
Alkene Epoxidation and Thioether Oxidation with Hydrogen Peroxide Catalyzed by Mesoporous Zirconium-Silicates

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
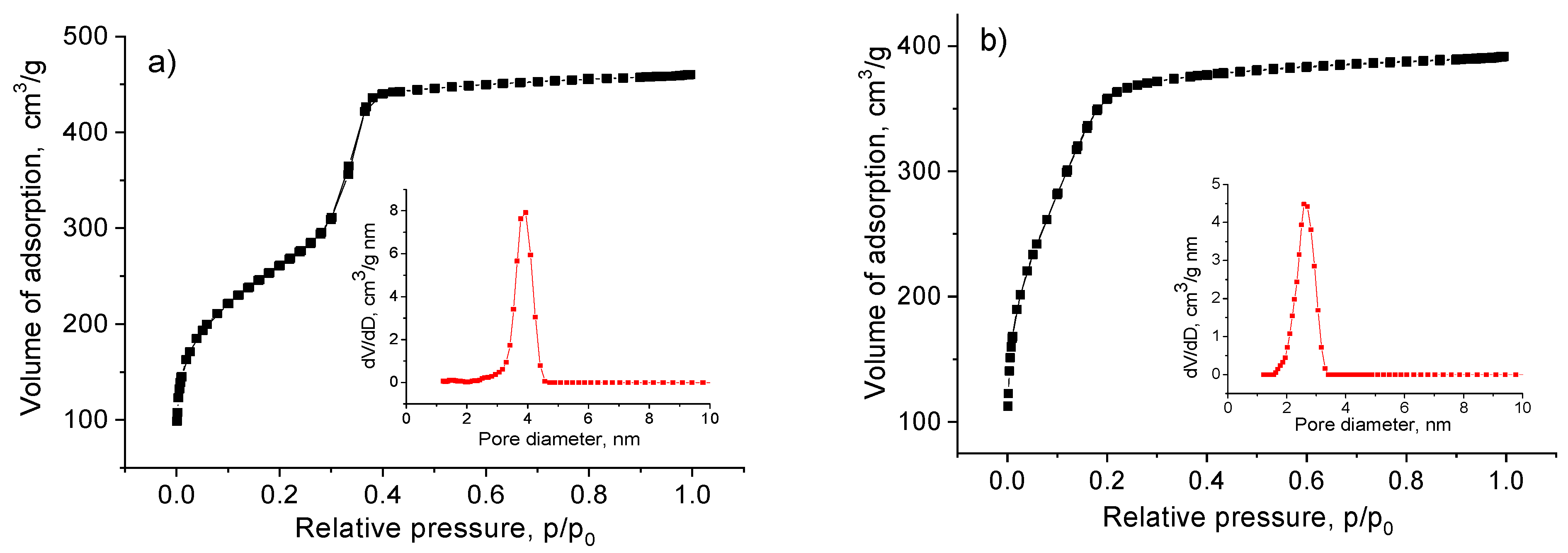
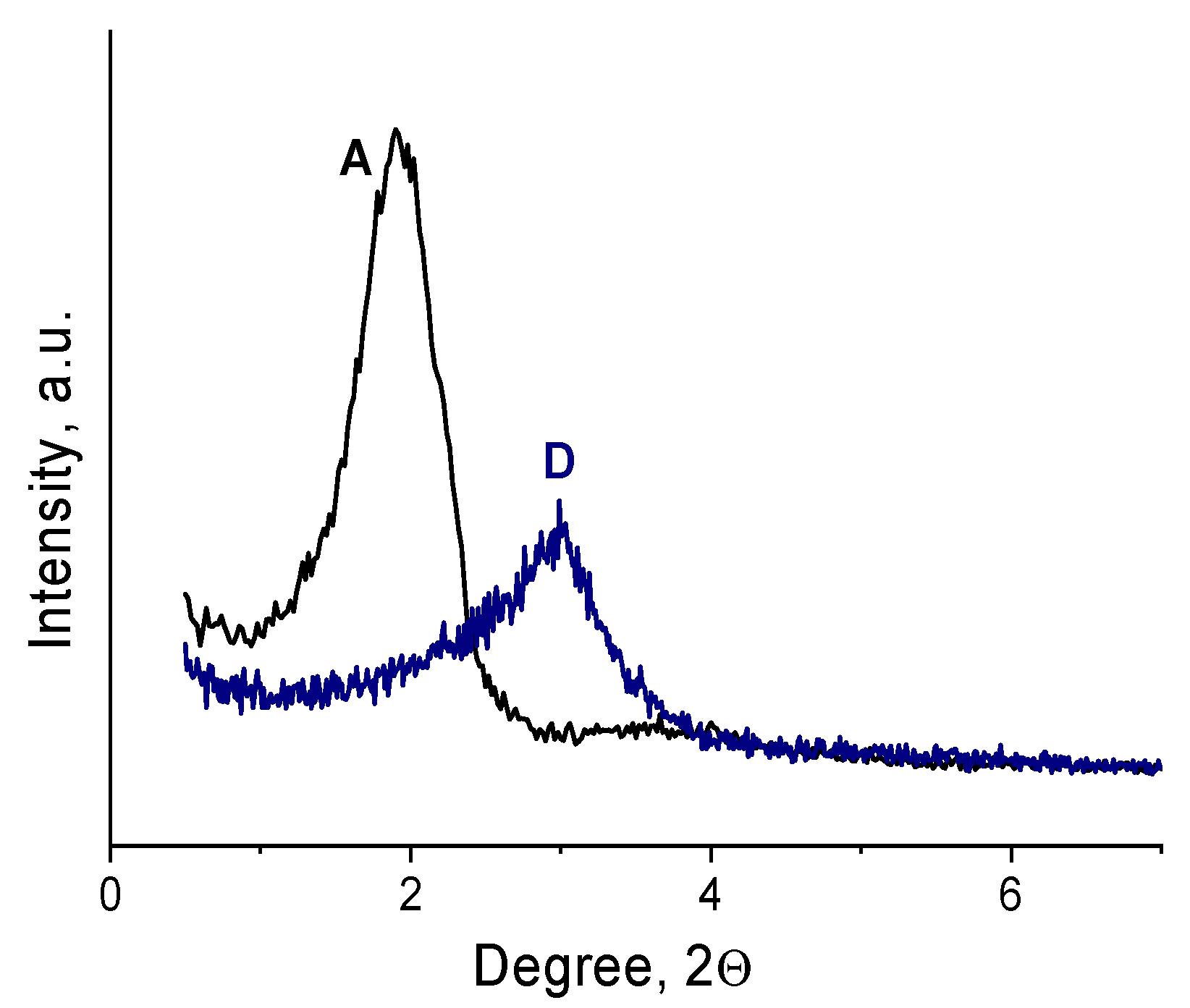
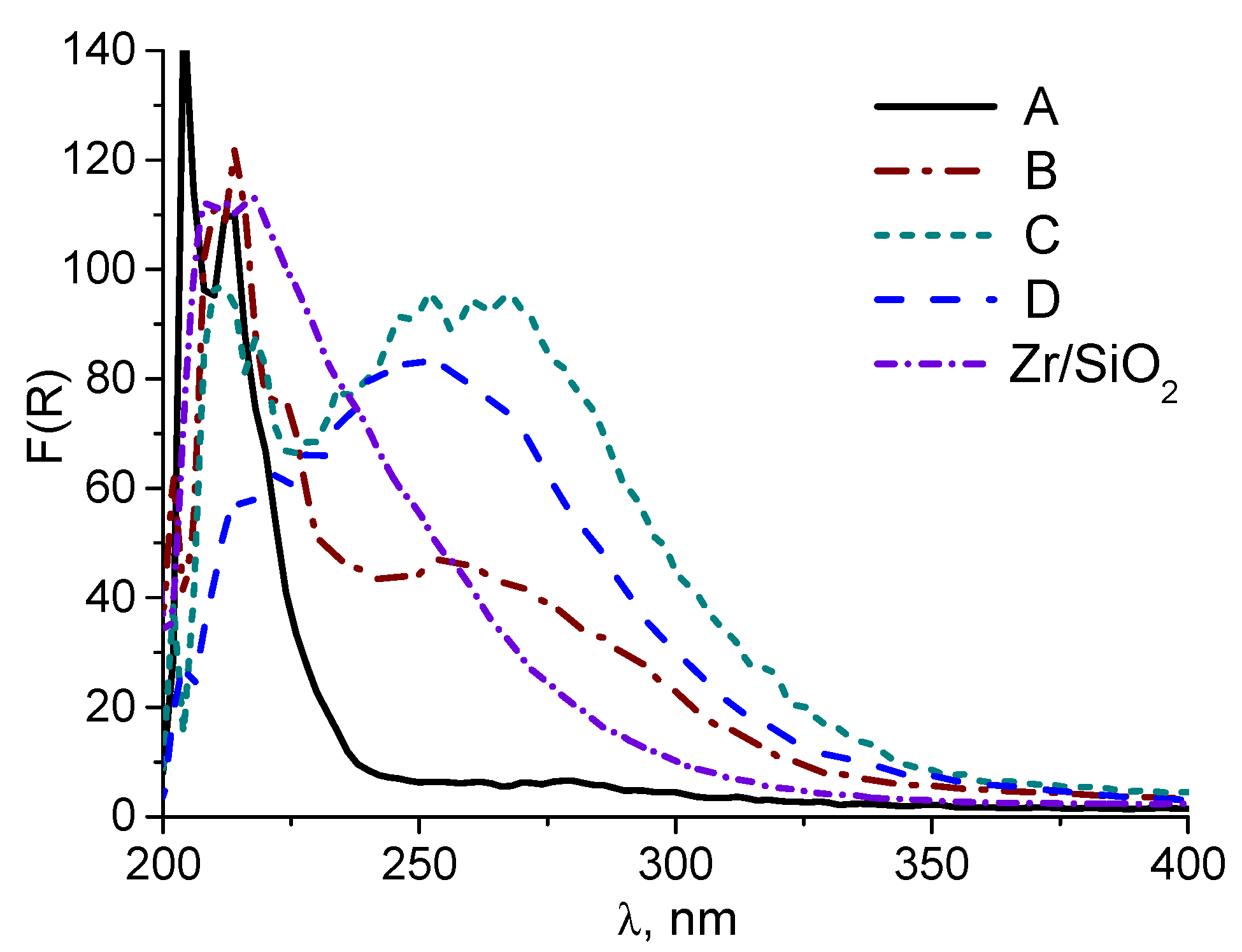
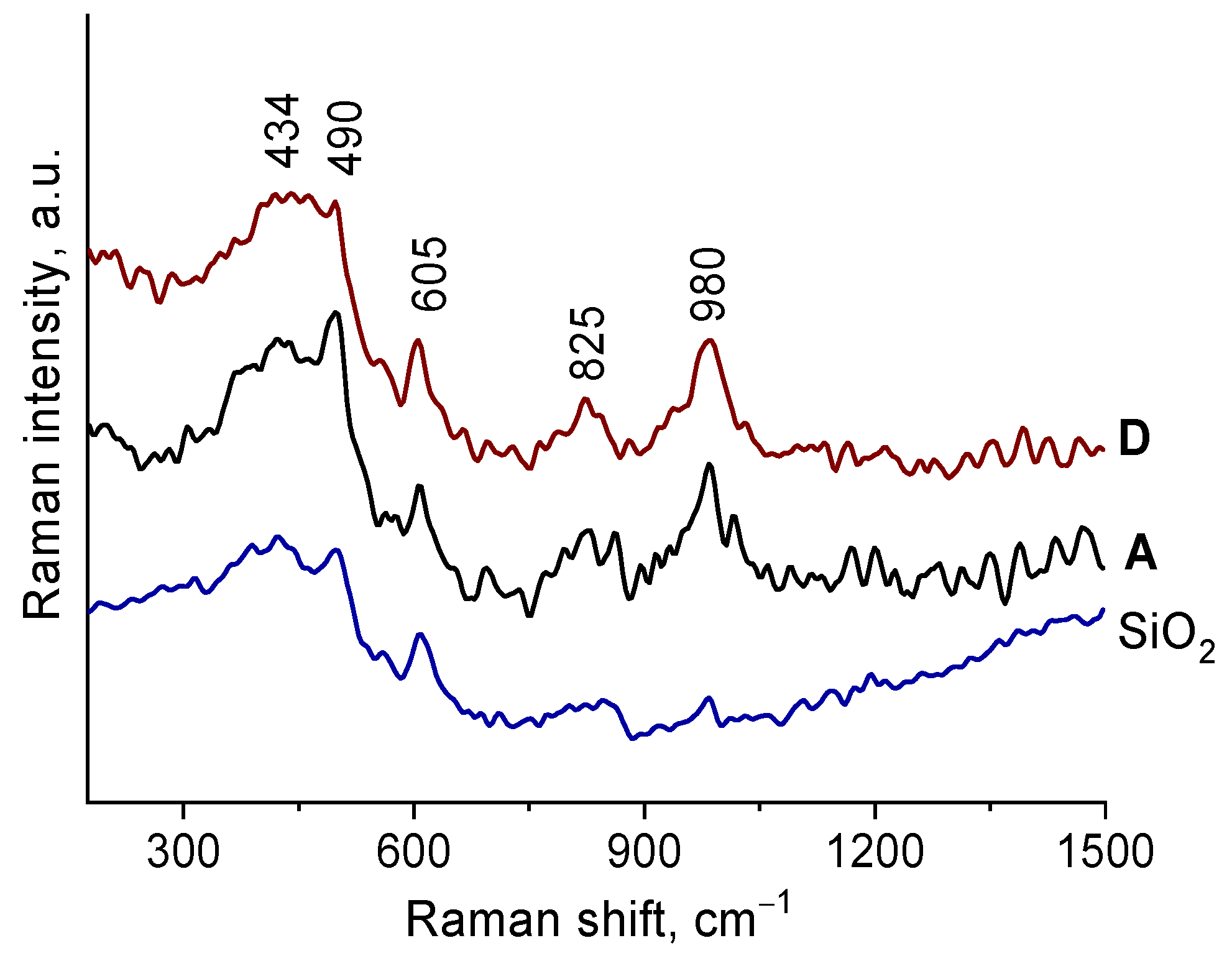
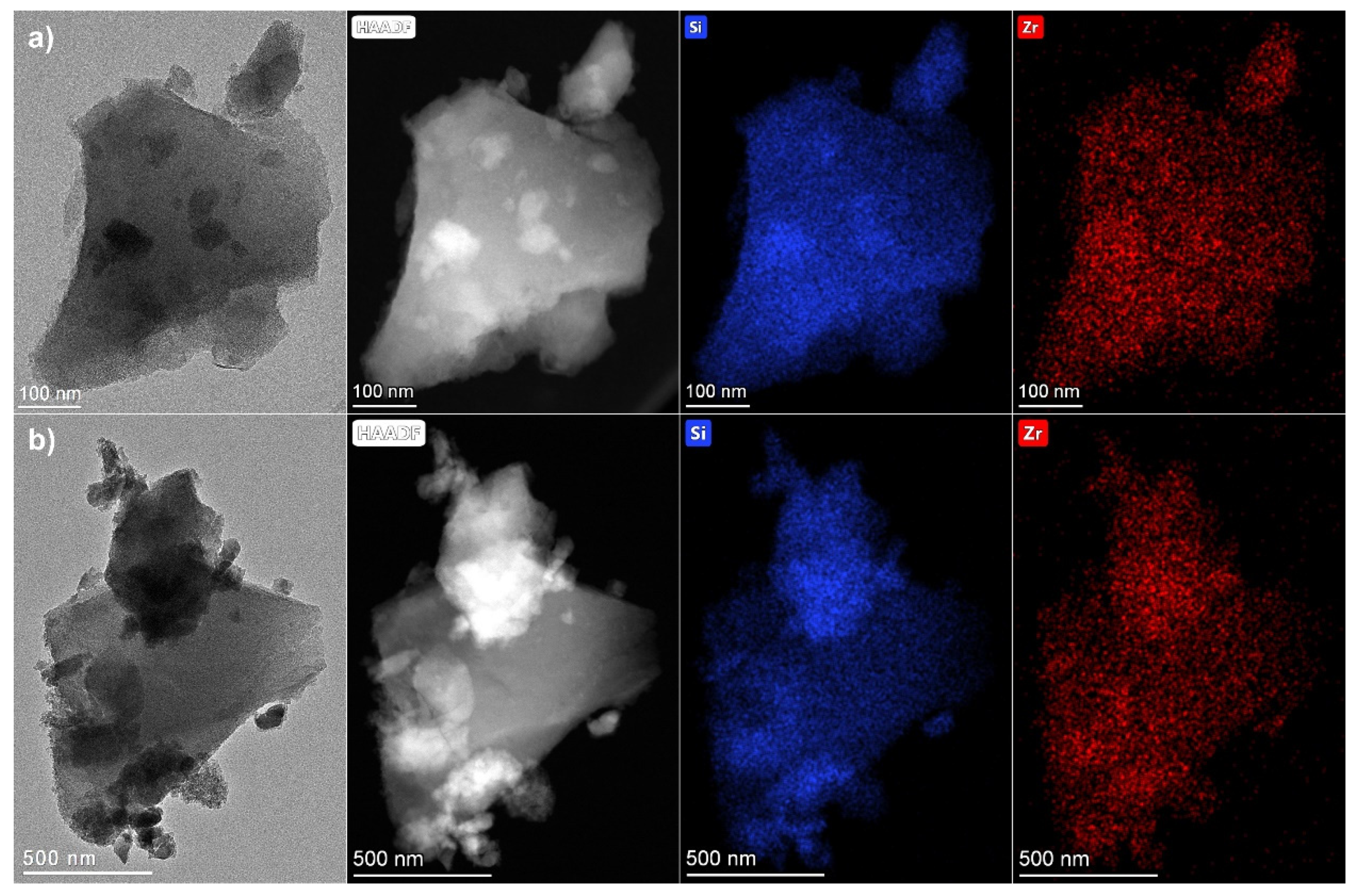
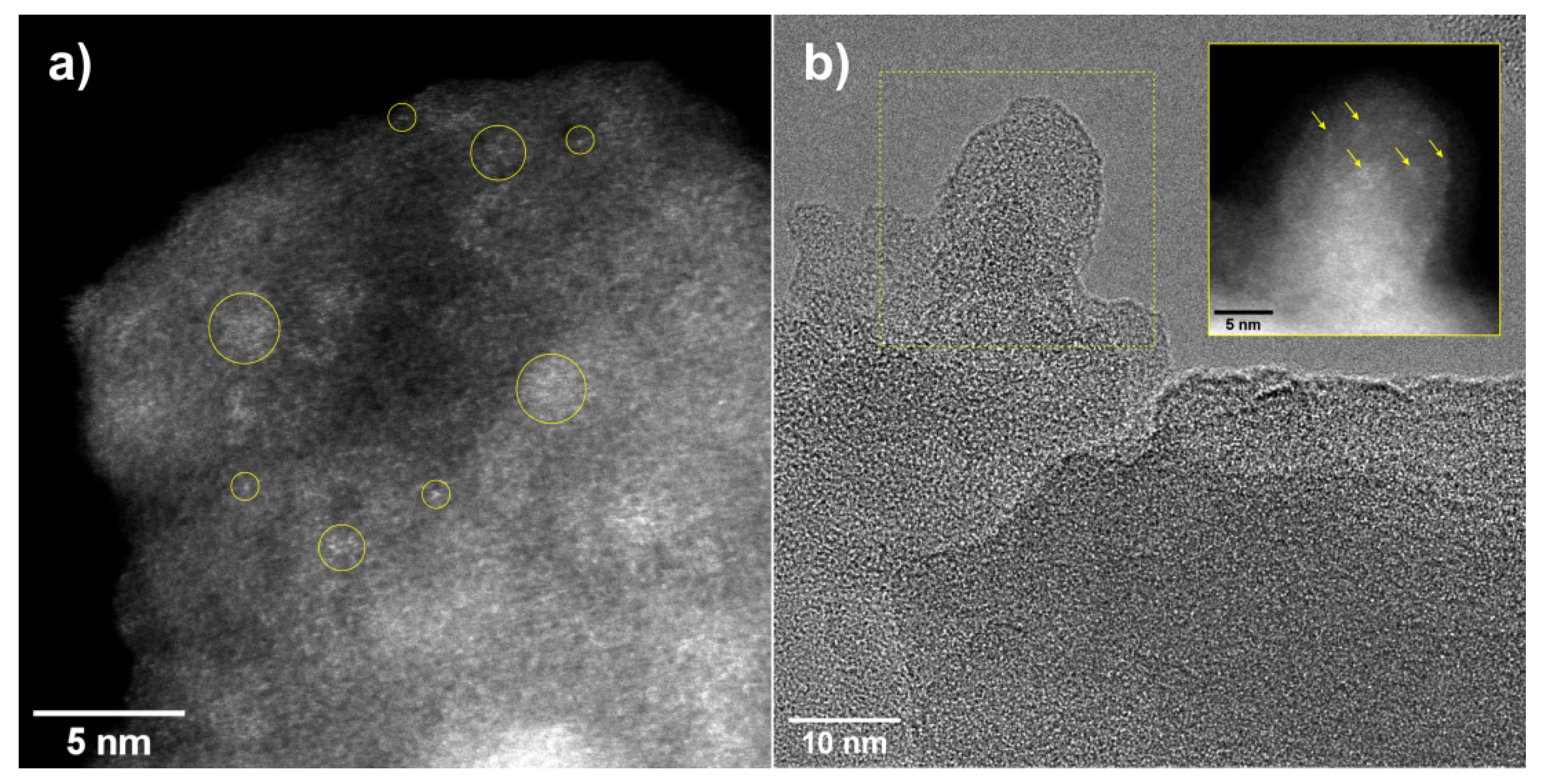
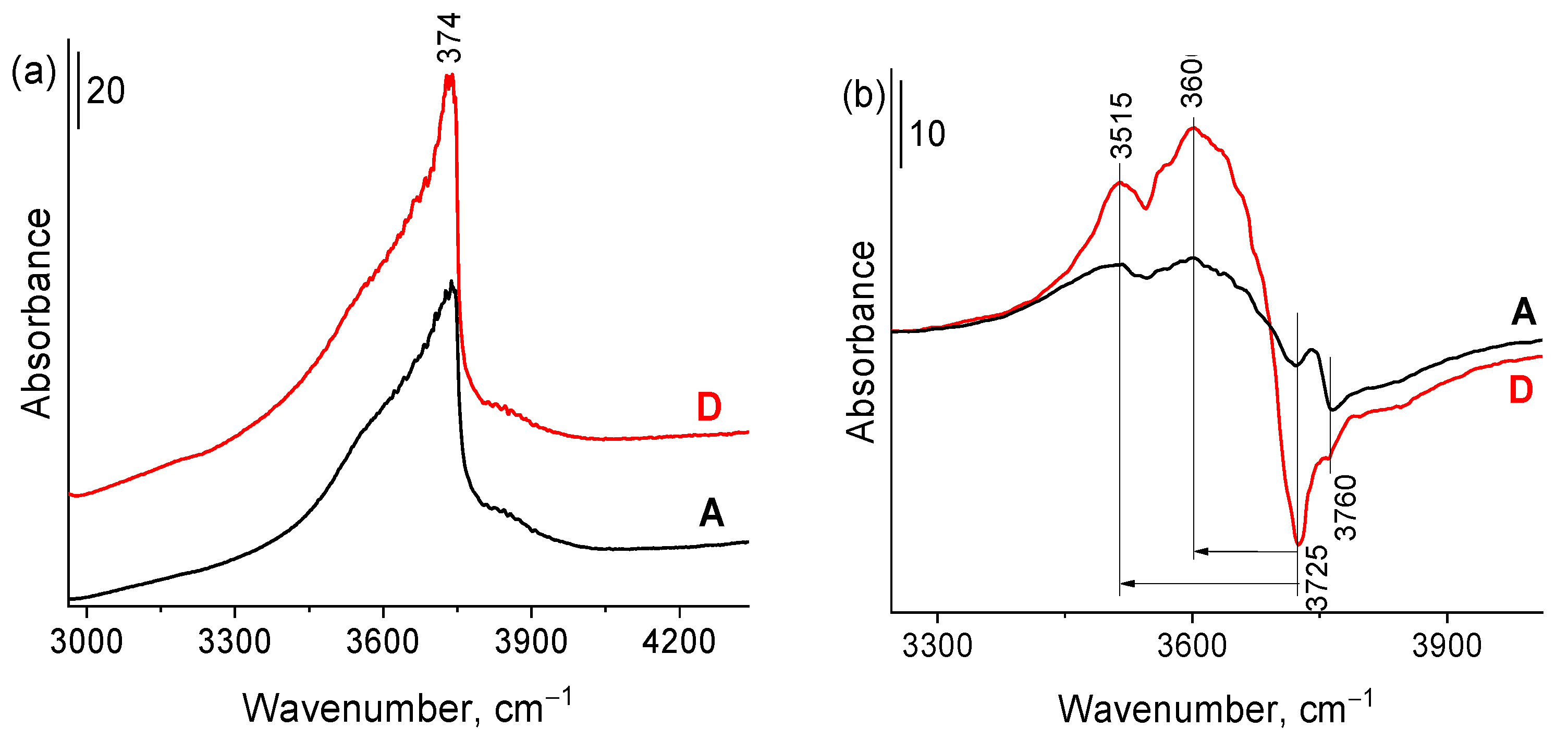
2.1. Synthesis and characterization of Zr-Si-Catalysts
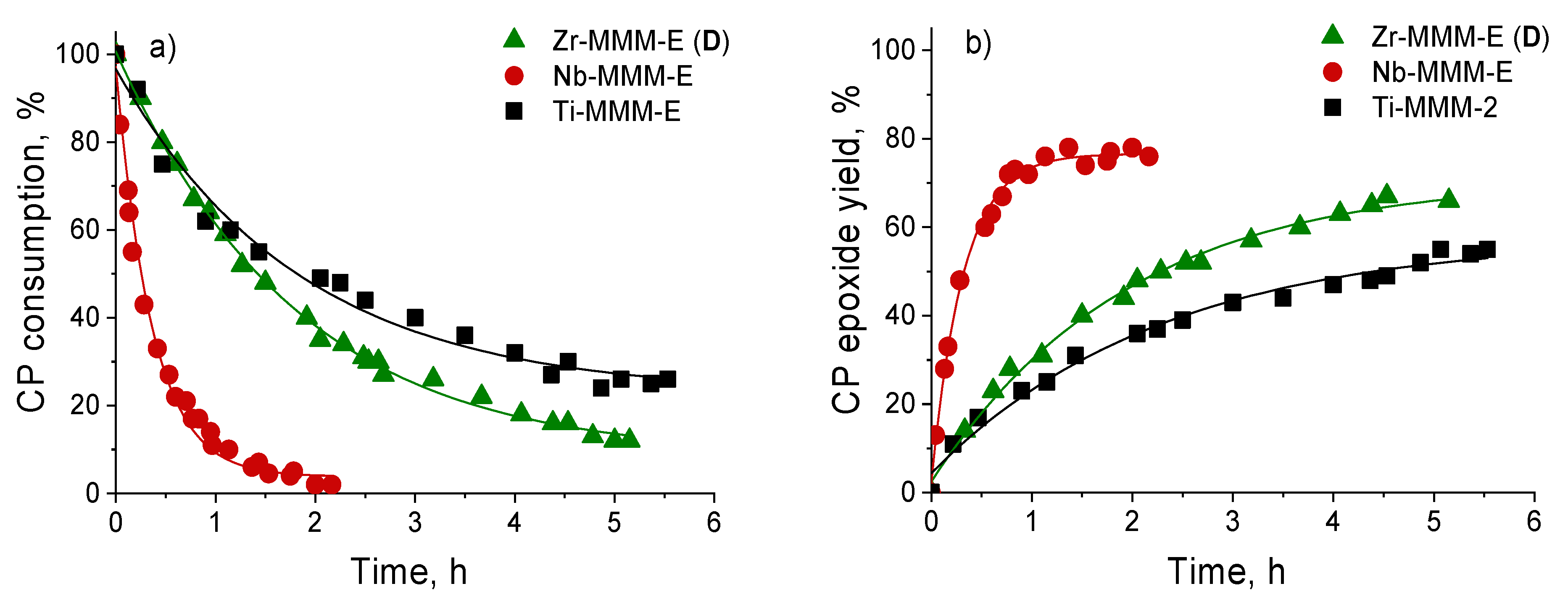
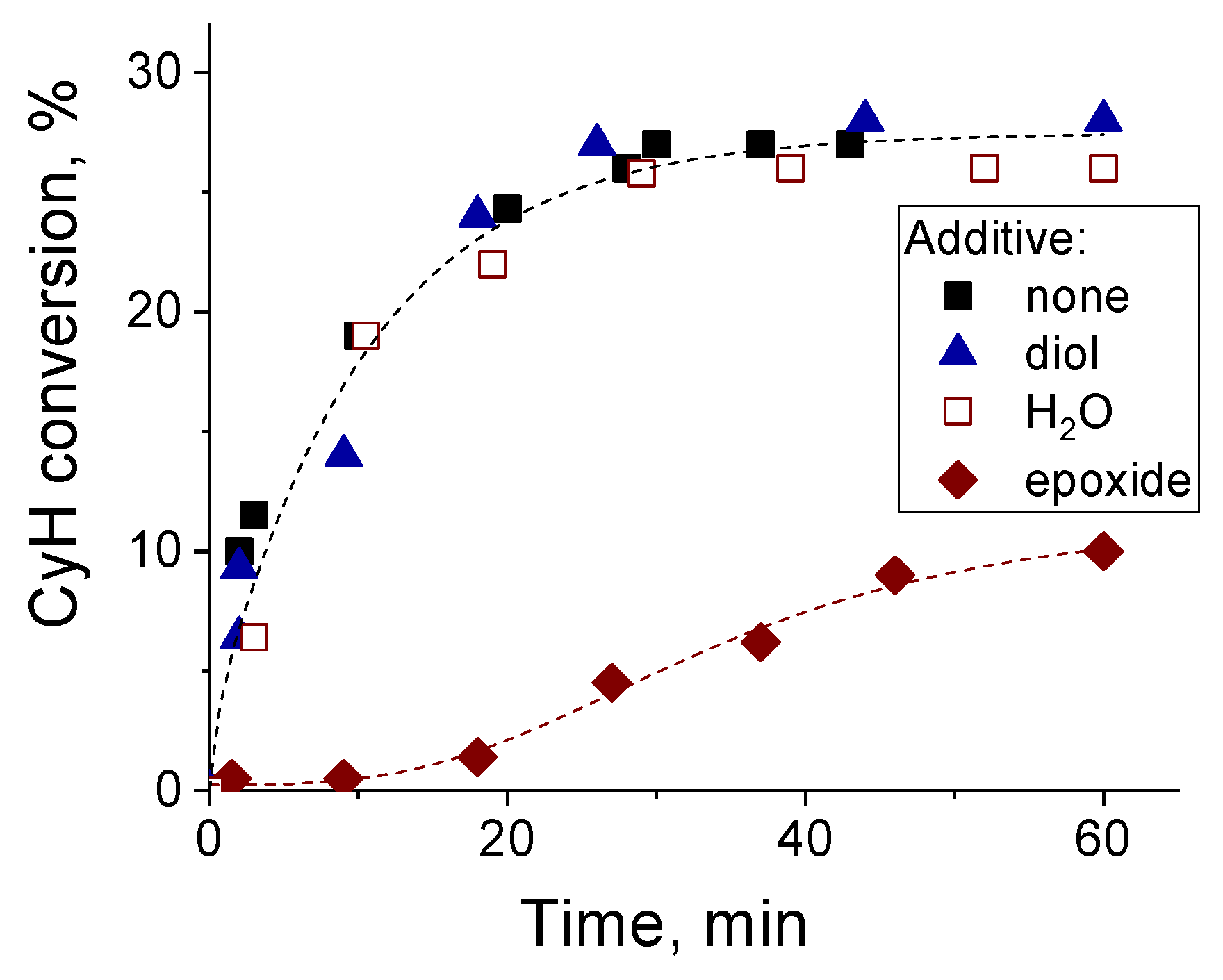
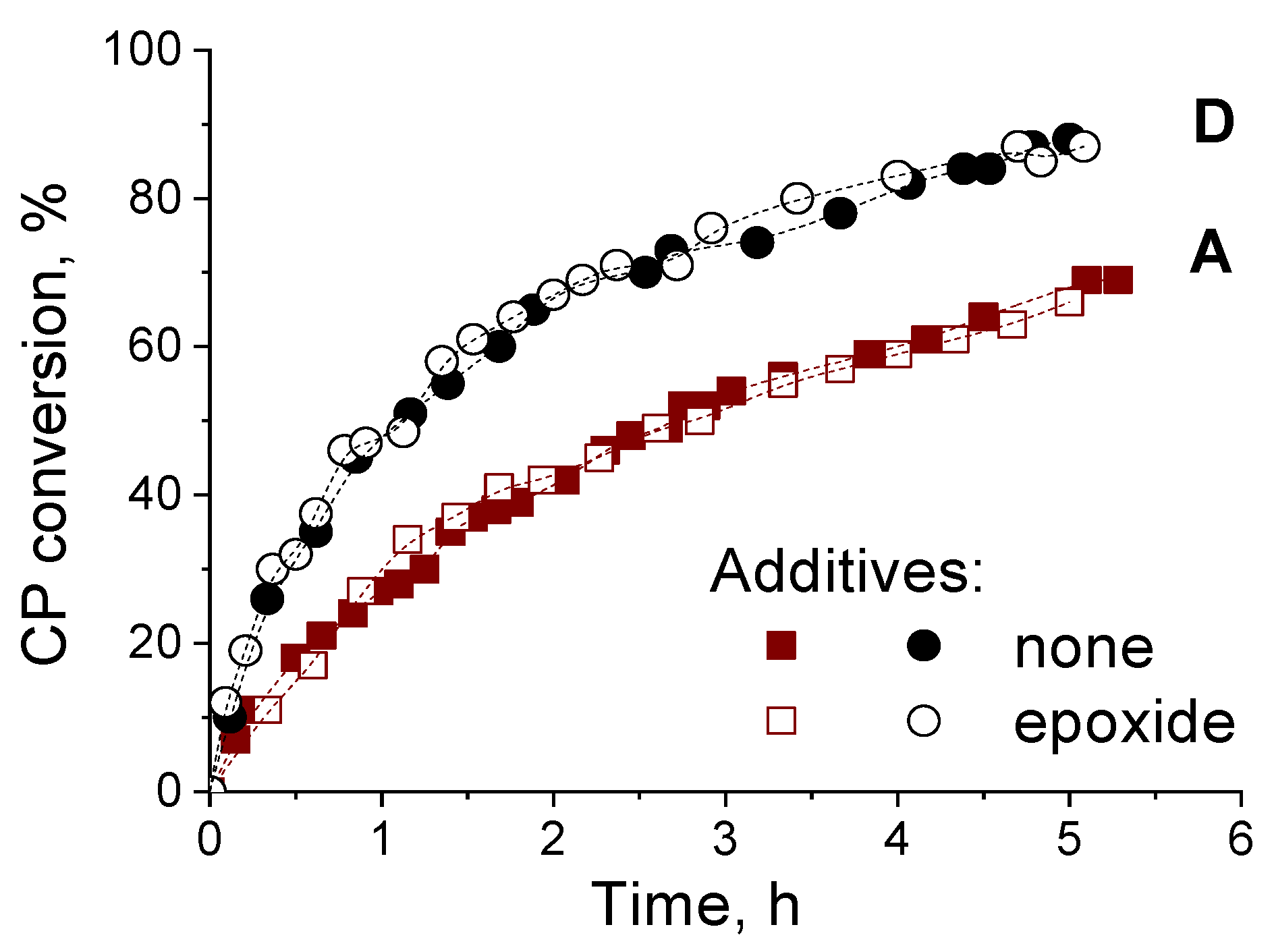
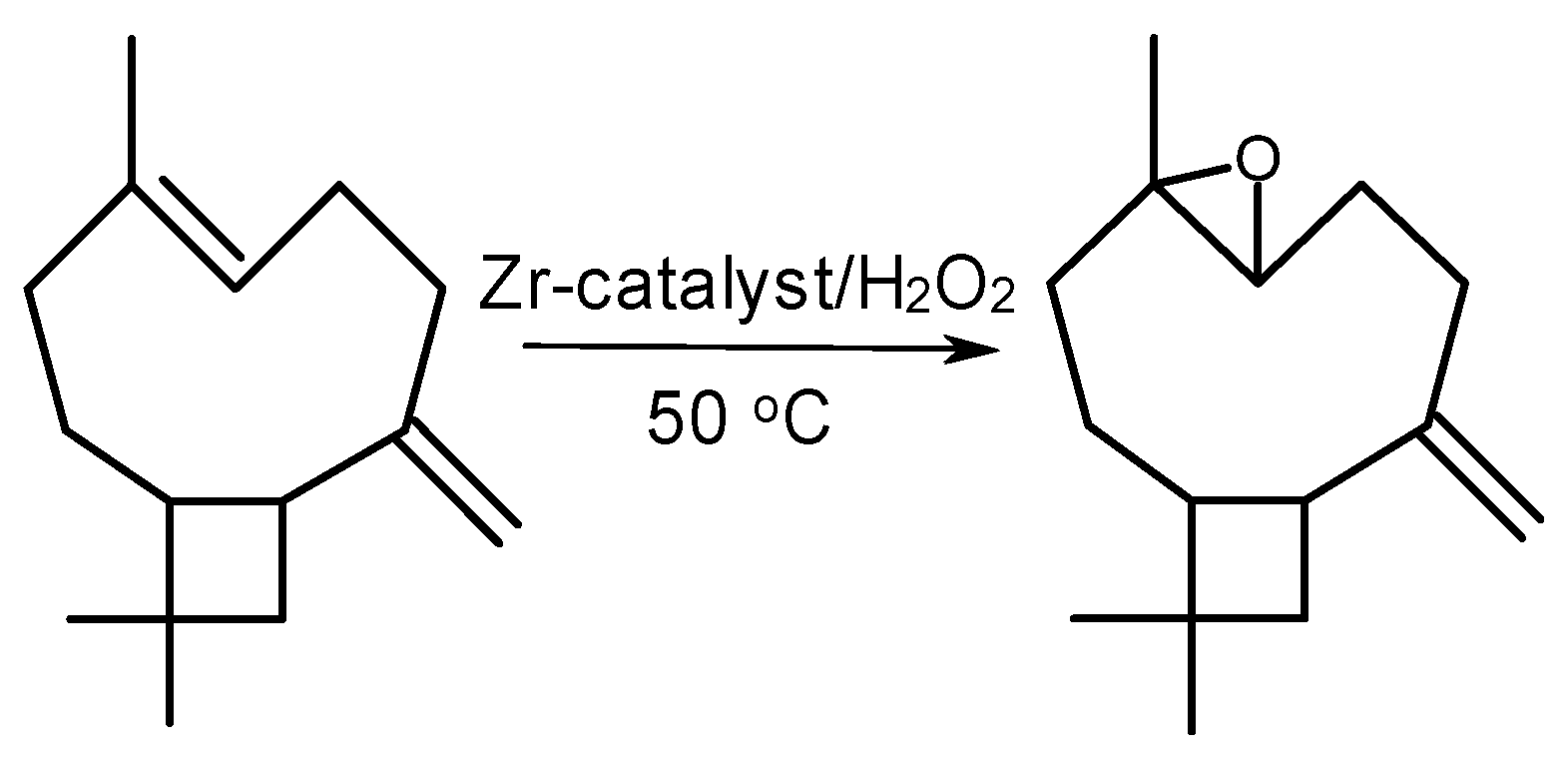
2.2. Catalytic Activity of Zr-Silicates in H2O2-Based Oxidation of Alkenes
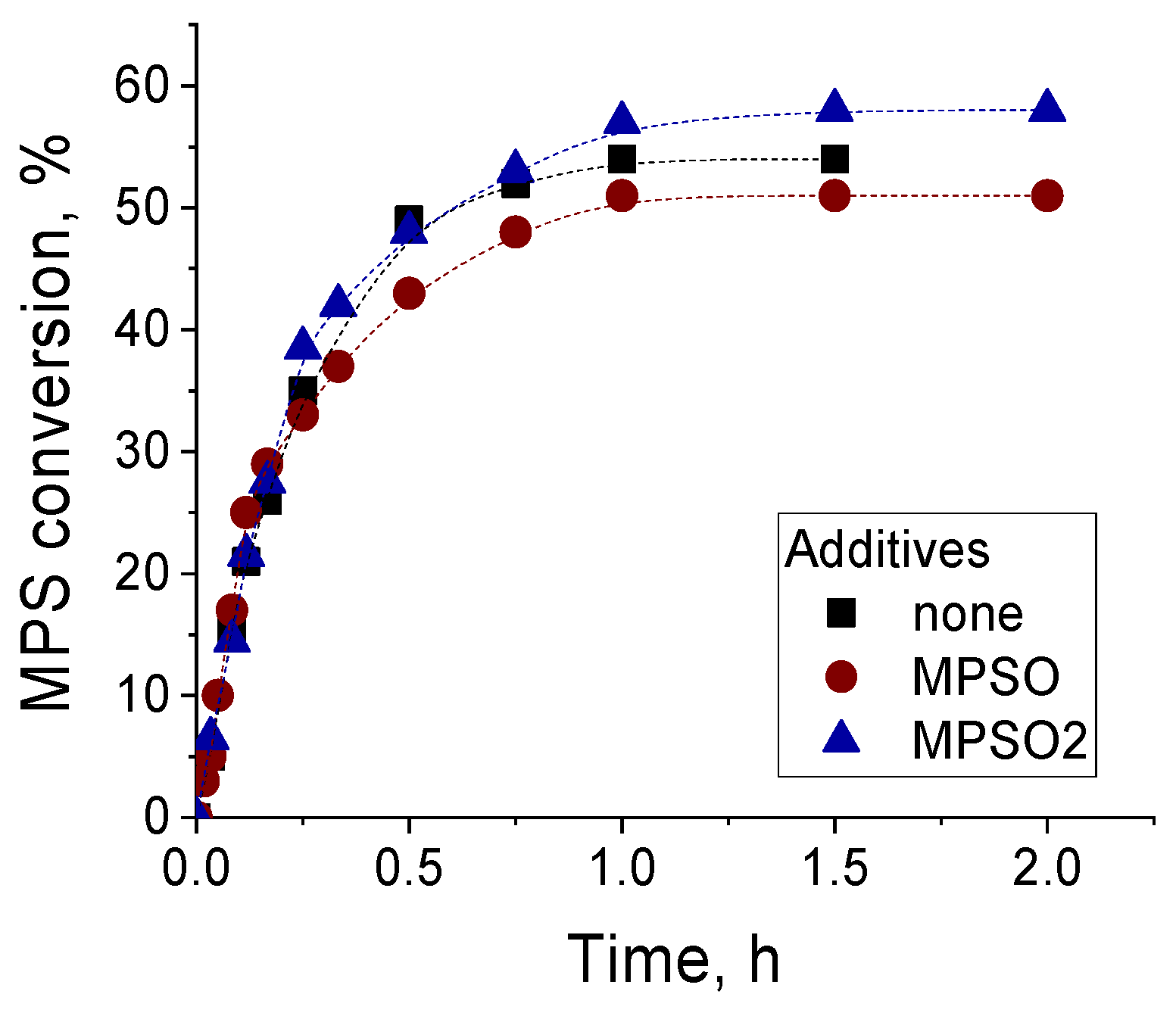
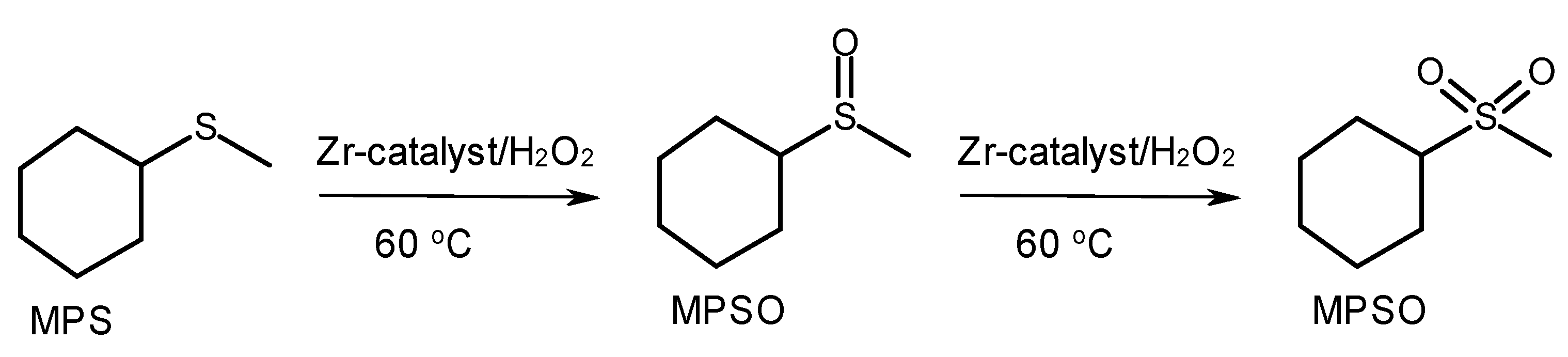
2.3. Catalytic Activity of Zr-silicates in H2O2-Based Oxidation of Methyl Phenyl Sulfide
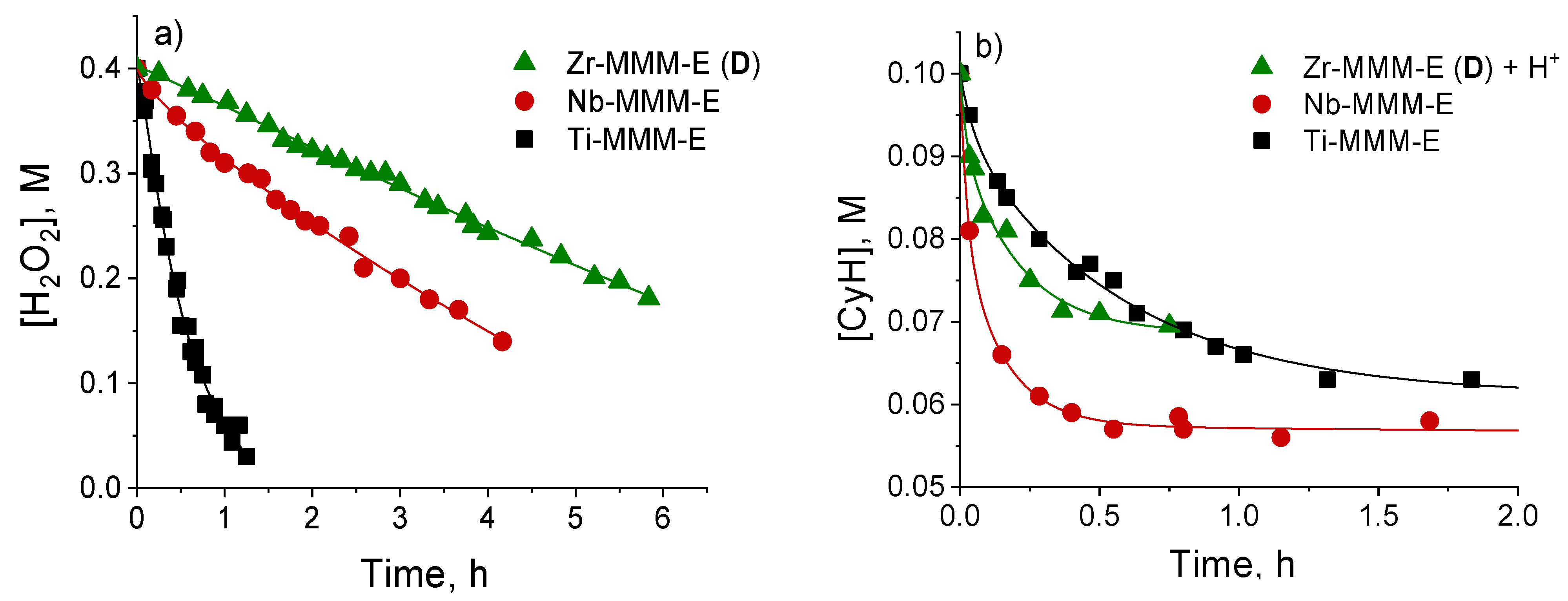
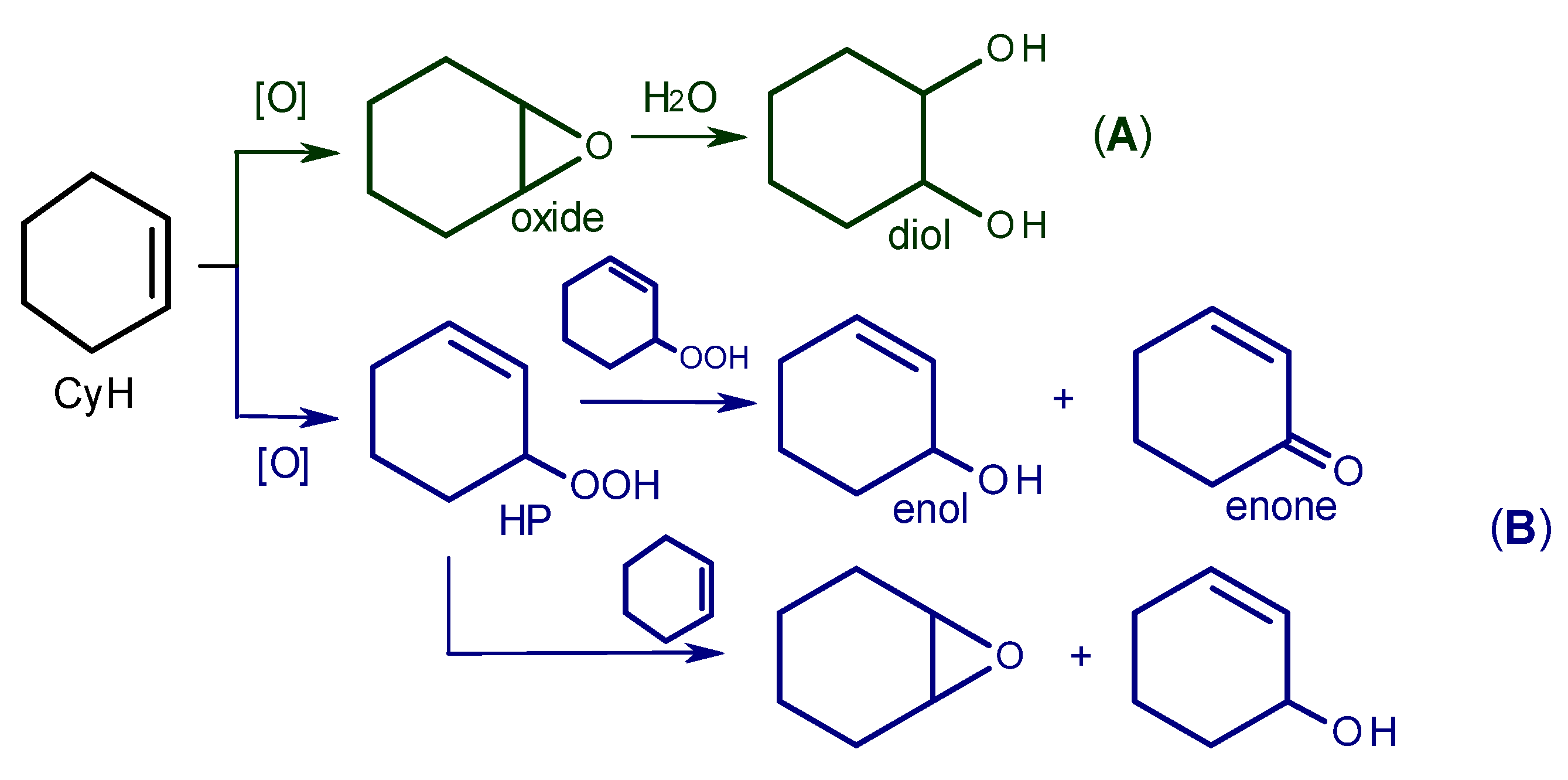
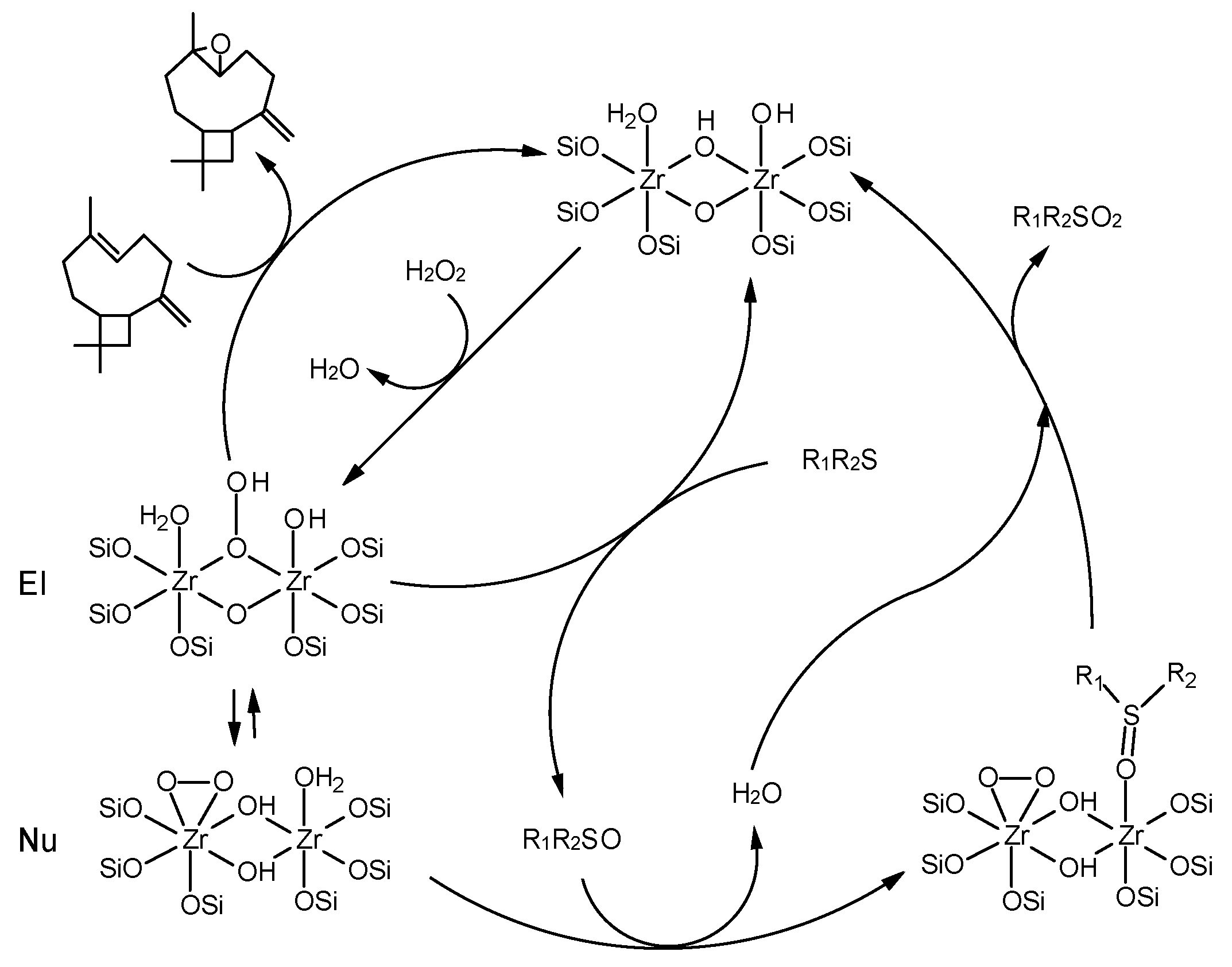
2.4. Mechanism of Alkene and Thioether Oxidation with H2O2 over Zr-Silicate Catalysts
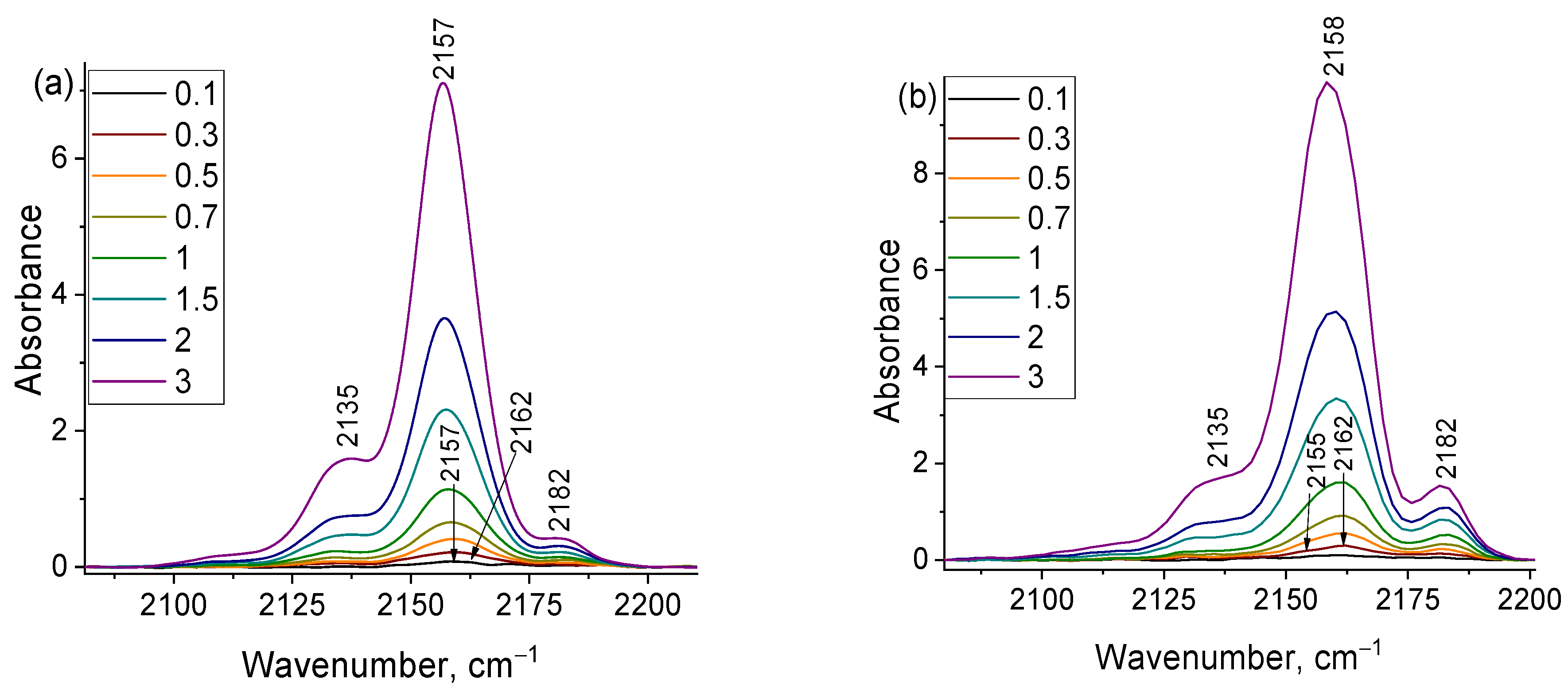
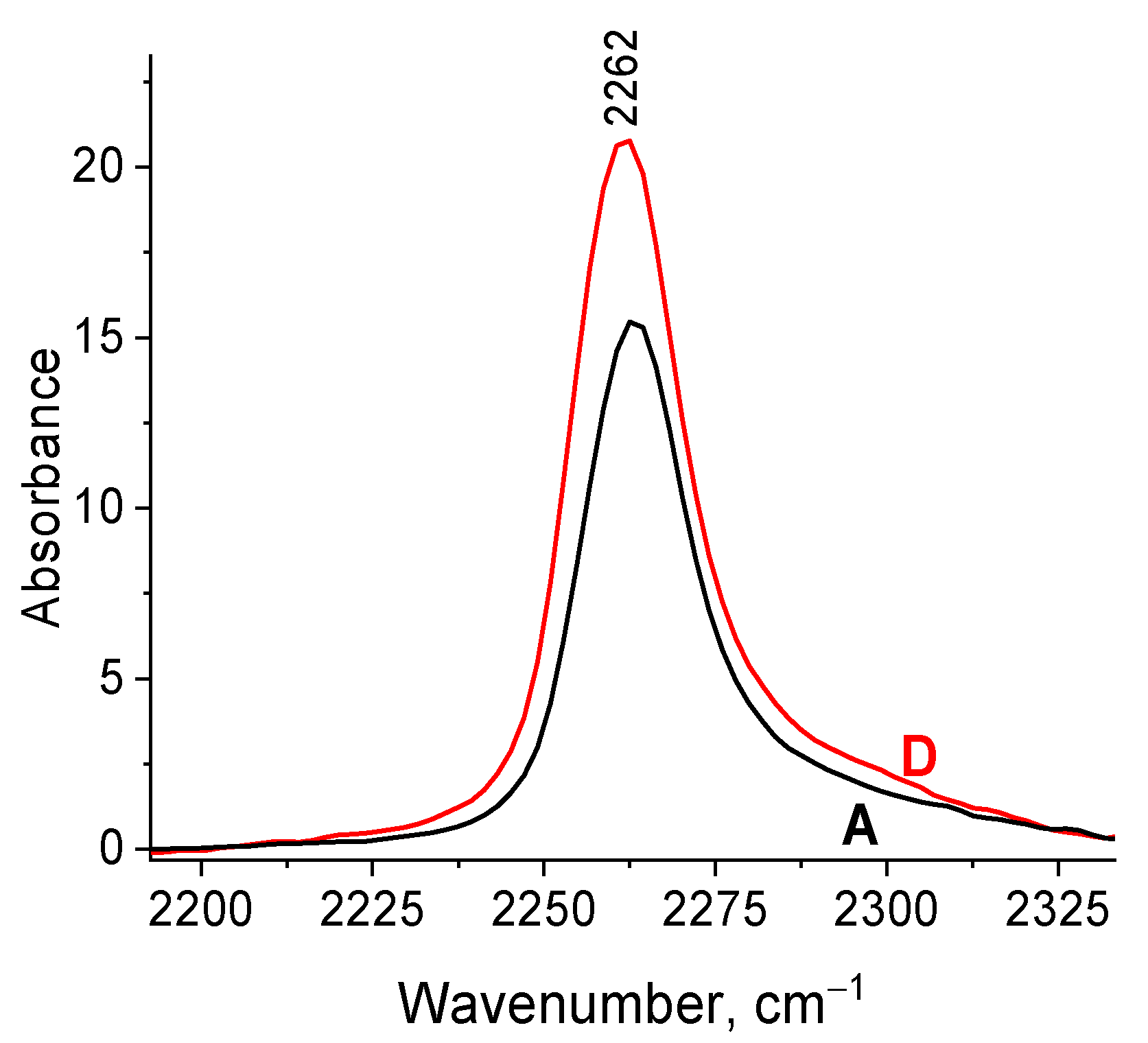
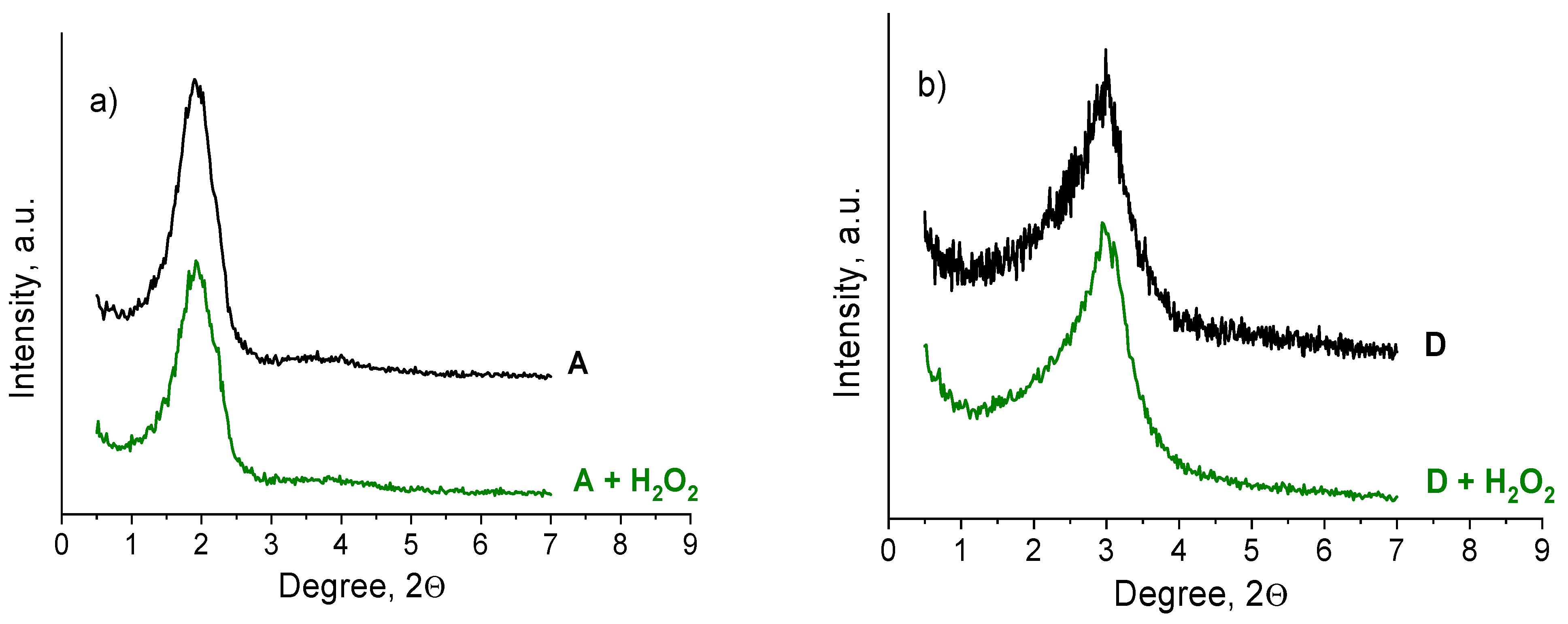
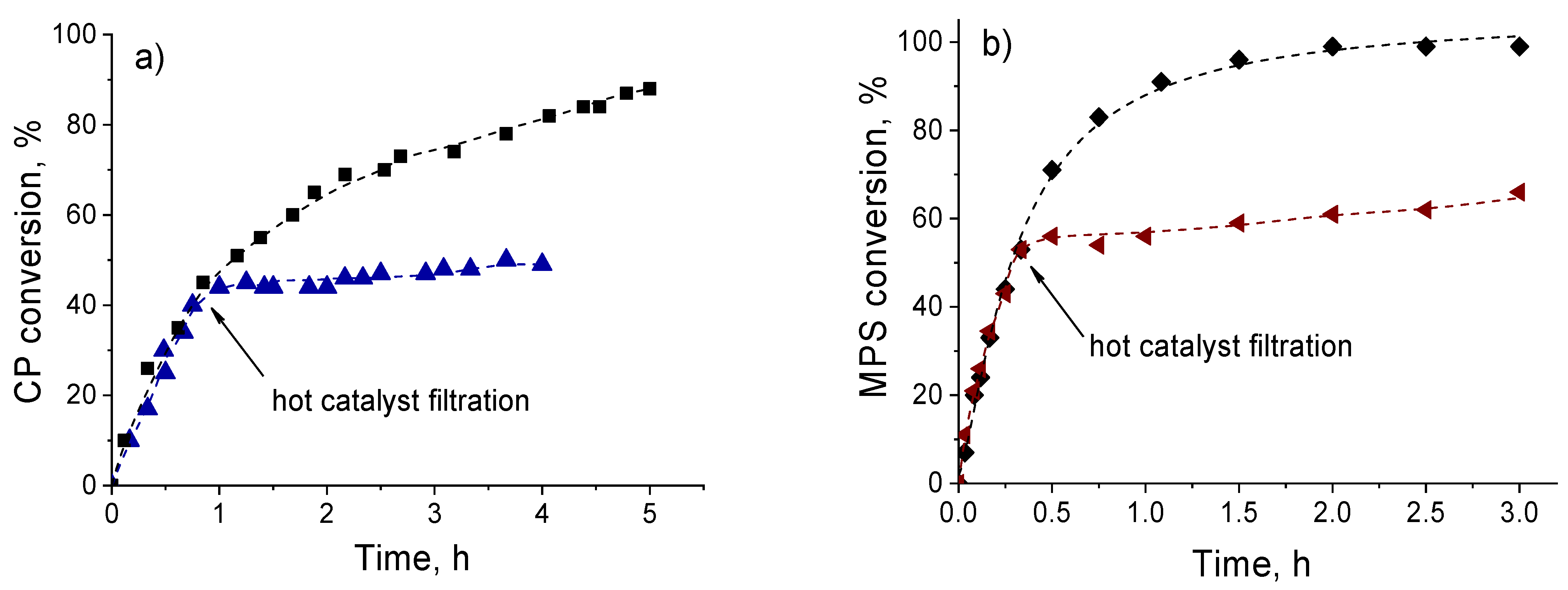
2.5. The reasons for Zr-Si Catalyst Deactivation
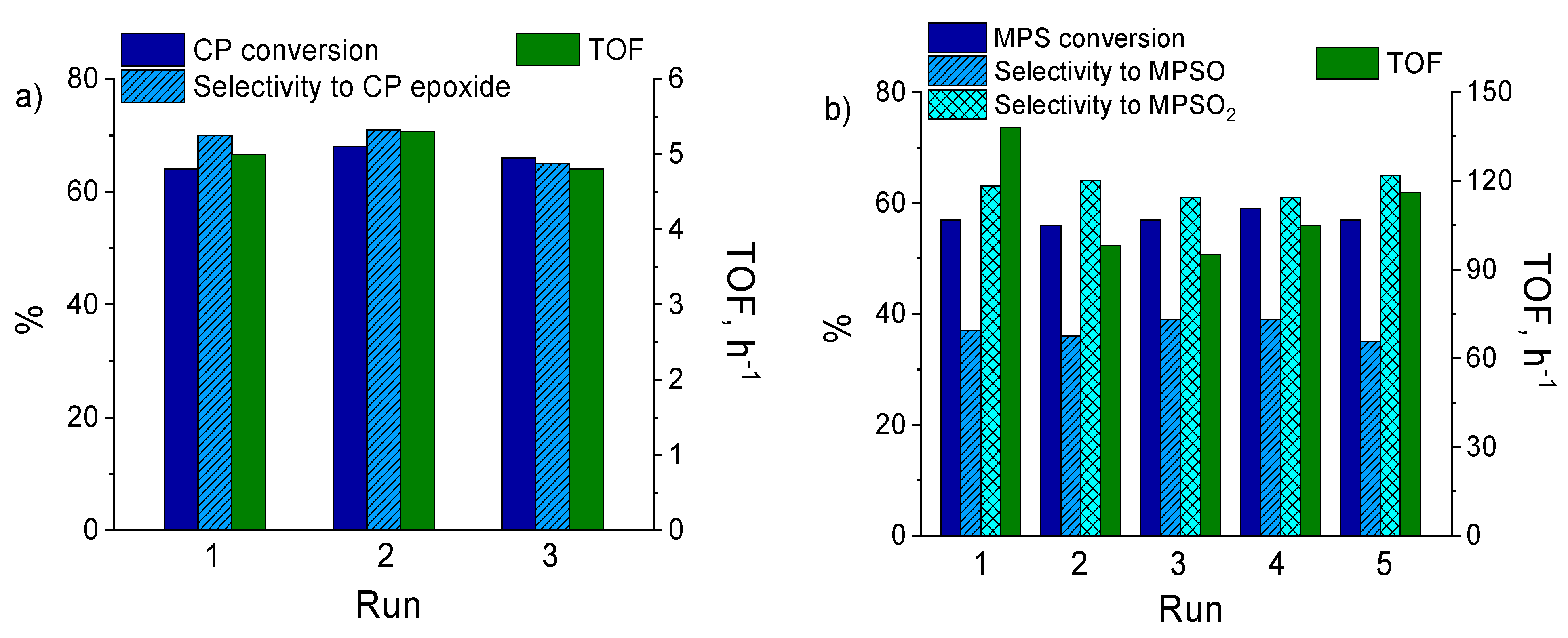
2.6. Catalyst Stability and Reusability
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation and Characterization
3.3. Catalytic Oxidations
3.4. Hydrogen Peroxide Decomposition
3.5. Sorption Studies
3.6. Hydrothermal Stability and Stability towards Aqueous H2O2
3.7. Instrumentation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sienel, G.; Rieth, R.; Rowbottom, K.T. Epoxides. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Clerici, M.G.; Kholdeeva, O.A. (Eds.) Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013; 546p. [Google Scholar] [CrossRef]
- Duprez, D.; Cavani, F. (Eds.) Handbook of Advanced Methods and Processes in Oxidation Catalysis; Imperial College Press: London, UK, 2014; 1036p. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Bekkum, H. Fine Chemicals through Heterogeneous Catalysis; Wiley: Weinheim, Germany, 2001; pp. 473–551. [Google Scholar] [CrossRef]
- Mizuno, N. (Ed.) Modern Heterogeneous Oxidation Catalysis: Design, Reactions and Characterization; Wiley-VCH: Weinheim, Germany, 2009; 356p. [Google Scholar] [CrossRef]
- Sheldon, R.A. E factors, green chemistry and catalysis: An odyssey. Chem. Comm. 2008, 10, 3352–3365. [Google Scholar] [CrossRef]
- Clerici, M.G.; Bellussi, G.; Romano, U. Synthesis of propylene oxide from propylene and hydrogen peroxide catalyzed by titanium silicalite. J. Catal. 1991, 129, 159–167. [Google Scholar] [CrossRef]
- Forlin, A.; Bergamo, M.; Lindner, J. Production of Propylene Oxide. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; Chapter 10.3; pp. 474–495. ISBN 978-0-470-94552-3. [Google Scholar] [CrossRef]
- Tanev, P.T.; Chibwe, M.; Pinnavaia, T. Titanium-Containing Mesoporous Molecular Sieves for Catalytic Oxidation of Aromatic Compounds. Nature 1994, 368, 321–323. [Google Scholar] [CrossRef]
- Sayari, A. Catalysis by Crystalline Mesoporous Molecular Sieves. Chem. Mater. 1996, 8, 1840–1852. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- Tuel, A. Modification of mesoporous silicas by incorporation of heteroelements in the framework. Micropor. Mesopor. Mater. 1999, 27, 151–169. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Selective Oxidations Catalyzed by Mesoporous Metal-Silicates. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; Chapter 4; pp. 127–219. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Recent Developments in Liquid-Phase Selective Oxidation Using Environmentally Benign Oxidants and Mesoporous Metal-Silicates. Catal. Sci. Technol. 2014, 4, 1869–1889. [Google Scholar] [CrossRef]
- Dal Santo, V.; Guidotti, M.; Psaro, R.; Marchese, L.; Carniato, F.; Bisio, C. Rational Design of Single-Site Heterogeneous Catalysts: Towards High Chemo-, Regio-and Stereoselectivity. Proceed. Royal Soc. A 2012, 468, 1904–1926. [Google Scholar] [CrossRef]
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Liang, J.; Liang, Z.; Zou, R.; Zhao, Y. Heterogeneous Catalysis in Zeolites, Mesoporous Silica, and Metal–Organic Frameworks. Adv. Mater. 2017, 29, 1701139. [Google Scholar] [CrossRef]
- Samantaray, M.K.; Pump, E.; Bendjeriou-Sedjerari, A.; D’Elia, A.; Pelletier, J.D.A.; Guidotti, M.; Psaro, R.; Basset, J.M. Surface organometallic chemistry in heterogeneous catalysis. Chem. Soc. Rev. 2018, 47, 8403–8437. [Google Scholar] [CrossRef] [PubMed]
- Kholdeeva, O.A.; Melgunov, M.S.; Shmakov, A.N.; Trukhan, N.N.; Kriventsov, V.V.; Zaikovskii, V.I.; Malishev, M.E.; Romannikov, V.N. A New Mesoporous Titanium-Silicate Ti-MMM-2: A Highly Active and Hydrothermally Stable Catalyst for H2O2-Based Selective Oxidations. Catal. Today 2004, 91–92, 205–209. [Google Scholar] [CrossRef]
- Xiao, F.-S. Ordered Mesoporous Materials with Improved Stability and Catalytic Activity. Topics Catal. 2005, 35, 9–24. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Tuel, A. Metallophthalocyanine Functionalized Silicas: Catalysts for the Selective Oxidation of Aromatic Compounds. Catal. Today 2000, 57, 45–59. [Google Scholar] [CrossRef]
- Xiao, F.S.; Han, Y.; Yu, Y.; Meng, X.; Yang, M.; Wu, S. Hydrothermally Stable Ordered Mesoporous Titanosilicates with Highly Active Catalytic Sites. J. Am. Chem. Soc. 2002, 124, 888–889. [Google Scholar] [CrossRef]
- Wu, P.; Tatsumi, T.; Komatsu, T.; Yashima, T. Postsynthesis, Characterization, and Catalytic Properties in Alkene Epoxidation of Hydrothermally Stable Mesoporous Ti-SBA-15. Chem. Mater. 2002, 14, 1657–1664. [Google Scholar] [CrossRef]
- Moliner, M.; Corma, A. Advances in the Synthesis of Titanosilicates: From the Medium Pore TS-1 Zeolite to Highly-Accessible Ordered Materials. Micropor. Mesopor. Mater. 2014, 189, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, M.; Gavrilova, E.; Galarneau, A.; Coq, B.; Psaro, R.; Ravasio, N. Epoxidation of Methyl Oleate with Hydrogen Peroxide. The Use of Ti-Containing Silica Solids as Efficient Heterogeneous Catalysts. Green Chem. 2011, 13, 1806–1811. [Google Scholar] [CrossRef]
- Guidotti, M.; Pirovano, C.; Ravasio, N.; Lázaro, B.; Fraile, J.M.; Mayoral, J.A.; Coq, B.; Galarneau, A. The Use of H2O2 over Titanium-Grafted Mesoporous Silica Catalysts: A Step Further towards Sustainable Epoxidation. Green Chem. 2009, 11, 1421–1427. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Kovalev, M.K.; Mel’gunov, M.S.; Shmakov, A.N.; Kholdeeva, O.A. User-Friendly Synthesis of Highly Selective and Recyclable Mesostructured Titanium-Silicate Catalysts for the Production of Bulky Benzoquinones. Catal. Sci. Technol. 2014, 4, 200–207. [Google Scholar] [CrossRef]
- Ziolek, M. Niobium-Containing Catalysts—The State of the Art. Catal. Today 2003, 78, 47–64. [Google Scholar] [CrossRef]
- Aronne, A.; Turco, M.; Bagnasco, G.; Ramis, G.; Santacesaria, E.; Di Serio, M.; Marenna, E.; Bevilacqua, M.; Cammarano, C.; Fanelli, E. Gel Derived Niobium–Silicon Mixed Oxides: Characterization and Catalytic Activity for Cyclooctene Epoxidation. Appl. Catal. A General 2008, 347, 179–185. [Google Scholar] [CrossRef]
- Gallo, A.; Tiozzo, C.; Psaro, R.; Carniato, F.; Guidotti, M. Niobium Metallocenes Deposited onto Mesoporous Silica via Dry Impregnation as Catalysts for Selective Epoxidation of Alkenes. J. Catal. 2013, 298, 77–83. [Google Scholar] [CrossRef]
- Ramanathan, A.; Maheswari, R.; Subramaniam, B. Facile Styrene Epoxidation with H2O2 over Novel Niobium Containing Cage Type Mesoporous Silicate, Nb-KIT-5. Top. Catal. 2015, 58, 314–324. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Maksimchuk, N.V.; Skobelev, I.Y.; Kaichev, V.V.; Kholdeeva, O.A. Mesoporous Niobium–Silicates Prepared by Evaporation–Induced Self–Assembly as Catalysts for Selective Oxidations with Aqueous H2O2. J. Catal. 2015, 332, 138–148. [Google Scholar] [CrossRef]
- Hajjami, N.E.; Thompson, A.B.; Notestein, J.M. Periodic Trends in Highly Dispersed Groups IV and V Supported Metal Oxide Catalysts for Alkene Epoxidation with H2O2. ACS Catal. 2015, 5, 5077–5088. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Maksimchuk, N.V.; Skobelev, I.Y. H2O2-Based Selective Oxidations: Nb(V) versus Ti(IV). Catal. Today 2019, 333, 63–70. [Google Scholar] [CrossRef]
- Zhang, Z.; Suo, J.; Zhang, X.; Li, S. Synthesis of Highly Active Tungsten-Containing MCM-41 Mesoporous Molecular, Sieve Catalyst. Chem. Commun. 1998, 2, 241–242. [Google Scholar] [CrossRef]
- Brégeault, J.-M.; Piquemal, J.-Y.; Briot, E.; Duprey, E.; Launay, F.; Salles, L.; Vennat, M.; Legrand, A.-P. New Approaches to Anchoring or Inserting Highly Dispersed Tungsten Oxo (Peroxo) Species in Mesoporous Silicates. Micropor. Mesopor. Mater. 2001, 44–45, 409–417. [Google Scholar] [CrossRef]
- Dai, W.-L.; Chen, H.; Cao, Y.; Li, H.; Xie, S.; Fan, K. Novel Economic and Green Approach to the Synthesis of Highly Active W-MCM41 Catalyst in Oxidative Cleavage of Cyclopentene. Chem. Commun. 2003, 7, 892–893. [Google Scholar] [CrossRef]
- Gao, R.; Yanga, X.; Dai, W.-L.; Le, Y.; Li, H.; Fan, K. High-Activity, Single-Site Mesoporous WO3-MCF Materials for the Catalytic Epoxidation of Cycloocta-1,5-diene with Aqueous Hydrogen Peroxide. J. Catal. 2008, 256, 259–267. [Google Scholar] [CrossRef]
- Yan, W.; Ramanathan, A.; Ghanta, M.; Subramaniam, B. Towards Highly Selective Ethylene Epoxidation Catalysts Using Hydrogen Peroxide and Tungsten- or Niobium-Incorporated Mesoporous Silicate (KIT-6). Catal. Sci. Technol. 2014, 4, 4433–4439. [Google Scholar] [CrossRef] [Green Version]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Zalomaeva, O.V.; Skobelev, I.Y.; Chesalov, Y.A.; Shmakov, A.A.; Kholdeeva, O.A. Tungsten-Incorporated Mesoporous Silicate W-MMM-E as Heterogeneous Catalyst for Liquid-Phase Oxidations with Aqueous H2O2. Catalysts 2018, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Ten Dam, J.; Badloe, D.; Ramanathan, A.; Djanashvili, K.; Kapteijn, F.; Hanefeld, U. Synthesis, Characterisation and Catalytic Performance of a Mesoporous Tungsten Silicate: W-TUD-1. Appl. Catal. A General 2013, 468, 150–159. [Google Scholar] [CrossRef]
- Gontier, S.; Tuel, A. Novel Zirconium Containing Mesoporous SIlicas for Oxidation Reactions in the liquid Phase. Appl. Catal. A Gen. 1996, 143, 125–135. [Google Scholar] [CrossRef]
- Besson, M.; Bonnet, M.C.; Gallezot, P.; Tkatchenko, I.; Tuel, A. Catalysis for Fine Chemicals: Towards Specificity with Polyphasic Media. Catal. Today 1999, 51, 547–560. [Google Scholar] [CrossRef]
- Morey, M.S.; Stucky, G.D.; Schwarz, S.; Fröba, M. Isomorphic Substitution and Postsynthesis Incorporation of Zirconium into MCM-48 Mesoporous Silica. J. Phys. Chem. B 1999, 103, 2037–2041. [Google Scholar] [CrossRef]
- Palazzi, C.; Oliva, L.; Signoretto, M.; Strukul, G. Microporous Zirconia–Silica Mixed Oxides Made by Sol–Gel as Catalysts for the Liquid-Phase Oxidation of Olefins with Hydrogen Peroxide. J. Catal. 2000, 194, 286–293. [Google Scholar] [CrossRef]
- Morandin, M.; Gavagnin, R.; Pinna, F.; Strukul, G. Oxidation of Cyclohexene with Hydrogen Peroxide Using Zirconia–Silica Mixed Oxides: Control of the Surface Hydrophilicity and Influence on the Activity of the Catalyst and Hydrogen Peroxide Efficiency. J. Catal. 2002, 212, 193–200. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Melgunov, M.S.; Mrowiec-Białoń, J.; Jarzębski, A.B.; Kholdeeva, O.A. H2O2-Based Allylic Oxidation of α-Pinene over Different Single Site Catalysts. J. Catal. 2005, 235, 175–183. [Google Scholar] [CrossRef]
- Chaudhari, K.; Bal, R.; Srinivas, D.; Chandwadkar, A.J.; Sivasanker, S. Redox Behavior and Selective Oxidation Properties of Mesoporous Titano- and Zirconosilicate MCM-41 Molecular Sieves. Microporous Mesoporous Mater. 2001, 50, 209–218. [Google Scholar] [CrossRef]
- Liu, J.M.; Liao, S.J.; Jiang, G.D.; Zhang, X.L.; Petrik, L. Preparation, Characterization and Catalytic Activity of Zr Embedded MSU-V with High Thermal and Hydrothermal Stability. Microporous Mesoporous Mater. 2006, 95, 306–311. [Google Scholar] [CrossRef]
- Quignard, F.; Choplin, A.; Teissier, R. A Molecular Route Towards Silica Supported Zirconium Catalysts Active for the Mild Oxidation of Olefins with H2O2. J. Mol. Catal. A Chem. 1997, 120, L27–L31. [Google Scholar] [CrossRef]
- Thornburg, N.E.; Notestein, J.M. Rate and Selectivity Control in Thioether and Alkene Oxidation with H2O2 over Phosphonate-Modified Niobium(V)–Silica Catalysts. ChemCatChem 2017, 9, 3714–3724. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Evtushok, V.Y.; Zalomaeva, O.V.; Maksimov, G.M.; Ivanchikova, I.D.; Chesalov, Y.A.; Eltsov, I.V.; Abramov, P.A.; Glazneva, T.S.; Yanshole, V.V.; et al. Activation of H2O2 over Zr(IV). Insights from Model Studies on Zr-Monosubstituted Lindqvist Tungstates. ACS Catal. 2021, 11, 10589–10603. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Cho, K.H.; Zalomaeva, O.V.; Evtushok, V.Y.; Larionov, K.P.; Glazneva, T.S.; Chang, J.-S.; Kholdeeva, O.A. Catalytic Performance of Zr-Based Metal-Organic Frameworks Zr-abtc and MIP-200 in Selective Oxidations with H2O2. Chem. Eur. J. 2021, 27, 6985–6992. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Maksimchuk, N.V. Metal-Organic Frameworks in Oxidation Catalysis with H2O2. Catalysts 2021, 11, 283. [Google Scholar] [CrossRef]
- Jaenicke, S.; Loh, W.L. Preparation of highly dispersed molybdenum on alumina by thermal decomposition of Mo(CO)6. Catal. Today 1999, 49, 123–130. [Google Scholar] [CrossRef]
- Debecker, D.P.; Stoyanova, M.; Rodemerck, U.; Eloy, P.; Leonard, A.; Su, B.L.; Gaigneaux, E.M. Thermal Spreading As an Alternative for the Wet Impregnation Method: Advantages and Downsides in the Preparation of MoO3/SiO2–Al2O3 Metathesis Catalysts. J. Phys. Chem. C 2010, 114, 18664–18673. [Google Scholar] [CrossRef]
- Tiozzo, C.; Bisio, C.; Carniato, F.; Marchese, L.; Gallo, A.; Ravasio, N.; Psaro, R.; Guidotti, M. Epoxidation with Hydrogen Peroxide of Unsaturated Fatty Acid Methyl Esters over Nb(V)-Silica Catalysts. Eur. J. Lipid Sci. Technol. 2013, 115, 86–93. [Google Scholar] [CrossRef]
- Nielsen, R.H.; Schlewitz, J.H.; Nielsen, H. Zirconium and Zirconium Compounds. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Schubert, U. Chemical Modification of Titanium Alkoxides for Sol–Gel Processing. J. Mater. Chem. 2005, 15, 3701–3715. [Google Scholar] [CrossRef]
- Tuel, A.; Gontier, S.; Teissier, R. Zirconium Containing Mesoporous Silicas: New Catalysts for Oxidation Reactions in the Liquid Phase. Chem. Commun. 1996, 651–652. [Google Scholar] [CrossRef]
- El Haskouri, J.; Cabrera, S.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Marcos, M.D.; Amorós, P. Atrane Precursors in the One-Pot Surfactant-Assisted Synthesis of High Zirconium Content Porous Silicas. Chem. Mater. 2002, 14, 5015–5022. [Google Scholar] [CrossRef]
- Newalkar, B.L.; Olanrewaju, J.; Komarneni, S. Microwave-Hydrothermal Synthesis and Characterization of Zirconium Substituted SBA-15 Mesoporous Silica. J. Phys. Chem. B 2001, 105, 8356–8360. [Google Scholar] [CrossRef]
- Ramanathan, A.; Castro Villalobos, M.; Kwakernaak, C.; Telalovic, S.; Hanefeld, U. Zr-TUD-1: A Lewis Acidic, Three-Dimensional, Mesoporous, Zirconium-Containing Catalyst. Chem. Eur. J. 2008, 14, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Maheswari, R.; Ramanathan, A.; Subramaniam, B. Evaporation-Induced Self-Assembly of Mesoporous Zirconium Silicates with Tunable Acidity and Facile Catalytic Dehydration Activity. Microporous Mesoporous Mater. 2016, 223, 46–52. [Google Scholar] [CrossRef]
- Barberis, P.; Merle-Méjean, T.; Quintard, P. On Raman Spectroscopy of Zirconium Oxide Films. J. Nucl. Mater. 1997, 246, 232–243. [Google Scholar] [CrossRef]
- Kontoyannis, C.G.; Orkoula, M. Quantitative Determination of the Cubic, Tetragonal and Monoclinic Phases in Partially Stabilized Zirconias by Raman Spectroscopy. J. Mater. Sci. 1994, 29, 5316–5320. [Google Scholar] [CrossRef]
- Morrow, B.A.; McFarlan, A.J. Infrared and Gravimetric Study of an Aerosil and a Precipitated Silica using Chemical and Hydrogen/Deuterium Exchange Probes. Langmuir 1991, 7, 1695–1701. [Google Scholar] [CrossRef]
- Miller, J.B.; Ko, E.I. Acidic Properties of Silica-Containing Mixed Oxide Aerogels: Preparation and Characterization of Zirconia–Silica and Comparison to Titania–Silica. J. Catal. 1996, 159, 58–68. [Google Scholar] [CrossRef]
- Flego, C.; Carluccio, L.; Rizzo, C.; Perego, C. Synthesis of Mesoporous SiO2–ZrO2 Mixed Oxides by Sol–Gel Method. Catal. Commun. 2001, 2, 43–48. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Lavalley, J.C. FT–IR Spectroscopic Study of CO Adsorption on Monoclinic Zirconia of Different Hydroxylation Degrees. Catal. Commun. 2001, 2, 129–133. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Di Ciero, S. IR Study of the Low Temperature Adsorption of CO on Tetragonal Zirconia and Sulfated Tetragonal Zirconia. Appl. Surf. Sci. 1998, 126, 107–128. [Google Scholar] [CrossRef]
- Paukshtis, E.A.; Yurchenko, E.N. Study of the Acid–Base Properties of Heterogeneous Catalysts by Infrared Spectroscopy. Russ. Chem. Rev. 1983, 52, 242–258. [Google Scholar] [CrossRef]
- Trukhan, N.N.; Panchenko, A.A.; Roduner, E.; Melgunov, M.S.; Kholdeeva, O.A.; Mrowiec-Bialon, J.; Jarzebski, A.B. FTIR Spectroscopic Study of Titanium-Containing Mesoporous Silicate Materials. Langmuir 2005, 21, 10545–10554. [Google Scholar] [CrossRef]
- Zecchina, A.; Bordiga, S.; Spoto, G.; Marchese, I.; Petrini, G.; Leofanti, G.; Padovan, M. Silicalite Characterization. 2. IR Spectroscopy of the Interaction of Carbon Monoxide with Internal and External Hydroxyl Groups. J. Phys. Chem. 1992, 96, 4991–4997. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Morita, T.; Salama, T.M.; Tanabe, K. Surface properties of ZrO2 dispersed on SiO2. Catal. Lett. 1990, 4, 1–6. [Google Scholar] [CrossRef]
- Chen, L.F.; Zhou, X.L.; Norena, L.E.; Wang, J.A.; Navarrete, J.; Salas, P.; Montoya, A.; Del Angel, P.; Llanos, M.E. Comparative Studies of Zr-Based MCM-41 and MCM-48 Mesoporous Molecular Sieves: Synthesis and Physicochemical Properties. Appl. Surf. Sci. 2006, 253, 2443–2451. [Google Scholar] [CrossRef]
- Ramanathan, A.; Subramaniam, B.; Maheswari, R.; Hanefeld, U. Synthesis and Characterization of Zirconium Incorporated Ultra Large Pore Mesoporous Silicate, Zr–KIT-6. Micropor. Mesopor. Mater. 2013, 167, 207–212. [Google Scholar] [CrossRef]
- Vasudevan, S.V.; Cai, J.; Bu, Q.; Mao, H. Ordered Mesoporous Zirconium Silicates as a Catalyst for Biofuel Precursors Synthesis. Mol. Catal. 2021, 516, 112003. [Google Scholar] [CrossRef]
- Nash, C.P.; Ramanathan, A.; Ruddy, D.A.; Behl, M.; Gjersing, E.; Griffin, M.; Zhu, H.; Subramaniam, B.; Schaidle, J.A.; Hensley, J.E. Mixed Alcohol Dehydration over Brønsted and Lewis Acidic Catalysts. Appl. Catal. A Gen. 2016, 510, 110–124. [Google Scholar] [CrossRef] [Green Version]
- Ivanchikova, I.D.; Skobelev, I.Y.; Maksimchuk, N.V.; Paukshtis, E.A.; Shashkov, M.V.; Kholdeeva, O.A. Toward Understanding the Unusual Reactivity of Mesoporous Niobium Silicates in Epoxidation of C=C Bonds with Hydrogen Peroxide. J. Catal. 2017, 356, 85–99. [Google Scholar] [CrossRef]
- Soltanov, R.I.; Paukshtis, E.A.; Yurchenko, E.N. IK-Study of Thermodynamic Characteristics of Carbon Monoxide Interaction with the Surface of Some Oxide Adsorbents. Kinet. Catal. 1982, 23, 164–170. [Google Scholar]
- Zalomaeva, O.V.; Evtushok, V.Y.; Ivanchikova, I.D.; Glazneva, T.S.; Chesalov, Y.A.; Larionov, K.P.; Skobelev, I.Y.; Kholdeeva, O.A. Nucleophilic versus Electrophilic Activation of Hydrogen Peroxide over Zr-Based Metal–Organic Frameworks. Inorg. Chem. 2020, 59, 10634–10649. [Google Scholar] [CrossRef] [PubMed]
- Paukshtis, E.A.; Kotsarenko, N.S.; Karakchiev, L.G. Investigation of Proton-Acceptor Properties of Oxide Surfaces by IR Spectroscopy of Hydrogen-Bonded Complexes. React. Kinet. Catal. Lett. 1979, 12, 315–319. [Google Scholar] [CrossRef]
- Paukshtis, E.A. Infrared Spectroscopy in Heterogeneous Acid–Base Catalysis; Novosibirsk: Nauka, Russia, 1992; 253p. (In Russian) [Google Scholar]
- Sheldon, R.A.; Kochi, J.K. Metal–Catalyzed Oxidations of Organic Compounds; Academic Press: New York, NY, USA, 1981; 446p. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Lee, J.S.; Solovyeva, M.V.; Cho, K.H.; Shmakov, A.N.; Chesalov, Y.A.; Chang, J.-S.; Kholdeeva, O.A. Protons Make Possible Heterolytic Activation of Hydrogen Peroxide over Zr-Based Metal-Organic Frameworks. ACS Catal. 2019, 9, 9699–9704. [Google Scholar] [CrossRef]
- Van Sickle, D.E.; Mayo, F.R.; Arluck, R.M. Liquid-Phase Oxidations of Cyclic Alkenes. J. Amer. Chem. Soc. 1965, 87, 4824–4832. [Google Scholar] [CrossRef]
- Botar, B.; Geletii, Y.V.; Kögerler, P.; Musaev, D.G.; Morokuma, K.; Weinstock, I.A.; Hill, C.L. The True Nature of the Di-iron(III) γ-Keggin Structure in Water: Catalytic Aerobic Oxidation and Chemistry of an Unsymmetrical Trimer. J. Amer. Chem. Soc. 2006, 128, 11268–11277. [Google Scholar] [CrossRef]
- Food Drug Administration. Rules and Regulations: Title 21—Food and Drugs, Food Additives, Synthetic Flavoring Substances and Adjuvants. Fed Regist 1973, 95, 12913. [Google Scholar]
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragrance and Flavor Materials; Wiley-VCH: New York, NY, USA, 1997; 278p. [Google Scholar]
- Guidotti, M. Institute CNR-ISTM Internal Report; CNR-ISTM: Milan, Italy, 2018. [Google Scholar]
- Di Furia, F.; Modena, G. Mechanism of Oxygen Transfer from Peroxo Species. Pure Appl. Chem. 1982, 54, 1853–1866. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Zr, a wt% | Zr(acac)4:HCl b | SBET, m2/g | Vp, c cm3/g | Dp, d nm |
|---|---|---|---|---|---|
| A | 2.46 | 1:0 | 995 | 0.62 | 3.8 |
| B | 2.33 | 1:1 | 1318 | 0.61 | 3.6 |
| C | 2.43 | 1:2 | 1376 | 0.58 | 3.5 |
| D | 2.50 | 1:7 | 1404 | 0.62 | 3.1 |
| Zr/SiO2 | 1.91 | - e | 408 | 0.66 | 2.0–8.0 f |
| Entry | Catalyst | Time, b h | CyH conv., % | Product Selectivity, c % | ||
|---|---|---|---|---|---|---|
| Epoxide | Diol | Allylic d | ||||
| 1 e | - | 1.5 | 6 | 16 | 16 | 65 |
| 2 f | H+ | 1.5 | 9 | 43 | 21 | 28 |
| 3 | A | 1.5 | 7 | 31 | 15 | 39 |
| 4 | A + H+ | 0.3 | 27 | 74 | 19 | 4 |
| 5 g | A + H+ | 0.3 | 27 | 78 | 15 | 4 |
| 6 h | A + NaOAc | 1 | 4 | 25 | 25 | 26 |
| 7 | B | 3 | 7 | 31 | 15 | 31 |
| 8 | B + H+ | 0.5 | 23 | 70 | 22 | 6 |
| 9 | C | 4 | 6 | 33 | 9 | 34 |
| 10 | C + H+ | 0.3 | 25 | 66 | 28 | 4 |
| 11 | D | 2 | 8 | 31 | 13 | 26 |
| 12 | D + H+ | 0.3 | 29 | 73 | 17 | 4 |
| 13 | Zr/SiO2 | 1.5 | 8 | 25 | 19 | 38 |
| 14 | Zr/SiO2 + H+ | 0.3 | 21 | 67 | 24 | 5 |
| Catalyst | Time, h | CyOct conv., % | Selectivity to Epoxide, b % |
|---|---|---|---|
| A | 2 | 13 | 31 |
| 5 | 27 | 19 | |
| A + H+ c | 2 | 28 | 14 |
| D | 2 | 20 | 50 |
| 5 | 40 | 38 | |
| Zr/SiO2 | 2 | 18 | 39 |
| 5 | 39 | 28 |
| Catalyst | Time, h | CP conv, % | Selectivity to Epoxide, b % |
|---|---|---|---|
| A | 2 | 40 | 78 |
| 5 | 69 | 78 | |
| D | 2 | 63 | 76 |
| 5 | 87 | 77 | |
| Dc,d | 2 | 45 | 75 |
| 5 | 64 | 70 | |
| Zr/SiO2 | 2 | 37 | 78 |
| 5 | 65 | 80 | |
| UiO-67 e | 4 | 44 | 92 |
| Catalyst | Time, b h | MPS conv, % | Product Selectivity, c % | H2O2 Efficiency, d % | |
|---|---|---|---|---|---|
| MPSO | MPSO2 | ||||
| A | 2 | 51 | 26 | 74 | 89 |
| B | 1.5 | 55 | 36 | 64 | 90 |
| C | 1.5 | 55 | 36 | 64 | 90 |
| D | 1 | 57 | 37 | 63 | 93 |
| Zr/SiO2 | 0.5 | 53 | 38 | 62 | 86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanchikova, I.D.; Zalomaeva, O.V.; Maksimchuk, N.V.; Stonkus, O.A.; Glazneva, T.S.; Chesalov, Y.A.; Shmakov, A.N.; Guidotti, M.; Kholdeeva, O.A. Alkene Epoxidation and Thioether Oxidation with Hydrogen Peroxide Catalyzed by Mesoporous Zirconium-Silicates. Catalysts 2022, 12, 742. https://doi.org/10.3390/catal12070742
Ivanchikova ID, Zalomaeva OV, Maksimchuk NV, Stonkus OA, Glazneva TS, Chesalov YA, Shmakov AN, Guidotti M, Kholdeeva OA. Alkene Epoxidation and Thioether Oxidation with Hydrogen Peroxide Catalyzed by Mesoporous Zirconium-Silicates. Catalysts. 2022; 12(7):742. https://doi.org/10.3390/catal12070742
Chicago/Turabian StyleIvanchikova, Irina D., Olga V. Zalomaeva, Nataliya V. Maksimchuk, Olga A. Stonkus, Tatiana S. Glazneva, Yurii A. Chesalov, Alexander N. Shmakov, Matteo Guidotti, and Oxana A. Kholdeeva. 2022. "Alkene Epoxidation and Thioether Oxidation with Hydrogen Peroxide Catalyzed by Mesoporous Zirconium-Silicates" Catalysts 12, no. 7: 742. https://doi.org/10.3390/catal12070742