.K. Catalytic oxidation. IV. Ethylene and propylene oxidation over gold. J. Phys. Chem. 1971, 75, 2914–2921. [Google Scholar] [CrossRef]
- Blues, E.T.; Bryce-Smith, D.; Lawston, I.W.; Wall, G.D. Gold(I) ketenide. J. Chem. Soc. Chem. Commun. 1974, 13, 513–514. [Google Scholar] [CrossRef]
- McEwan, L.; Julius, M.; Roberts, S.; Fletcher, J.C.Q. A review of the use of gold catalysts in selective hydrogenation reactions. Gold Bull. 2010, 43, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Delannoy, L.; Chantry, R.L.; Casale, S.; Li, Z.Y.; Borensztein, Y.; Louis, C. HRTEM and STEM-HAADF characterisation of Au–TiO2 and Au–Al2O3 catalysts for a better understanding of the parameters influencing their properties in CO oxidation. Phys. Chem. Chem. Phys. 2013, 15, 3473. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Jin, M.; Lu, J.; Wei, Z.; Li, C. Direct gas-phase epoxidation of propylene to propylene oxide using air as oxidant on supported gold catalyst. J. Nat. Gas Chem. 2008, 17, 184–190. [Google Scholar] [CrossRef]
- Thornburg, N.E.; Thompson, A.B.; Notestein, J.M. Periodic Trends in Highly Dispersed Groups IV and V Supported Metal Oxide Catalysts for Alkene Epoxidation with H2O2. ACS Catal. 2015, 5, 5077–5088. [Google Scholar] [CrossRef]
- Zaki, M.I.; Hussein, G.A.M.; Mansour, S.A.A.; El-Ammawy, H.A. Adsorption and surface reactions of pyridine on pure and doped ceria catalysts as studied by infrared spectroscopy. J. Mol. Catal. 1989, 51, 209–220. [Google Scholar] [CrossRef]
- Longo, A.; de Boed, E.J.J.; Mammen, N.; van der Linden, M.; Honkala, K.; Häkkinen, H.; de Jongh, P.E.; Donoeva, B. Towards Atomically Precise Supported Catalysts from Monolayer-Protected Clusters: The Critical Role of the Support. Chem. Eur. J. 2020, 26, 7051–7058. [Google Scholar] [CrossRef]
- Erdem, B.; Hunsicker, R.A.; Simmons, G.W.; David Sudol, E.; Dimonie, V.L.; El-Aasser, M.S. XPS and FTIR surface characterization of TiO2 particles used in polymer encapsulation. Langmuir 2001, 17, 2664–2669. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. A Physicochem. Eng. Asp. 2000, 173, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Nguefack, M.; Popa, A.F.; Rossignol, S.; Kappenstein, C. Preparation of alumina through a sol-gel process. Synthesis, characterization, thermal evolution and model of intermediate boehmite. Phys. Chem. Chem. Phys. 2003, 5, 4279–4289. [Google Scholar] [CrossRef]
- Hietala, J.; Root, A.; Knuuttila, P. The surface acidity of pure and modified aluminas in Re/Al2O3 metathesis catalysts as studied by 1H MAS NMR spectroscopy and its importance in the ethenolysis of 1,5-cyclooctadiene. J. Catal. 1994, 150, 46–55. [Google Scholar] [CrossRef]
- McHale, J.M.; Auroux, A.; Perrotta, A.J.; Navrotsky, A. Surface energies and thermodynamic phase stability in nanocrystalline aluminas. Science 1997, 277, 788–789. [Google Scholar] [CrossRef] [Green Version]
- Bigey, C.; Hilaire, L.; Maire, G. Catalysis on Pd/WO3 and Pd/WO2: Effect of the modifications of the surface states due to redox treatments on the skeletal rearrangement of hydrocarbons. Part I. Physical and chemical characterizations of catalysts by BET, TPR, XRD, XAS, and XPS. J. Catal. 1999, 184, 406–420. [Google Scholar] [CrossRef]
- Arends, I.W.C.E.; Sheldon, R.A. Activities and stabilities of heterogeneous catalysts in selective liquid phase oxidations: Recent developments. Appl. Catal. A Gen. 2001, 212, 175–187. [Google Scholar] [CrossRef]
- Robinson, M.W.C.; Pillinger, K.S.; Graham, A.E. Highly efficient Meinwald rearrangement reactions of epoxides catalyzed by copper tetrafluoroborate. Tetrahedron Lett. 2006, 47, 5919–5921. [Google Scholar] [CrossRef]
- Cumaranatunge, L.; Delgass, W.N. Enhancement of Au capture efficiency and activity of Au/TS-1 catalysts for propylene epoxidation. J. Catal. 2005, 232, 38–42. [Google Scholar] [CrossRef]
- Feng, X.; Yang, J.; Duan, X.; Cao, Y.; Chen, B.; Chen, W.; Lin, D.; Qian, G.; Chen, D.; Yang, C.; et al. Enhanced Catalytic Performance for Propene Epoxidation with H2 and O2 over Bimetallic Au–Ag/Uncalcined Titanium Silicate-1 Catalysts. ACS Catal. 2018, 8, 7799–7808. [Google Scholar] [CrossRef]
- Cozzolino, M.; Di Serio, M.; Tesser, R.; Santacesaria, E. Grafting of titanium alkoxides on high-surface SiO2 support: An advanced technique for the preparation of nanostructured TiO2/SiO2 catalysts. Appl. Catal. A Gen. 2007, 325, 256–262. [Google Scholar] [CrossRef]
- Chen, J.; Halin, S.J.A.; Pidko, E.A.; Verhoeven, M.W.G.M.T.; Ferrandez, D.M.P.; Hensen, E.J.M.; Schouten, J.C.; Nijhuis, T.A. Enhancement of Catalyst Performance in the Direct Propene Epoxidation: A Study into Gold-Titanium Synergy. ChemCatChem 2013, 5, 467–478. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, G.; Lu, Y. Propylene epoxidation with O2 and H2: A high-performance Au/TS-1 catalyst prepared via a deposition-precipitation method using urea. Catal. Sci. Technol. 2013, 3, 2906–2909. [Google Scholar] [CrossRef]
- Lee, W.S.; Cem Akatay, M.; Stach, E.A.; Ribeiro, F.H.; Nicholas Delgass, W. Gas-phase epoxidation of propylene in the presence of H2 and O2 over small gold ensembles in uncalcined TS-1. J. Catal. 2014, 313, 104–112. [Google Scholar] [CrossRef]
- Chowdhury, B.; Bravo-Suárez, J.J.; Daté, M.; Tsubota, S.; Haruta, M. Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts. Angew. Chemie Int. Ed. 2006, 45, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, G.; Hu, H. Synthesis, characterization and catalytic performance of titanium silicalite-1 prepared in the presence of nonionic surfactants. Mater. Chem. Phys. 2006, 100, 162–167. [Google Scholar] [CrossRef]
- Maschmeyer, T.; Rey, F.; Sankar, G.; Thomas, J.M. Heterogeneous catalysts obtained by grafting metallocene complexes onto mesoporous silica. Nature 1995, 378, 159–162. [Google Scholar] [CrossRef]
- Block, B.P.; Bailar, J.C. The Reaction of Gold(III) with Some Bidentate Coördinating Groups1. J. Am. Chem. Soc. 1951, 73, 4722–4725. [Google Scholar] [CrossRef]















| Catalyst | Au Loading (wt%) a | Gold Particle Size (nm) b | PZC (pH) |
|---|---|---|---|
| Au/CeO2 | 1.89 | 2.0 ± 0.7 | 6.2 |
| Au/WO3 | 0.82 | 5.2 ± 1.5 | 3.5 |
| Au/TiO2 | 0.10 | 2.7 ± 0.6 | 5.5 |
| Au/SiO2 | 0.16 | 2.3 ± 0.7 | 6.7 |
| Au/γ-Al2O3 | 0.08 | n.d. | 7.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Boed, E.J.J.; Folmer, B.J.; Tang, M.; Donoeva, B.; de Jongh, P.E. Influence of the Support on Propene Oxidation over Gold Catalysts. Catalysts 2022, 12, 327. https://doi.org/10.3390/catal12030327
de Boed EJJ, Folmer BJ, Tang M, Donoeva B, de Jongh PE. Influence of the Support on Propene Oxidation over Gold Catalysts. Catalysts. 2022; 12(3):327. https://doi.org/10.3390/catal12030327
Chicago/Turabian Stylede Boed, Ewoud J. J., Bryan J. Folmer, Min Tang, Baira Donoeva, and Petra E. de Jongh. 2022. "Influence of the Support on Propene Oxidation over Gold Catalysts" Catalysts 12, no. 3: 327. https://doi.org/10.3390/catal12030327