
Regiodivergent Organocatalytic Reactions

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
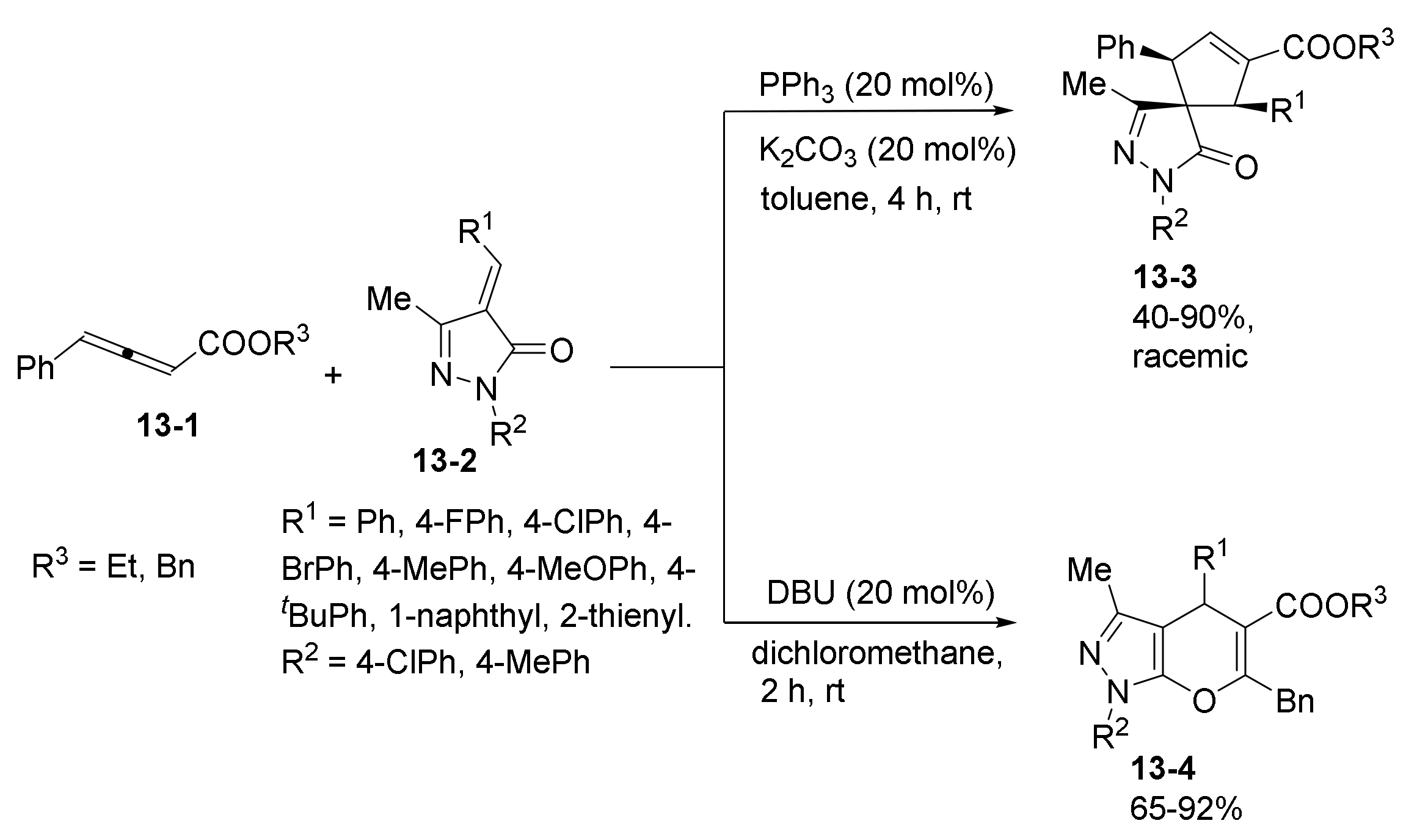
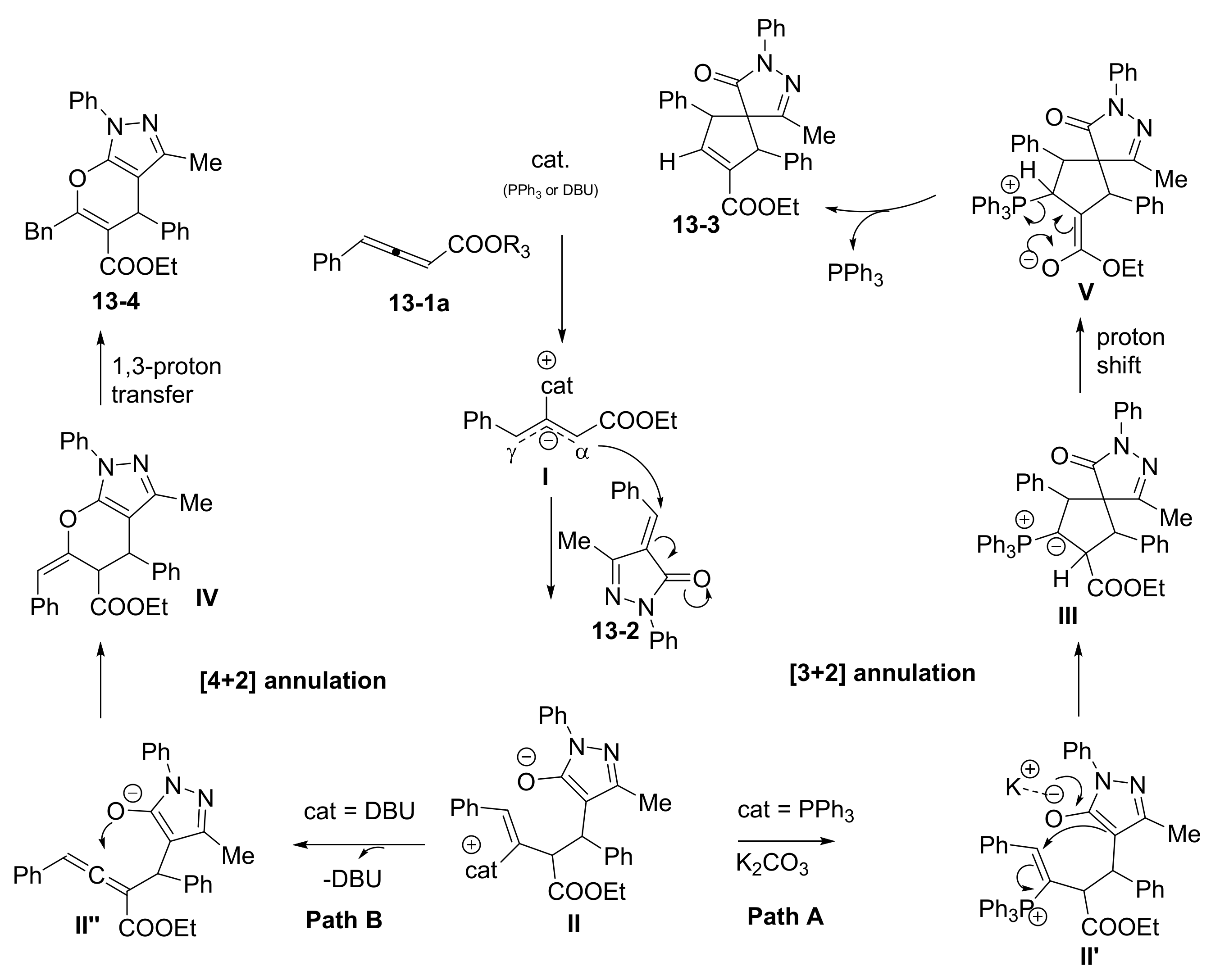
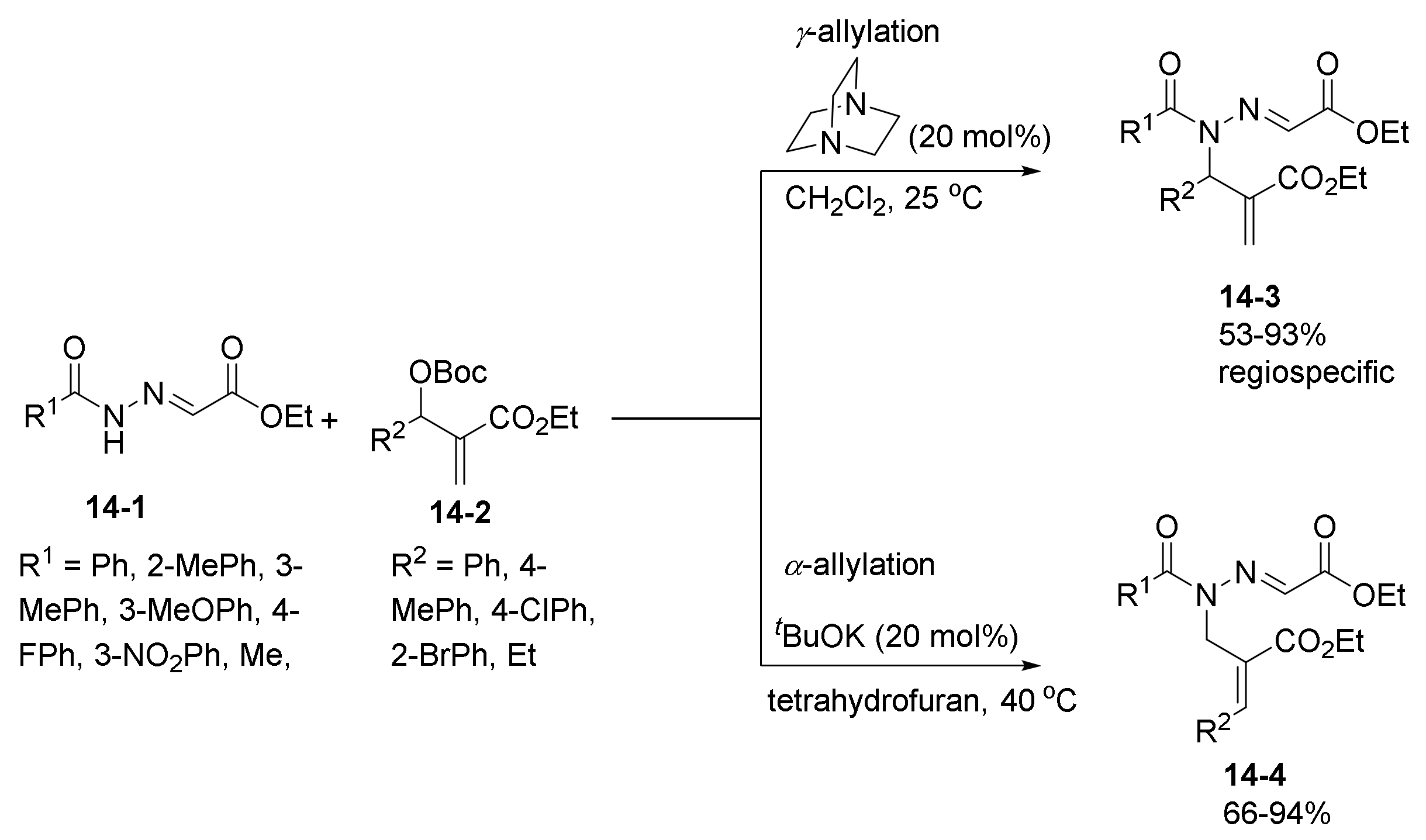
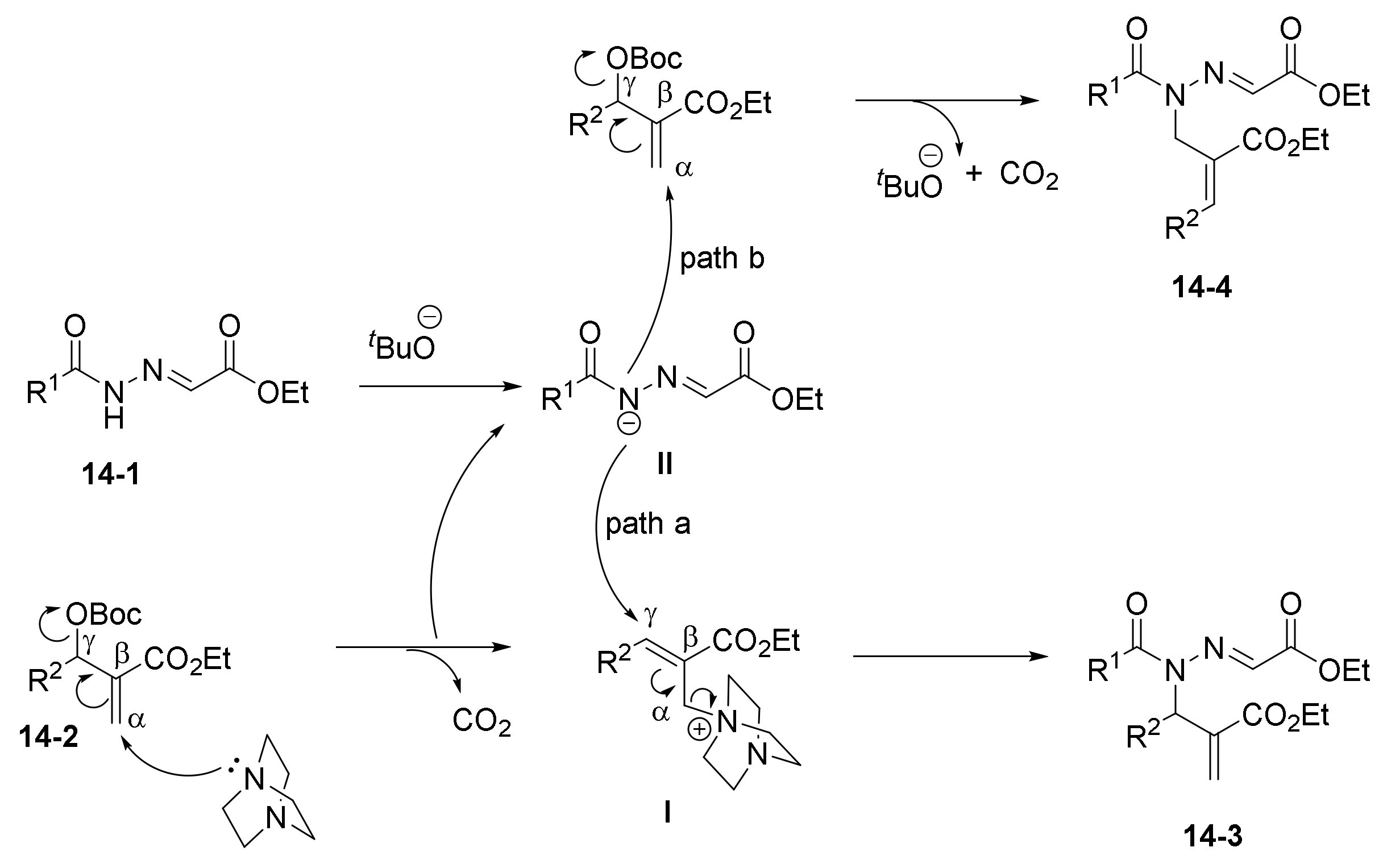
1. Introduction
2. Lewis Base Catalysts
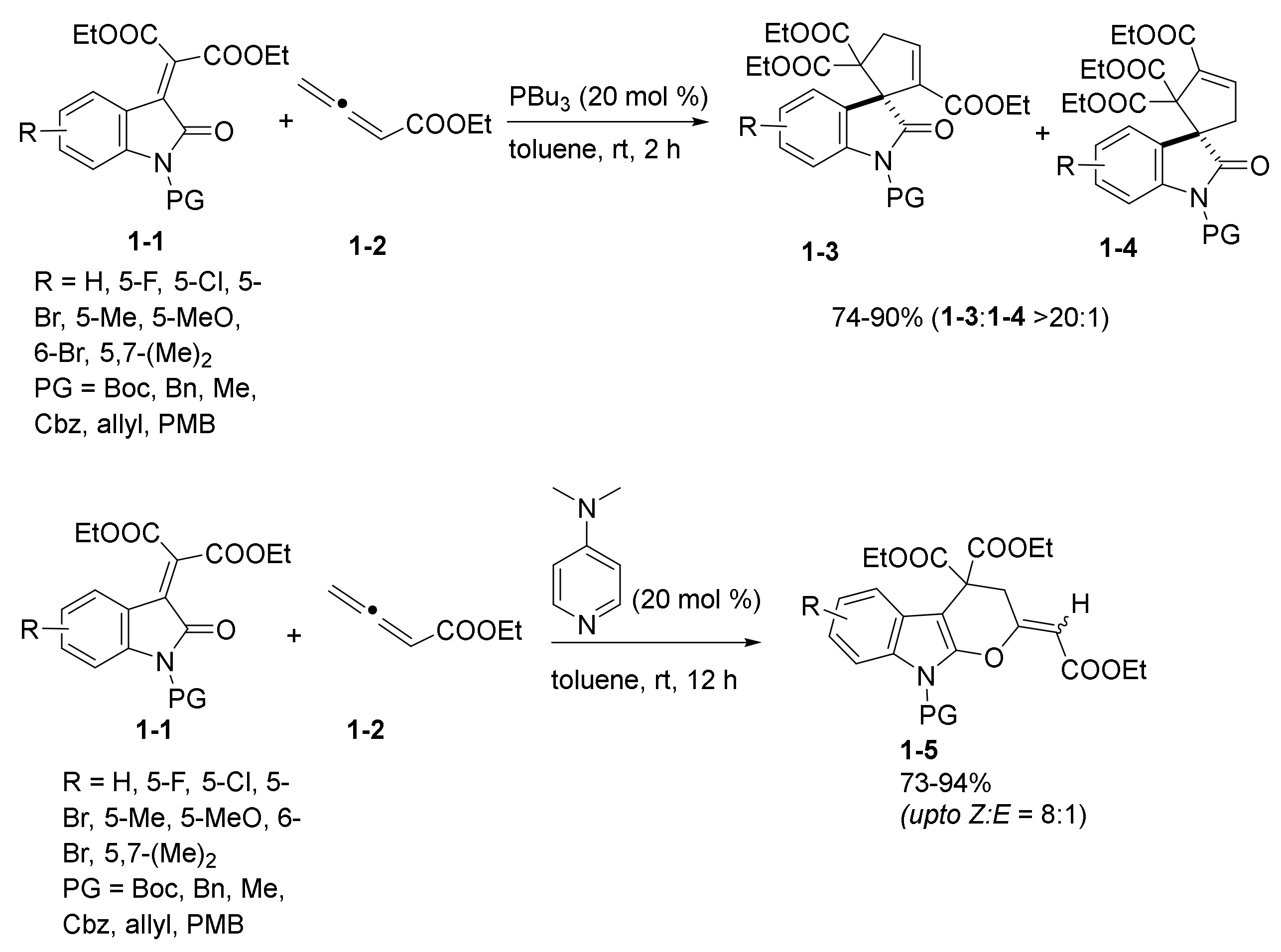
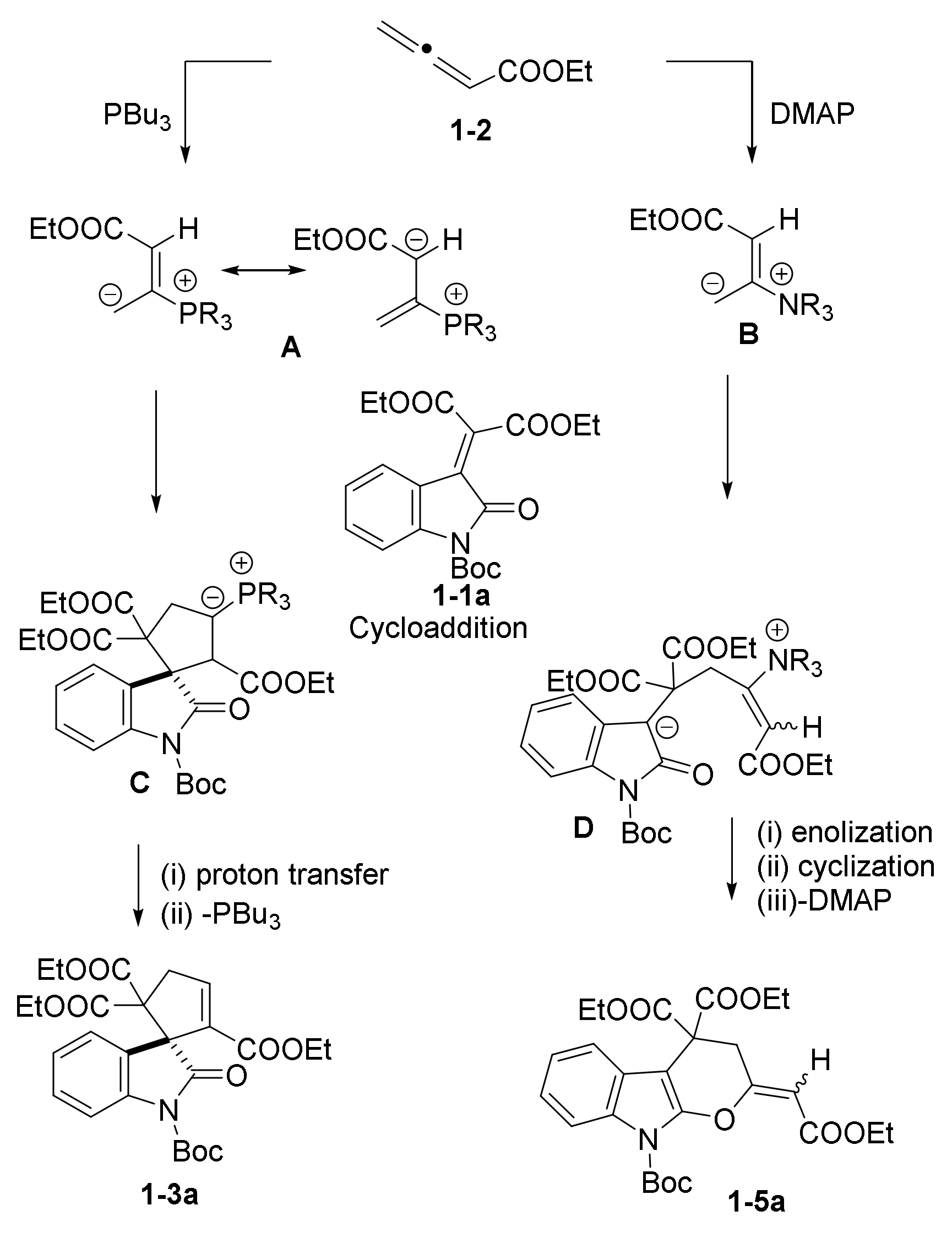
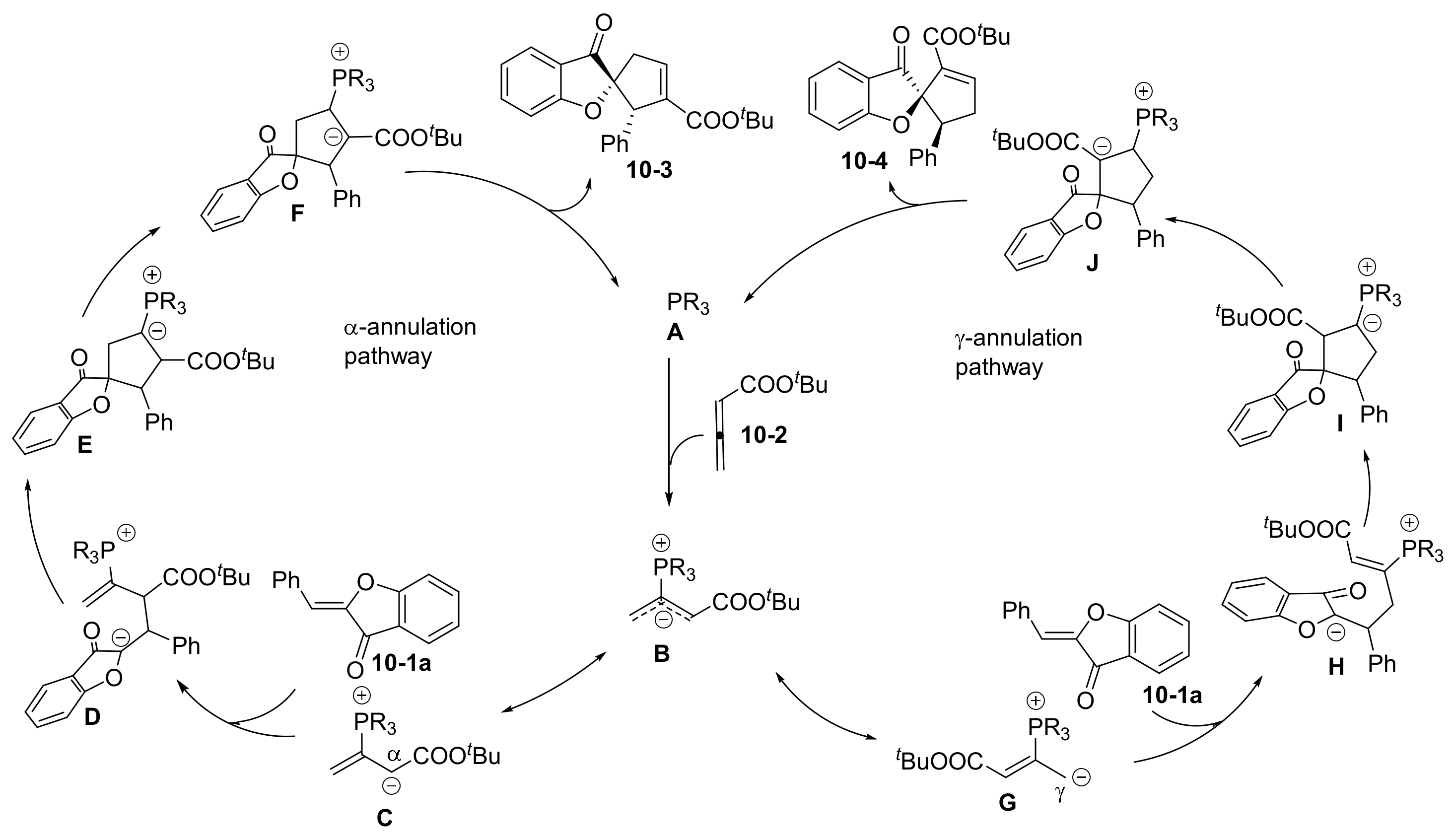
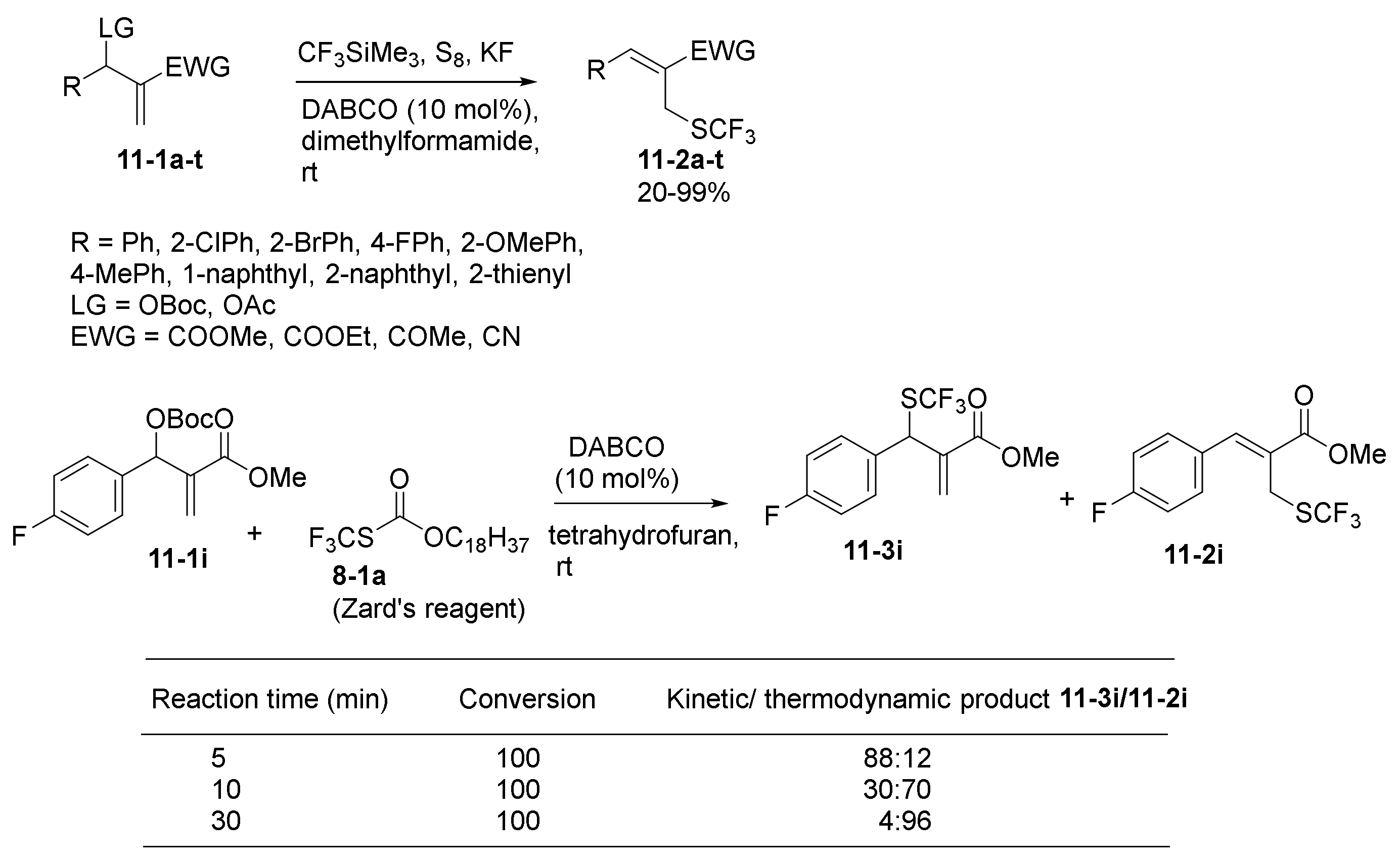
2.1. Phosphine and Amine Bases
2.2. Cinchona Alkaloids
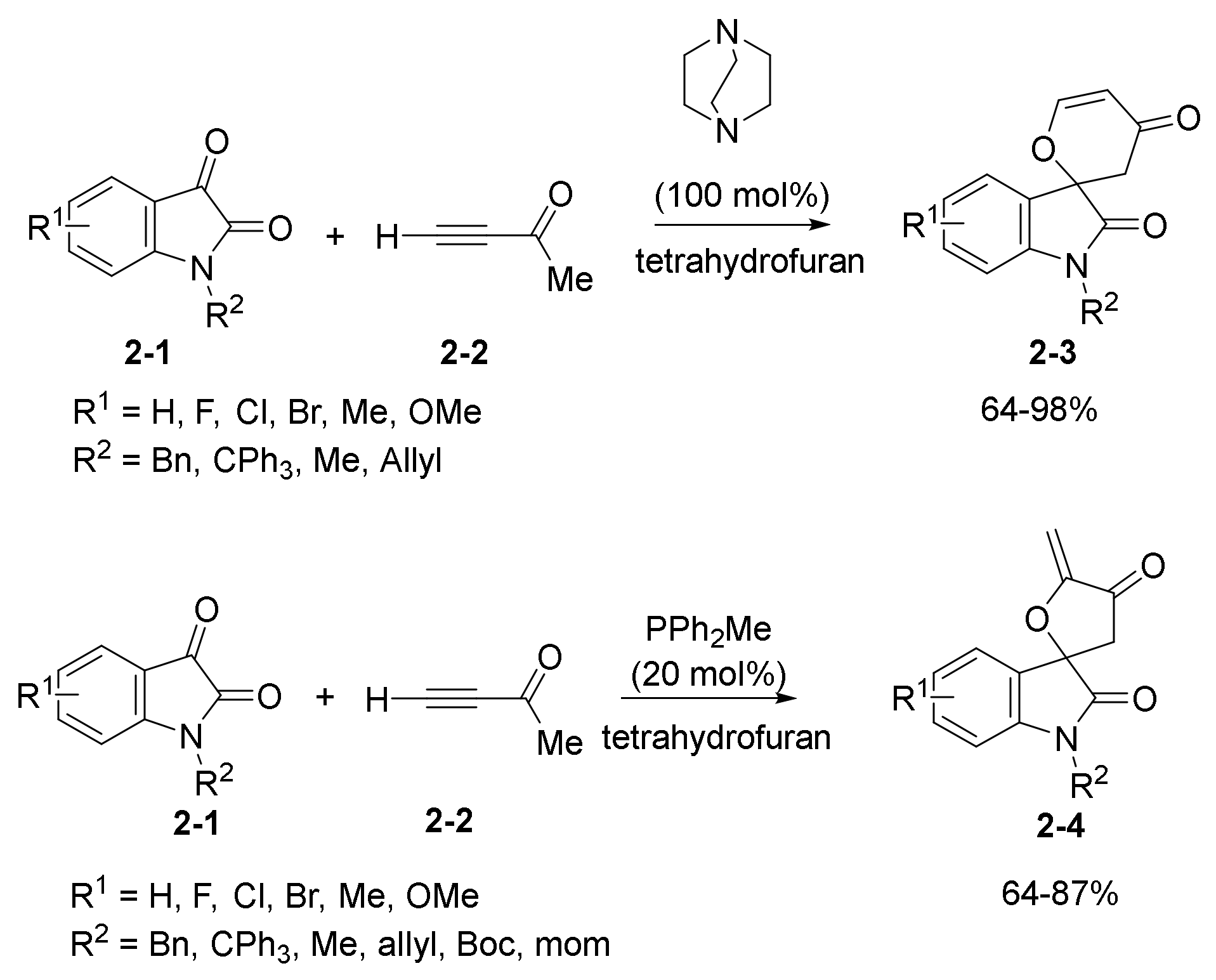
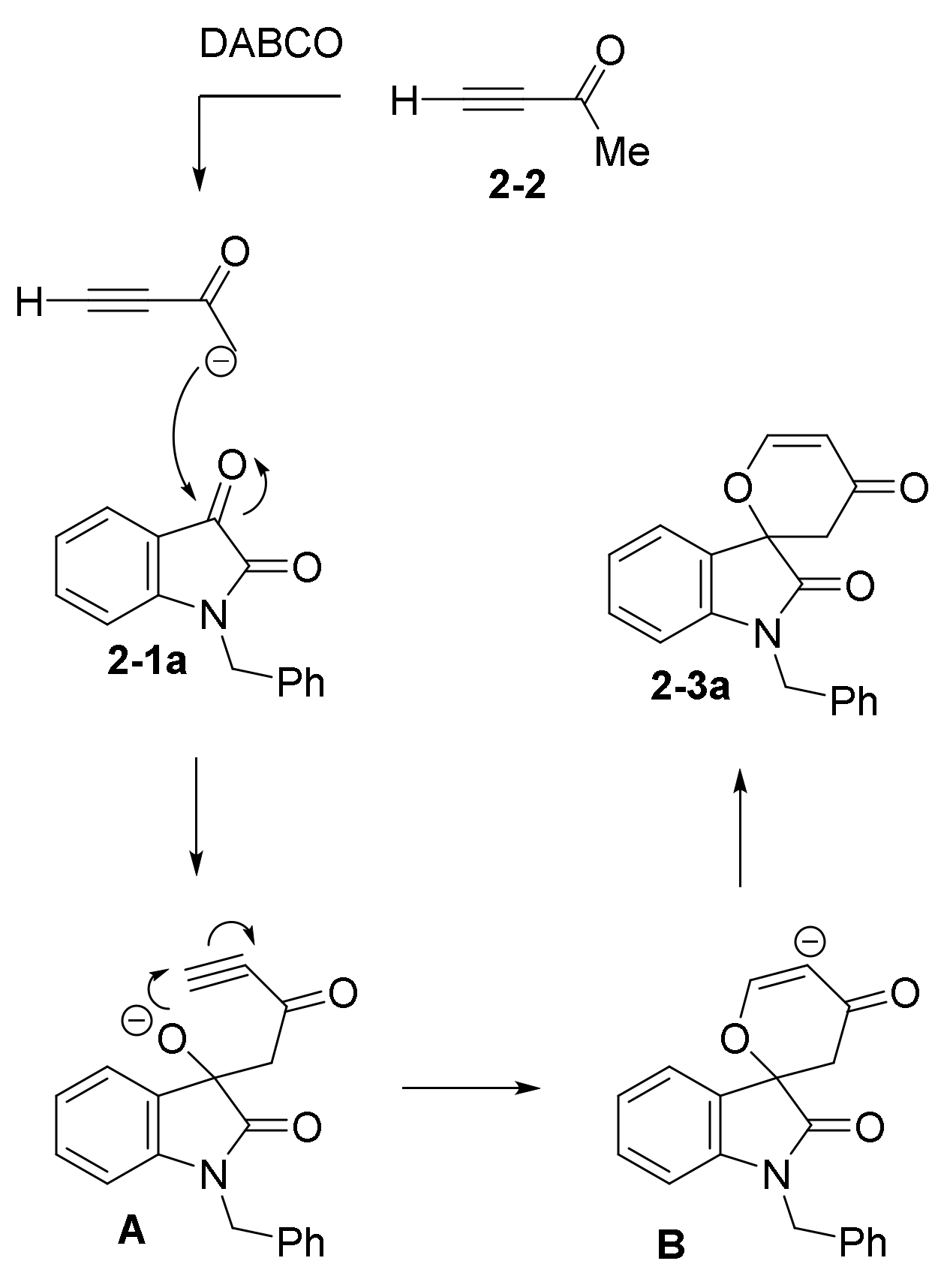
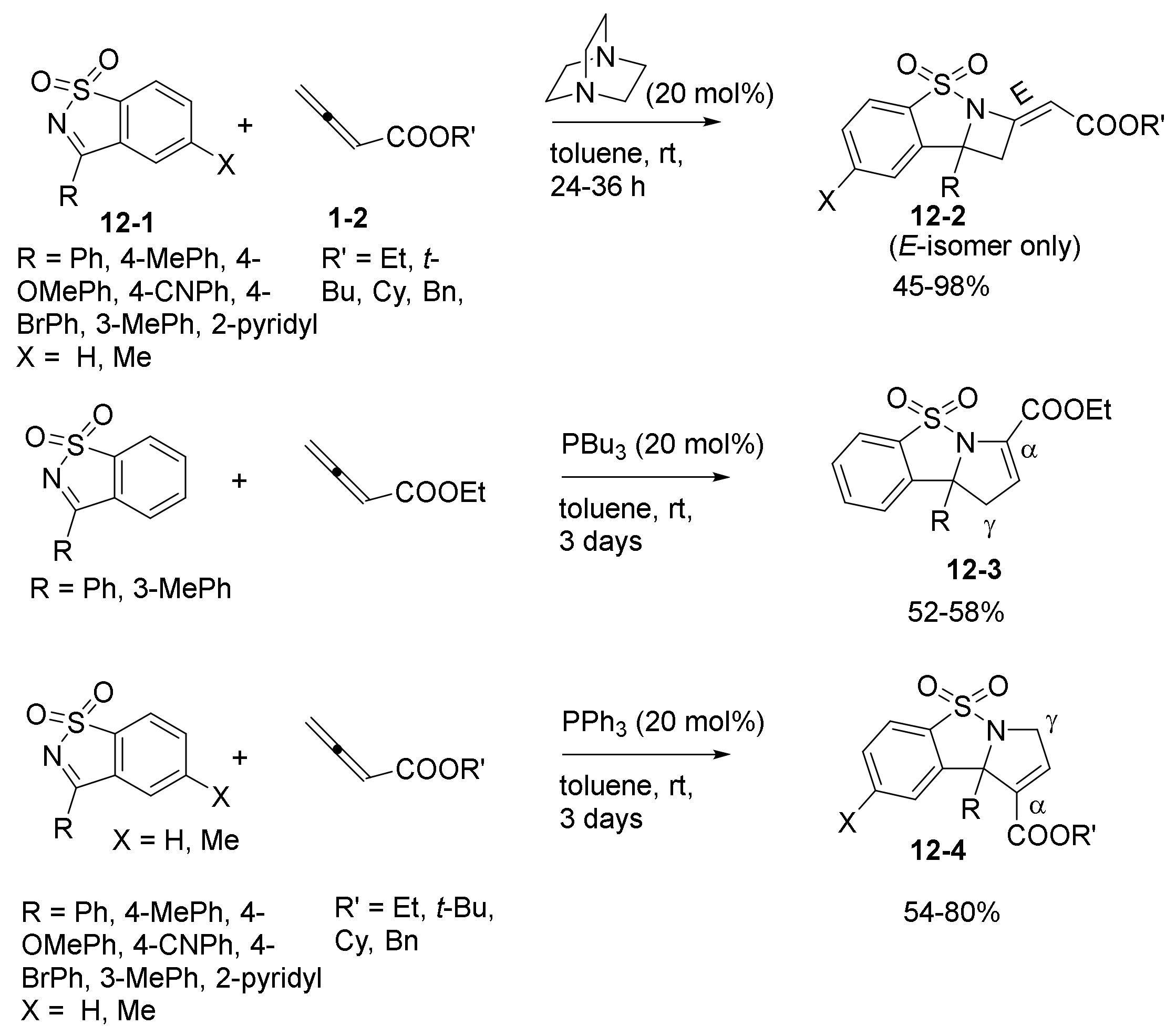
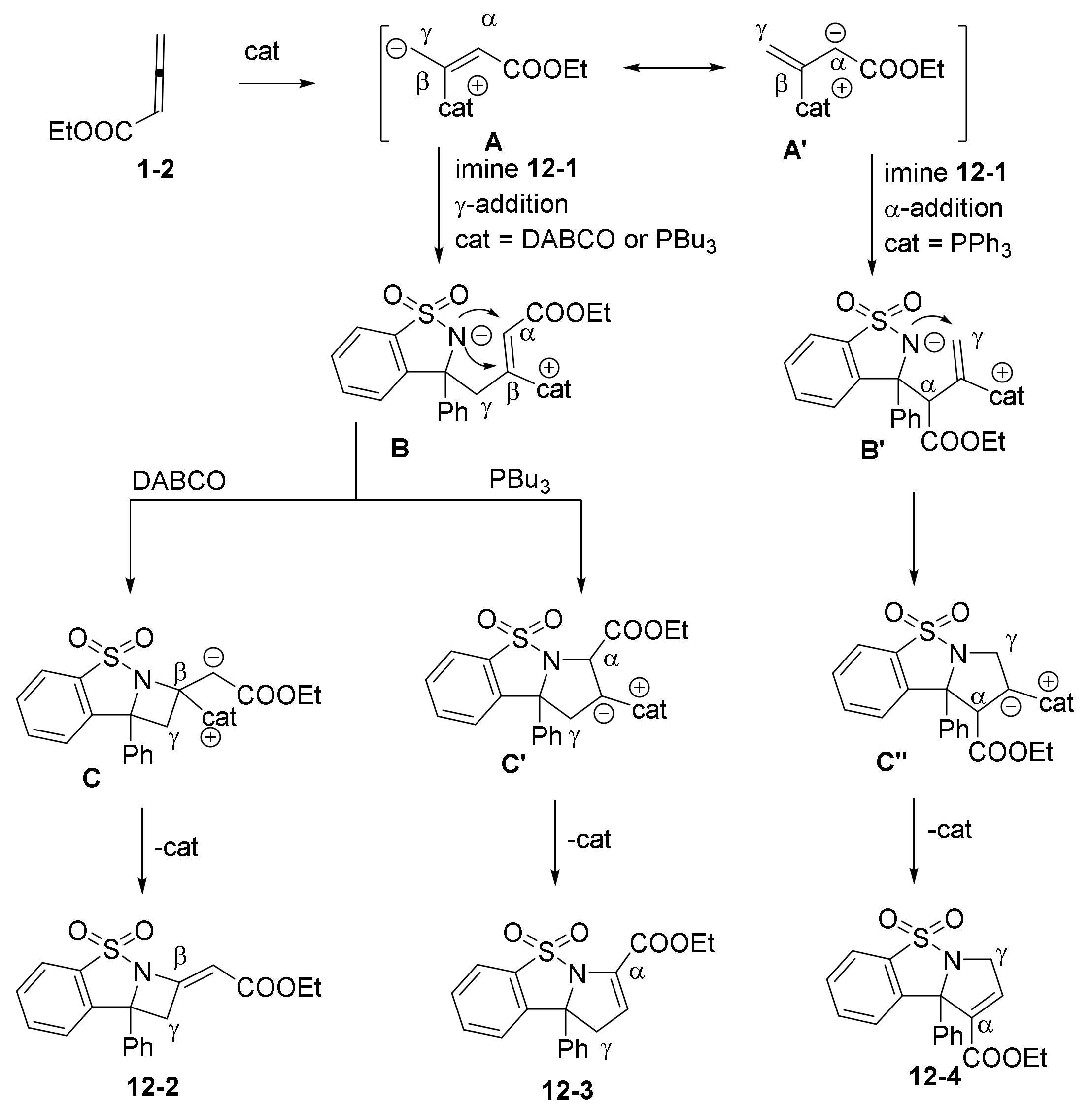
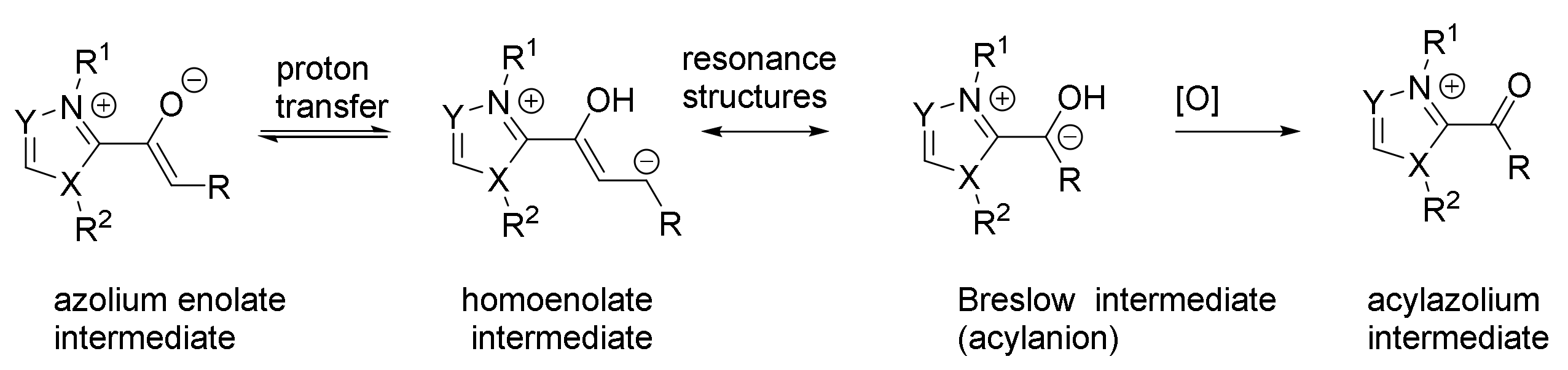
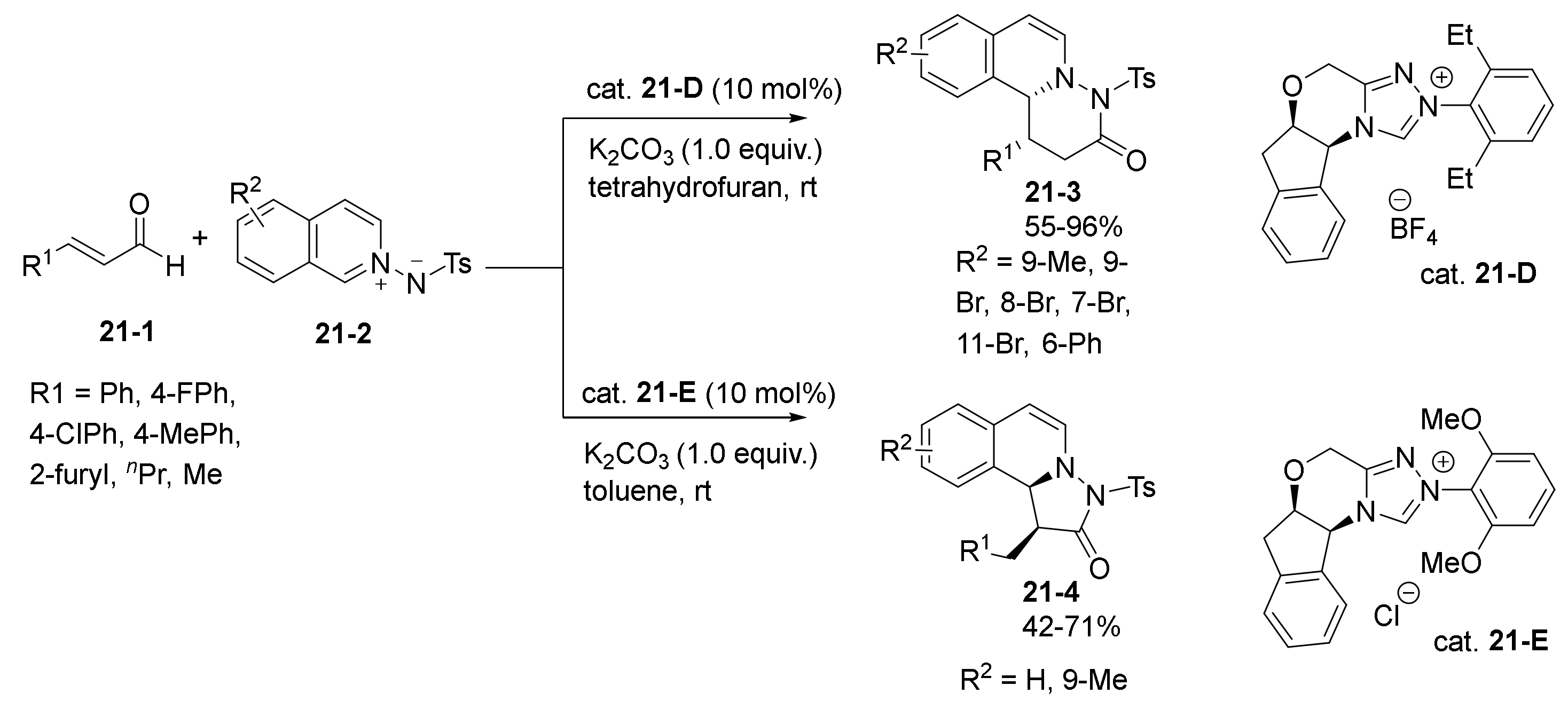
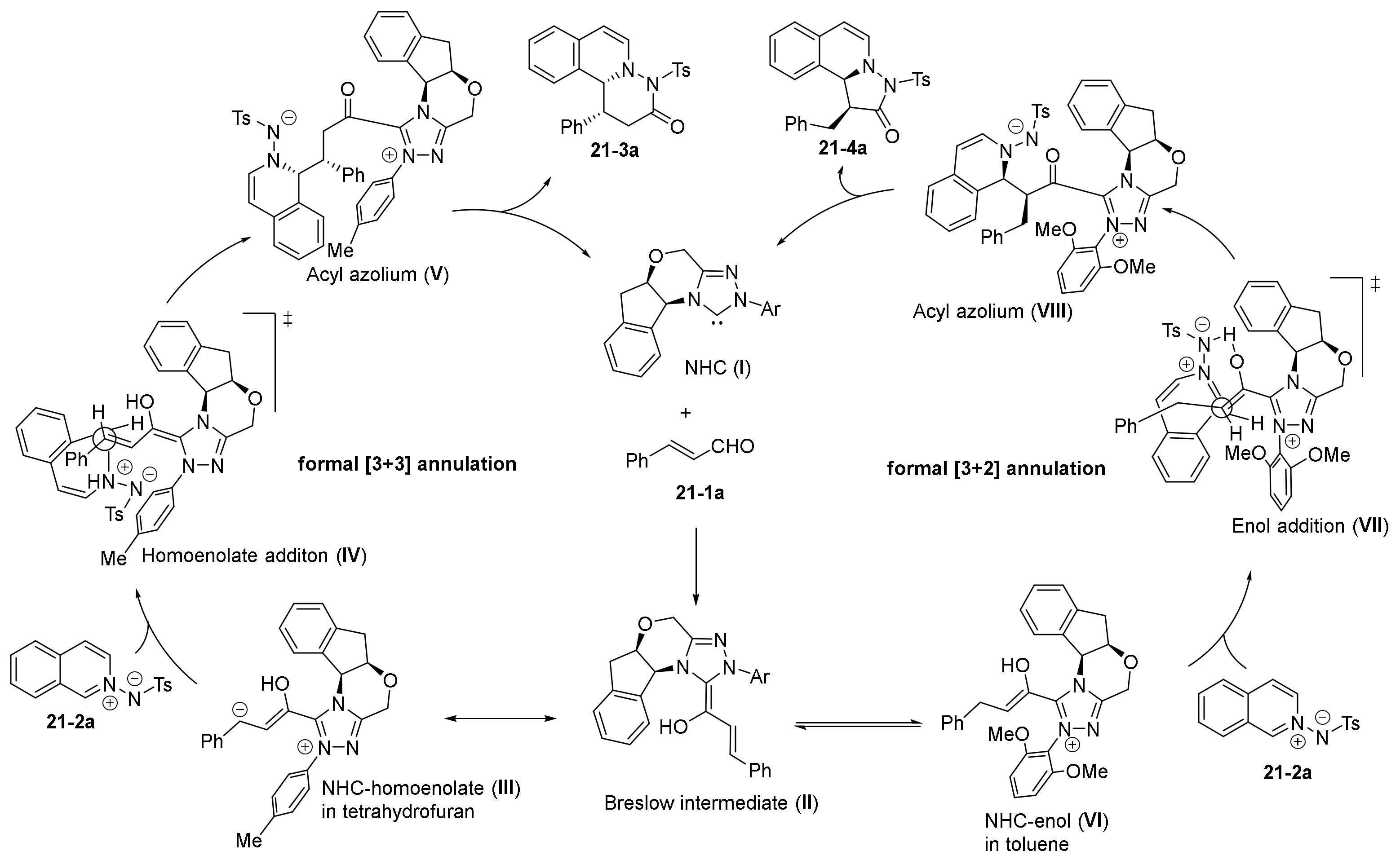
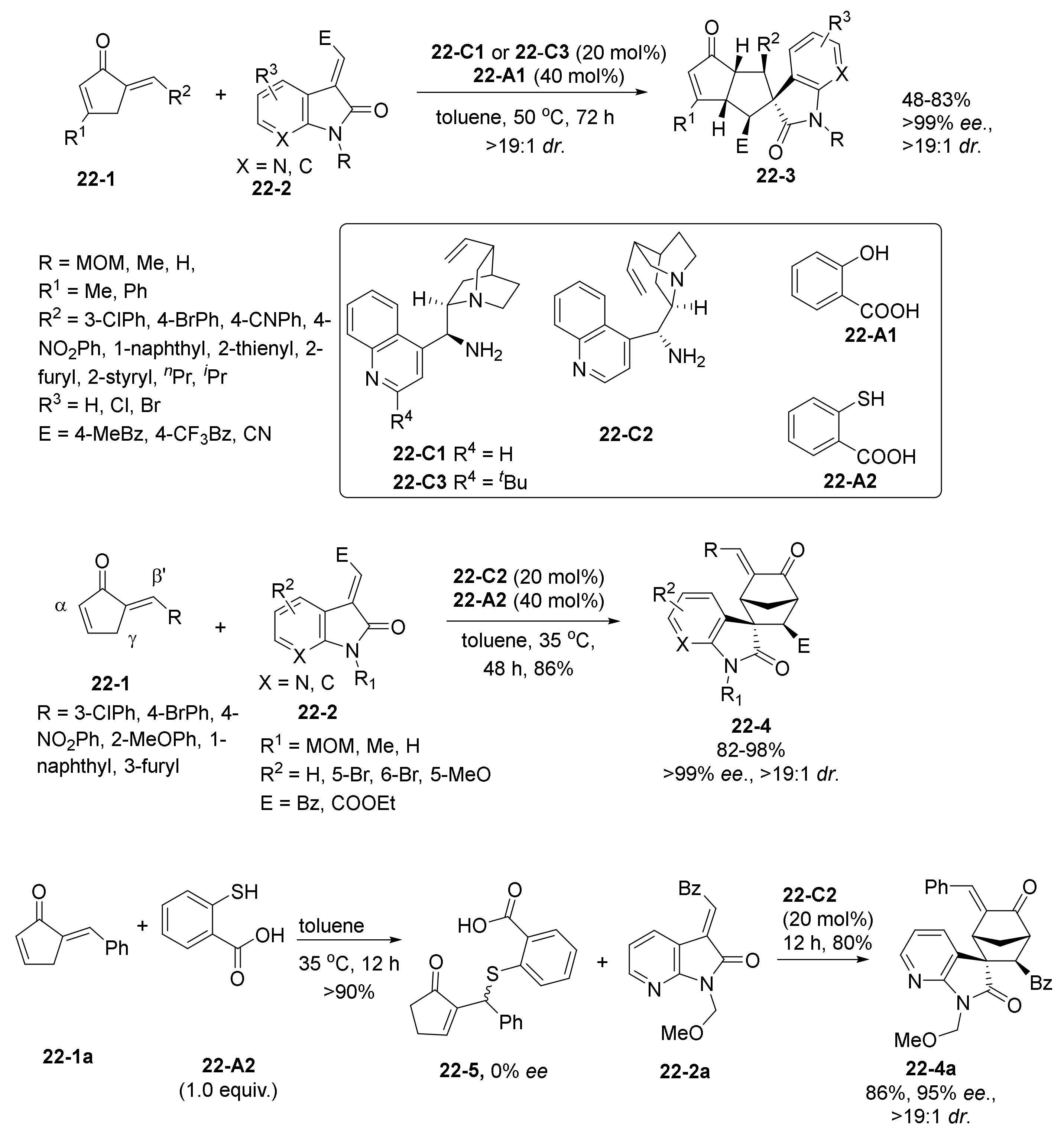
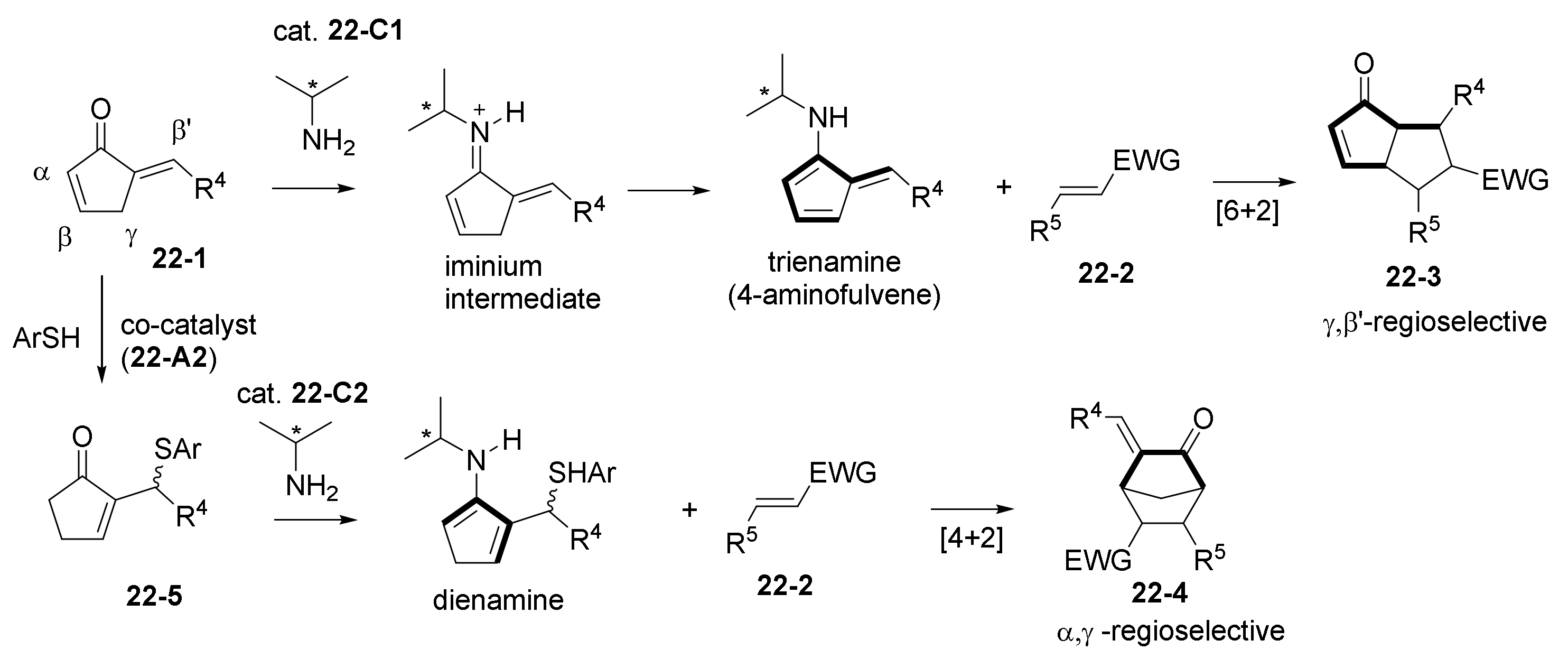
2.3. NHC Catalysts
3. Amine Catalysts
4. Brønsted Acid Catalysts
4.1. Phosphoric Acid Catalysts
4.2. p-Toluenesulfonic Acid Catalyst
5. Hydrogen Bond-Donating Catalysts
5.1. Thiourea Catalyst
5.2. Squaramide Catalyst
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xiao, X.; Shao, B.-X.; Lu, Y.-J.; Cao, Q.-Q.; Xia, C.-N.; Chen, F.-E. Recent Advances in Asymmetric Organomulticatalysis. Adv. Synth. Catal. 2021, 363, 352–387. [Google Scholar] [CrossRef]
- Milton, J.P.; Fossey, J.S. Azetidines and their applications in asymmetric catalysis. Tetrahedron 2021, 77, 131767. [Google Scholar] [CrossRef]
- Das, T.; Mohapatra, S.; Mishra, N.P.; Nayak, S.; Raiguru, B.P. Recent Advances in Organocatalytic Asymmetric Michael Addition Reactions to α, β-Unsaturated Nitroolefins. ChemistrySelect 2021, 6, 3745–3781. [Google Scholar] [CrossRef]
- Yin, Y.; Zhao, X.; Qiao, B.; Jiang, Z. Cooperative photoredox and chiral hydrogen-bonding catalysis. Org. Chem. Front. 2020, 7, 1283–1296. [Google Scholar] [CrossRef]
- Skrzyńska, A.; Frankowski, S.; Albrecht, Ł. Cyclic 1-Azadienes in the Organocatalytic Inverse-Electron-Demand Aza-Diels-Alder Cycloadditions. Asian J. Org. Chem. 2020, 9, 1688–1700. [Google Scholar] [CrossRef]
- Ding, P.-G.; Zhou, F.; Wang, X.; Zhao, Q.-H.; Yu, J.-S.; Zhou, J. H-bond donor-directed switching of diastereoselectivity in the Michael addition of α-azido ketones to nitroolefins. Chem. Sci. 2020, 11, 3852–3861. [Google Scholar] [CrossRef] [Green Version]
- He, X.-H.; Ji, Y.-L.; Peng, C.; Han, B. Organocatalytic Asymmetric Synthesis of Cyclic Compounds Bearing a Trifluoromethylated Stereogenic Center: Recent Developments. Adv. Synth. Catal. 2019, 361, 1923–1957. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Ye, X.; Zhao, X.; Jiang, Z. Organocatalytic asymmetric formal arylation of benzofuran-2(3H)-ones with cooperative visible light photocatalysis. Chem. Commun. 2016, 52, 13955–13958. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.G.; Juaristi, E. Recent efforts directed to the development of more sustainable asymmetric organocatalysis. Chem. Commun. 2012, 48, 5396–5409. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium Catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Tu, M.-S.; Chen, K.-W.; Wu, P.; Zhang, Y.-C.; Liu, X.-Q.; Shi, F. Advances in organocatalytic asymmetric reactions of vinylindoles: Powerful access to enantioenriched indole derivatives. Org. Chem. Front. 2021, 8, 2643–2672. [Google Scholar] [CrossRef]
- Laviós, A.; Sanz-Marco, A.; Vila, C.; Blay, G.; Pedro, J.R. Asymmetric Organocatalytic Synthesis of aza-Spirocyclic Compounds from Isothiocyanates and Isocyanides. Eur. J. Org. Chem. 2021, 2021, 2268–2284. [Google Scholar] [CrossRef]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef]
- Li, X.; Guo, L.; Peng, C.; Han, B. Organocatalytic Asymmetric Cascade Reactions Based on Gamma-Nitro Carbonyl Compound. Chem. Rec. 2019, 19, 394–423. [Google Scholar] [CrossRef]
- Wang, Y.; Cobo, A.A.; Franz, A.K. Recent advances in organocatalytic asymmetric multicomponent cascade reactions for enantioselective synthesis of spirooxindoles. Org. Chem. Front. 2021, 8, 4315–4348. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, H.; Peng, T.; Yang, D. Conjugated ynones in catalytic enantioselective reactions. Org. Biomol. Chem. 2021, 19, 2110–2145. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liang, S.; Luan, Y.; Chen, X.; Zhao, H.; Huang, A.; Li, P.; Li, W. Organocatalytic regio-, diastereo- and enantioselective γ-additions of isoxazol-5(4H)-ones to β,γ-alkynyl-α-imino esters for the synthesis of axially chiral tetrasubstituted α-amino allenoates. Org. Chem. Front. 2021, 8, 1243–1248. [Google Scholar] [CrossRef]
- Guo, X.; Chen, X.; Cheng, Y.; Chang, X.; Li, X.; Li, P. Organocatalytic enantioselective [2+4]-annulation of γ-substituted allenoates with N-acyldiazenes for the synthesis of optically active 1,3,4-oxadiazines. Org. Biomol. Chem. 2021, 19, 1727–1731. [Google Scholar] [CrossRef]
- Yang, Q.-Q.; Yin, X.; He, X.-L.; Du, W.; Chen, Y.-C. Asymmetric Formal [5+3] Cycloadditions with Unmodified Morita–Baylis–Hillman Alcohols via Double Activation Catalysis. ACS Catal. 2019, 9, 1258–1263. [Google Scholar] [CrossRef]
- Nanjo, T.; Zhang, X.; Tokuhiro, Y.; Takemoto, Y. Divergent and Scalable Synthesis of α-Hydroxy/Keto-β-amino Acid Analogues by the Catalytic Enantioselective Addition of Glyoxylate Cyanohydrin to Imines. ACS Catal. 2019, 9, 10087–10092. [Google Scholar] [CrossRef]
- Kostenko, A.A.; Kucherenko, A.S.; Komogortsev, A.N.; Lichitsky, B.V.; Zlotin, S.G. Asymmetric Michael addition between kojic acid derivatives and unsaturated ketoesters promoted by C2-symmetric organocatalysts. Org. Biomol. Chem. 2018, 16, 9314–9318. [Google Scholar] [CrossRef] [PubMed]
- He, X.-L.; Zhao, H.-R.; Duan, C.-Q.; Du, W.; Chen, Y.-C. Remote Asymmetric Oxa-Diels–Alder Reaction of 5-Allylic Furfurals via Dearomatizative Tetraenamine Catalysis. Org. Lett. 2018, 20, 804–807. [Google Scholar] [CrossRef]
- Bisai, V.; Bisai, A.; Singh, V.K. Enantioselective organocatalytic aldol reaction using small organic molecules. Tetrahedron 2012, 68, 4541–4580. [Google Scholar] [CrossRef]
- Terada, M. Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 2010, 1929–1982. [Google Scholar] [CrossRef]
- Terada, M. Binaphthol-derived phosphoric acid as a versatile catalyst for enantioselective carbon–carbon bond forming reactions. Chem. Commun. 2008, 35, 4097–4112. [Google Scholar] [CrossRef]
- Yang, G.-F.; Li, G.-X.; Huang, J.; Fu, D.-Q.; Nie, X.-K.; Cui, X.; Zhao, J.-Z.; Tang, Z. Regioselective, Diastereoselective, and Enantioselective One-Pot Tandem Reaction Based on an in Situ Formed Reductant: Preparation of 2,3-Disubstituted 1,5-Benzodiazepine. J. Org. Chem. 2021, 86, 5110–5119. [Google Scholar] [CrossRef]
- Kutwal, M.S.; Padmaja, V.M.D.; Appayee, C. Regio- and Enantioselective α,γ-Dialkylation of α,β-Unsaturated Aldehydes Through Cascade Organocatalysis. Eur. J. Org. Chem. 2020, 2020, 2720–2724. [Google Scholar] [CrossRef]
- Gui, H.-Z.; Meng, Z.; Xiao, Z.-S.; Yang, Z.-R.; Wei, Y.; Shi, M. Stereo- and Regioselective Construction of Spirooxindoles Having Continuous Spiral Rings via Asymmetric [3+2] Cyclization of 3-Isothiocyanato Oxindoles with Thioaurone Derivatives. Eur. J. Org. Chem. 2020, 2020, 6614–6622. [Google Scholar] [CrossRef]
- Padmaja, V.M.D.; Jangra, S.; Appayee, C. Highly regioselective α-alkylation of α,β,γ,δ-unsaturated aldehydes. Org. Biomol. Chem. 2019, 17, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Kutwal, M.S.; Dev, S.; Appayee, C. Catalytic Regioselective γ-Methylenation of α,β-Unsaturated Aldehydes Using Formaldehyde via Vinylogous Aldol Condensation. Org. Lett. 2019, 21, 2509–2513. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, Q.-Q.; Chen, Z.; Ouyang, Q.; Du, W.; Chen, Y.-C. Regio- and Diastereodivergent [4+2] Cycloadditions with Cyclic 2,4-Dienones. Org. Lett. 2018, 20, 236–239. [Google Scholar] [CrossRef]
- Kutwal, M.S.; Appayee, C. Highly Regio- and Enantioselective γ-Alkylation of Linear α,β-Unsaturated Aldehydes. Eur. J. Org. Chem. 2017, 2017, 4230–4234. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Jasiński, M.; Wojciechowski, J.; Mlostoń, G.; Albrecht, Ł. The first organocatalytic, ortho-regioselective inverse-electron-demand hetero-Diels–Alder reaction. Chem. Commun. 2017, 53, 11472–11475. [Google Scholar] [CrossRef] [Green Version]
- Arimitsu, S.; Yonamine, T.; Higashi, M. Cinchona-Based Primary Amine Catalyzed a Proximal Functionalization of Dienamines: Asymmetric α-Fluorination of α-Branched Enals. ACS Catal. 2017, 7, 4736–4740. [Google Scholar] [CrossRef]
- Stiller, J.; Marqués-López, E.; Herrera, R.P.; Fröhlich, R.; Strohmann, C.; Christmann, M. Enantioselective α- and γ-Alkylation of α,β-Unsaturated Aldehydes Using Dienamine Activation. Org. Lett. 2011, 13, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Guillena, G.; Hita, M.d.C.; Nájera, C. Organocatalyzed direct aldol condensation using ւ-proline and BINAM-prolinamides: Regio-, diastereo-, and enantioselective controlled synthesis of 1,2-diols. Tetrahedron Asymmetry 2006, 17, 1027–1031. [Google Scholar] [CrossRef]
- Kong, X.; Yu, F.; Chen, Z.; Gong, F.; Yang, S.; Liu, J.; Luo, B.; Fang, X. Catalytic chemodivergent annulations between α-diketones and alkynyl α-diketones. Sci. China Chem. 2021, 64, 991–998. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.-D.; Cheng, J.-P. Diazaphosphinyl radical-catalyzed deoxygenation of α-carboxy ketones: A new protocol for chemo-selective C–O bond scission via mechanism regulation. Chem. Sci. 2020, 11, 8476–8481. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhao, J.; Zhao, M.; Chong, R.; Li, X.; Qiao, Y. Mechanism, Chemoselectivity, and Stereoselectivity of NHC-Catalyzed Asymmetric Desymmetrization of Enal-Tethered Cyclohexadienones. Eur. J. Org. Chem. 2020, 2020, 3726–3733. [Google Scholar] [CrossRef]
- Boruah, D.J.; Maurya, R.A.; Yuvaraj, P. Chemo-selective Synthesis of [indoline-3,4′-isoxazolo[5,4-b]pyridine Fused spirooxindole Derivatives via Brønsted Acid Catalysed Three–Component Tandem Knoevenagel/Michael Addition Reaction. Results Chem. 2020, 2, 100064. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Chemodivergent reactions. Chem. Soc. Rev. 2020, 49, 7101–7166. [Google Scholar] [CrossRef]
- Liu, W.; Zou, L.; Fu, B.; Wang, X.; Wang, K.; Sun, Z.; Peng, F.; Wang, W.; Shao, Z. A Multifaceted Directing Group Switching Ynones as Michael Donors in Chemo-, Enantio-, and γ-Selective 1,4-Conjugate Additions with Nitroolefins. J. Org. Chem. 2016, 81, 8296–8305. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, X.; Li, F.; Wu, J.; Wang, J. Chemoselective N-Heterocyclic Carbene-Catalyzed Cascade of Enals with Nitroalkenes. Org. Lett. 2015, 17, 3588–3591. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Rong, Z.-Q.; Zhao, Y. Stereoselective synthesis of ε-lactones or spiro-heterocycles through NHC-catalyzed annulation: Divergent reactivity by catalyst control. Chem. Commun. 2014, 50, 15309–15312. [Google Scholar] [CrossRef]
- Ping, L.; Chung, D.S.; Bouffard, J.; Lee, S.G. Transition metal-catalyzed site- and regio-divergent C-H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Hong, S.W.; Oh, K.H.; Park, J.K. Divergent C–H Annulation for Multifused N-Heterocycles: Regio- and Stereospecific Cyclizations of N-Alkynylindoles. Angew. Chem. Int. Ed. 2017, 56, 13387–13391. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-P.; Wu, X.-F. Ligand-Controlled Copper-Catalyzed Regiodivergent Carbonylative Synthesis of α-Amino Ketones and α-Boryl Amides from Imines and Alkyl Iodides. Angew. Chem. Int. Ed. 2021, 60, 695–700. [Google Scholar] [CrossRef]
- Jiang, W.-S.; Ji, D.-W.; Zhang, W.-S.; Zhang, G.; Min, X.-T.; Hu, Y.-C.; Jiang, X.-L.; Chen, Q.-A. Orthogonal Regulation of Nucleophilic and Electrophilic Sites in Pd-Catalyzed Regiodivergent Couplings between Indazoles and Isoprene. Angew. Chem. Int. Ed. 2021, 60, 8321–8328. [Google Scholar] [CrossRef]
- Li, X.; Liang, G.; Shi, Z.-J. Regio-Divergent C-H Alkynylation with Janus Directing Strategy via Ir(III) Catalysis. Chin. J. Chem. 2020, 38, 929–934. [Google Scholar] [CrossRef]
- Kuai, C.-S.; Ji, D.-W.; Zhao, C.-Y.; Liu, H.; Hu, Y.-C.; Chen, Q.-A. Ligand-Regulated Regiodivergent Hydrosilylation of Isoprene under Iron Catalysis. Angew. Chem. Int. Ed. 2020, 59, 19115–19120. [Google Scholar] [CrossRef]
- Nájera, C.; Beletskaya, I.P.; Yus, M. Metal-catalyzed regiodivergent organic reactions. Chem. Soc. Rev. 2019, 48, 4515–4618. [Google Scholar] [CrossRef] [PubMed]
- Zhan, G.; Du, W.; Chen, Y.-C. Switchable divergent asymmetric synthesis via organocatalysis. Chem. Soc. Rev. 2017, 46, 1675–1692. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Babu, K.R.; Wu, Y.; Li, Y.; Tang, Y.; Xu, S. Phosphine-Catalyzed Cross-Coupling of Benzyl Halides and Fumarates. Org. Lett. 2021, 23, 4570–4574. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-S.; Yin, H.; Wang, Y.-F.; Wang, C.-J.; Han, H.-T.; Man, T.-T.; Xu, D.-Q. Catalytic Asymmetric Conjugate Addition/Hydroalkoxylation Sequence: Expeditious Access to Enantioenriched Eight-Membered Cyclic Ether Derivatives. Org. Lett. 2021, 23, 2471–2476. [Google Scholar] [CrossRef]
- Li, H.; He, Z. Chiral phosphine-catalyzed asymmetric [4+1] annulation of polar dienes with allylic derivatives: Enantioselective synthesis of substituted cyclopentenes. Tetrahedron Lett. 2021, 67, 152863. [Google Scholar] [CrossRef]
- Chen, G.-S.; Fang, Y.-B.; Ren, Z.; Tian, X.; Liu, Y.-L. Reaction condition-dependent divergent synthesis of spirooxindoles and bisoxindoles. Org. Chem. Front. 2021, 8, 3820–3828. [Google Scholar] [CrossRef]
- Zhang, Z.-B.; Yang, Y.; Yu, Z.-X.; Xia, J.-B. Lewis Base-Catalyzed Amino-Acylation of Arylallenes via C–N Bond Cleavage: Reaction Development and Mechanistic Studies. ACS Catal. 2020, 10, 5419–5429. [Google Scholar] [CrossRef]
- Deng, Q.; Mu, F.; Qiao, Y.; Wei, D. A theoretical review for novel Lewis base amine/imine-catalyzed reactions. Org. Biomol. Chem. 2020, 18, 6781–6800. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Sun, X.; Miao, Z. A PPh3-catalyzed sequential annulation reaction to construct cyclopentane-fused dihydropyrazolone-pyrrolidinediones. Org. Biomol. Chem. 2020, 18, 5577–5581. [Google Scholar] [CrossRef]
- Chen, L.; He, J. DABCO-Catalyzed Michael/Alkylation Cascade Reactions Involving α-Substituted Ammonium Ylides for the Construction of Spirocyclopropyl Oxindoles: Access to the Powerful Chemical Leads against HIV-1. J. Org. Chem. 2020, 85, 5203–5219. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Dai, C.; Huang, Y. DBU-Catalyzed Desymmetrization of Cyclohexadienones: Access to Vicinal Diamine-Containing Heterocycles. Org. Lett. 2018, 20, 5006–5009. [Google Scholar] [CrossRef]
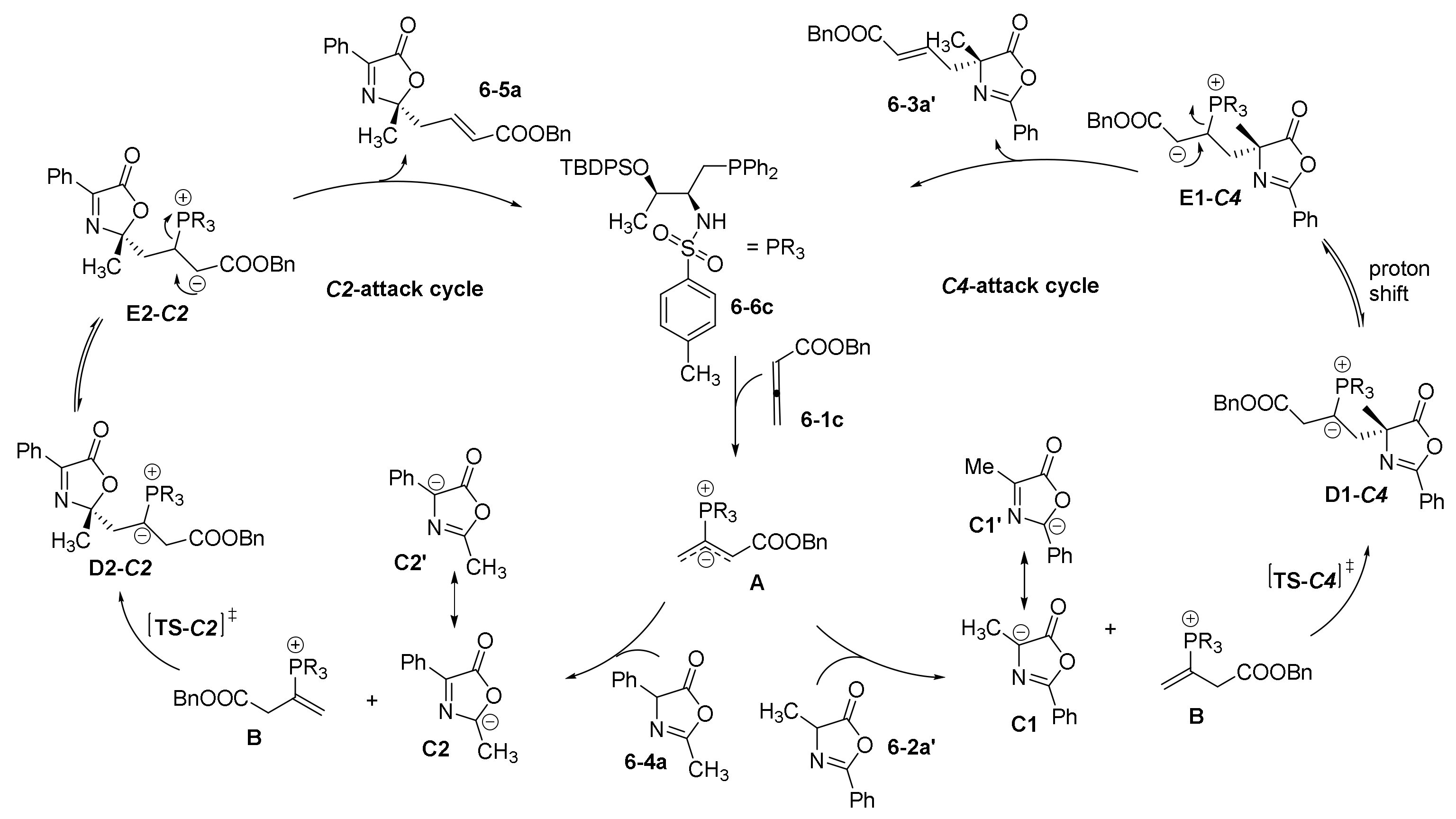
- Guo, H.; Fan, Y.C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef] [PubMed]
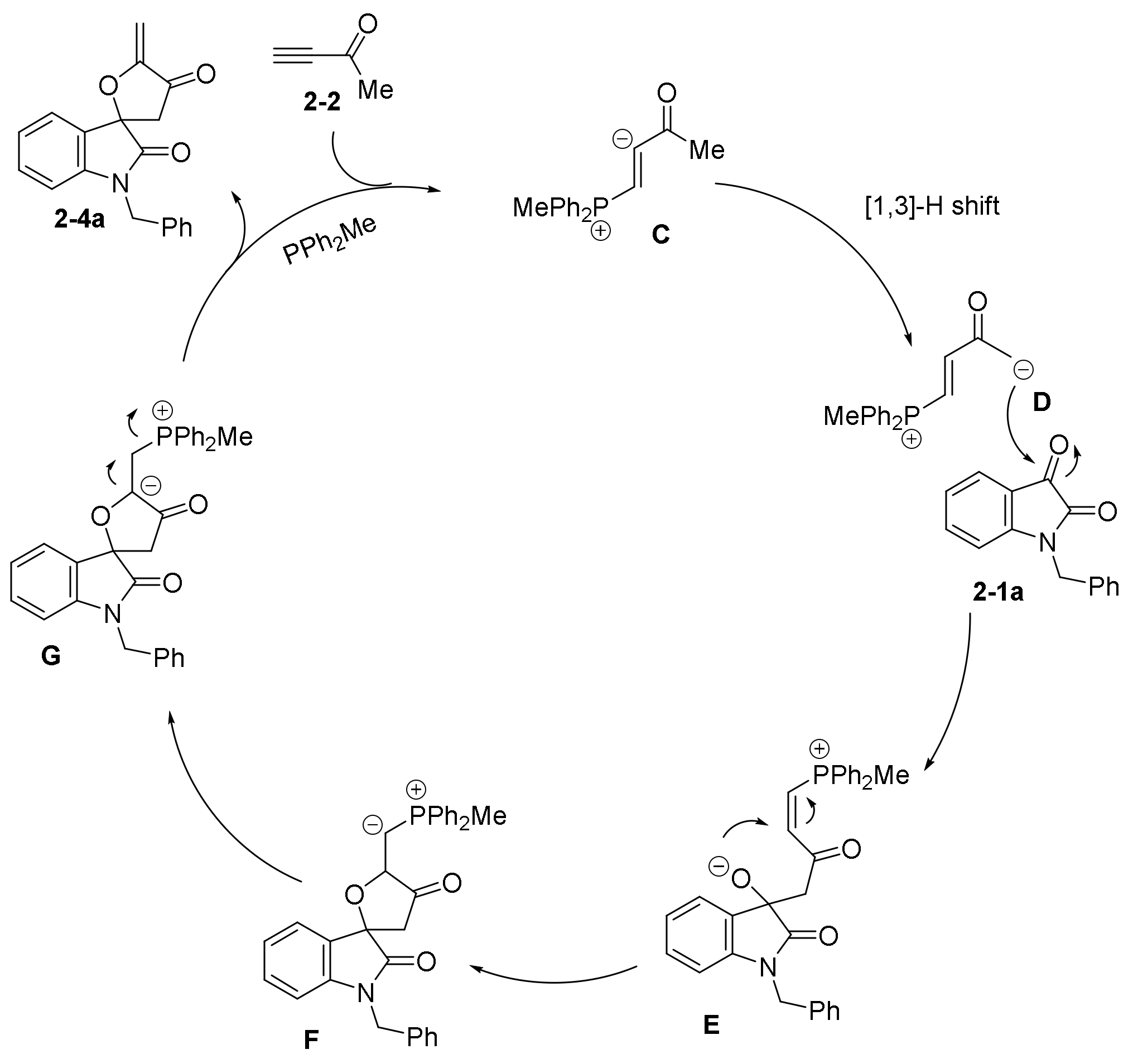
- Zhang, X.-C.; Cao, S.-H.; Wei, Y.; Shi, M. Phosphine- and Nitrogen-Containing Lewis Base Catalyzed Highly Regioselective and Geometric Selective Cyclization of Isatin Derived Electron-Deficient Alkenes with Ethyl 2,3-Butadienoate. Org. Lett. 2011, 13, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Shi, M. Nitrogen- and Phosphorus-Containing Lewis Base Catalyzed [4+2] and [3+2] Annulation Reactions of Isatins with But-3-yn-2-one. Eur. J. Org. Chem. 2012, 2012, 581–586. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Wei, Y.; Shi, M. Phosphine-Catalyzed Intermolecular Annulations of Fluorinated ortho-Aminophenones with Alkynones—The Switchable [4+2] or [4+2]/[3+2] Cycloaddition. Adv. Synth. Catal. 2019, 361, 2129–2135. [Google Scholar] [CrossRef]
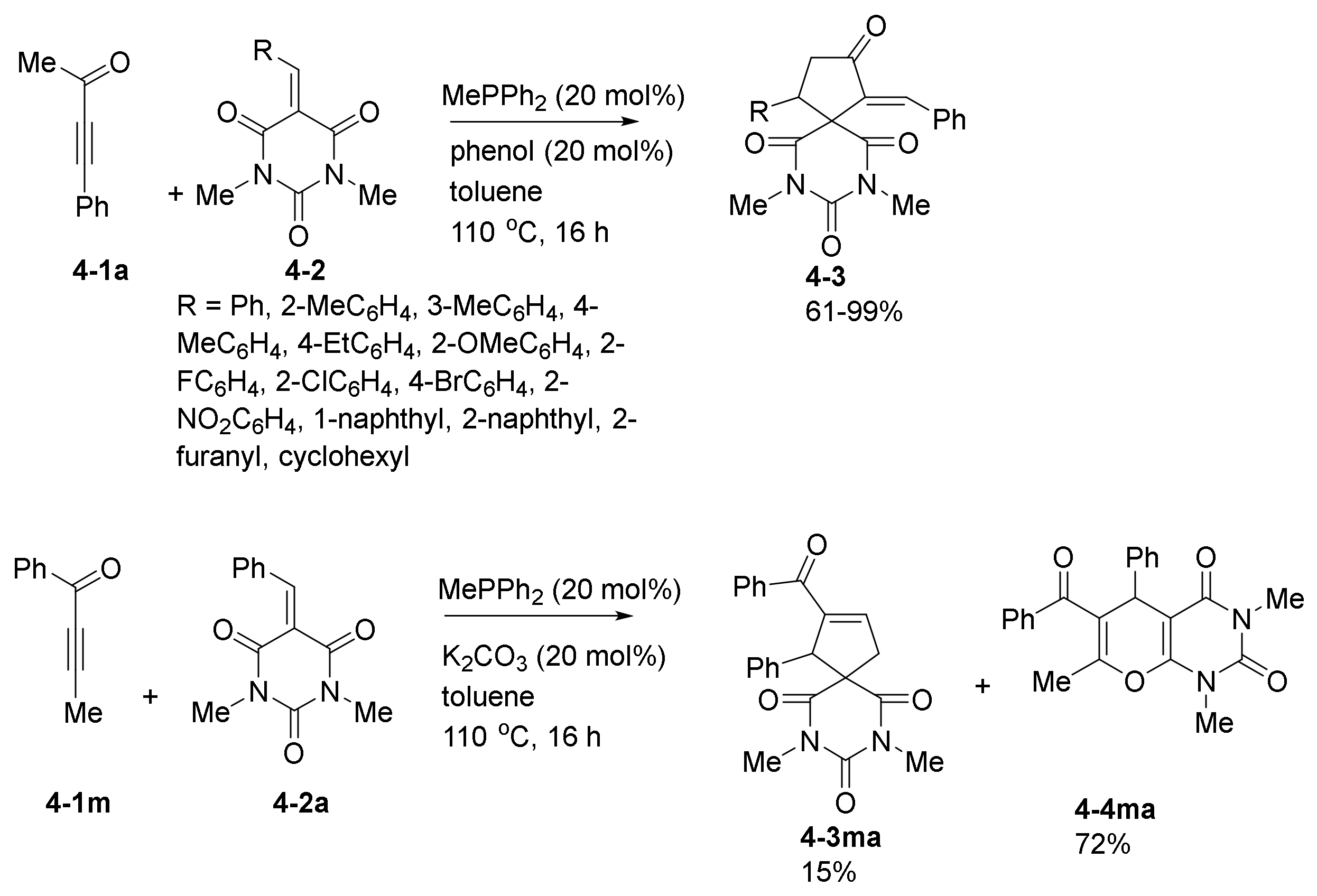
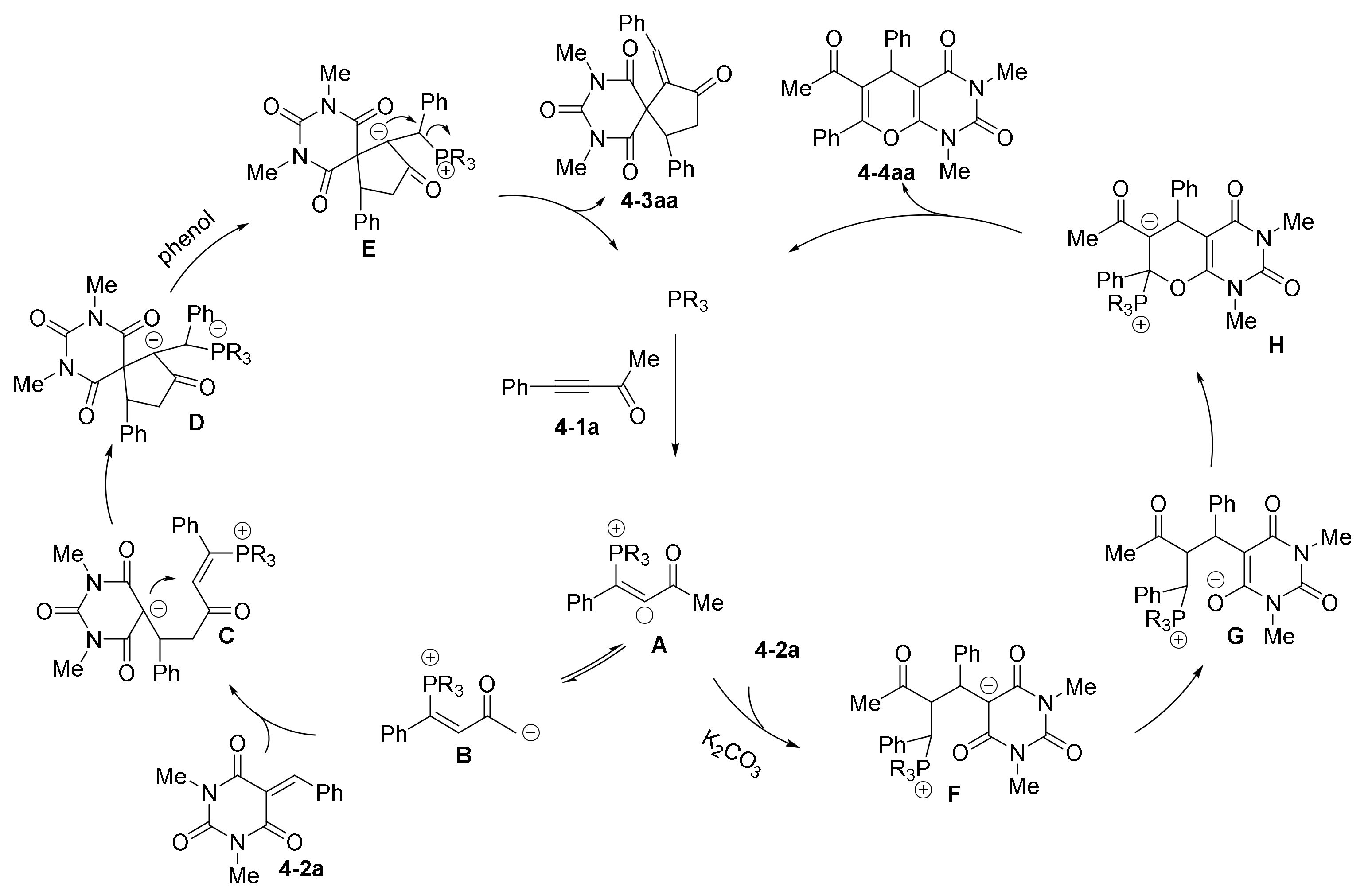
- Gao, X.; Li, Z.; Yang, W.; Liu, Y.; Chen, W.; Zhang, C.; Zheng, L.; Guo, H. Phosphine-catalyzed [3+2] and [4+2] annulation reactions of ynones with barbiturate-derived alkenes. Org. Biomol. Chem. 2017, 15, 5298–5307. [Google Scholar] [CrossRef]
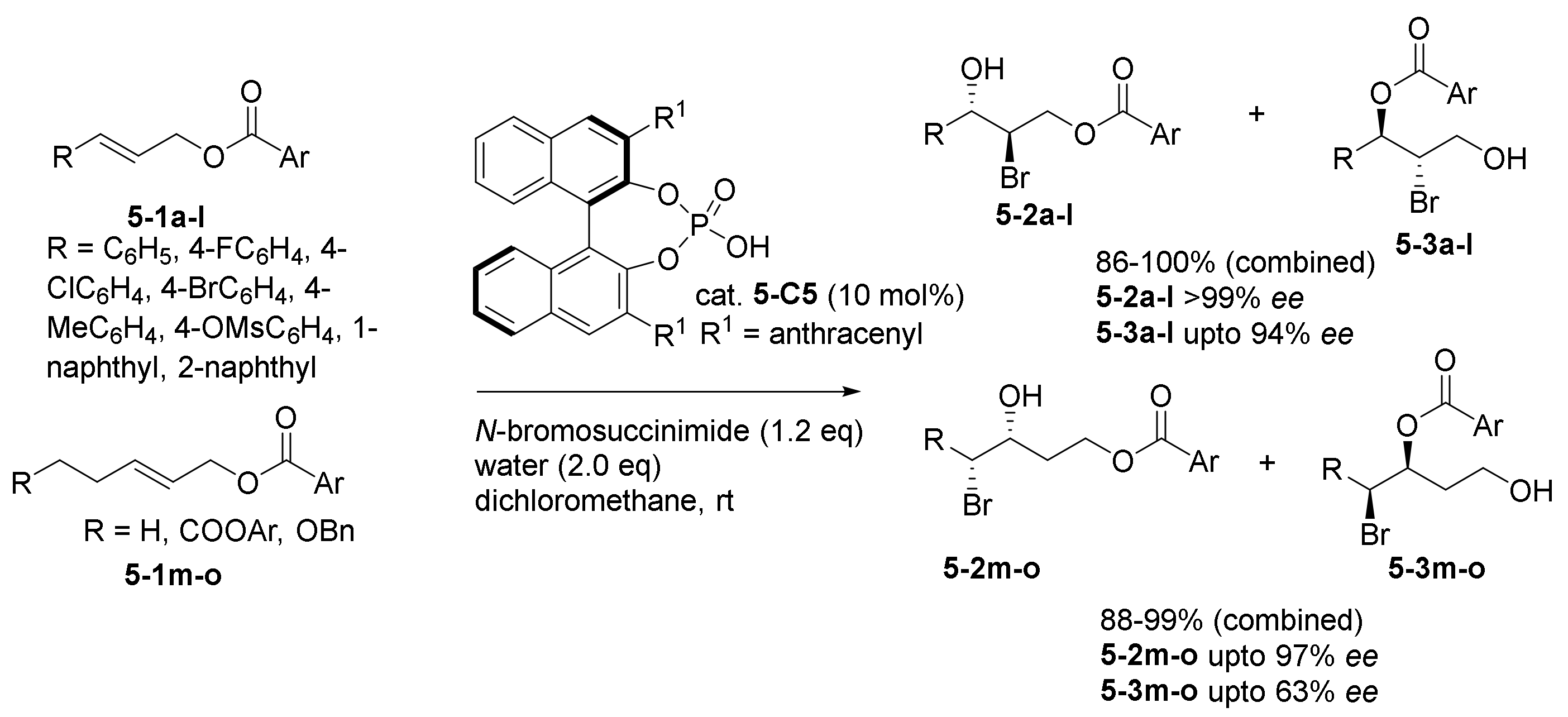
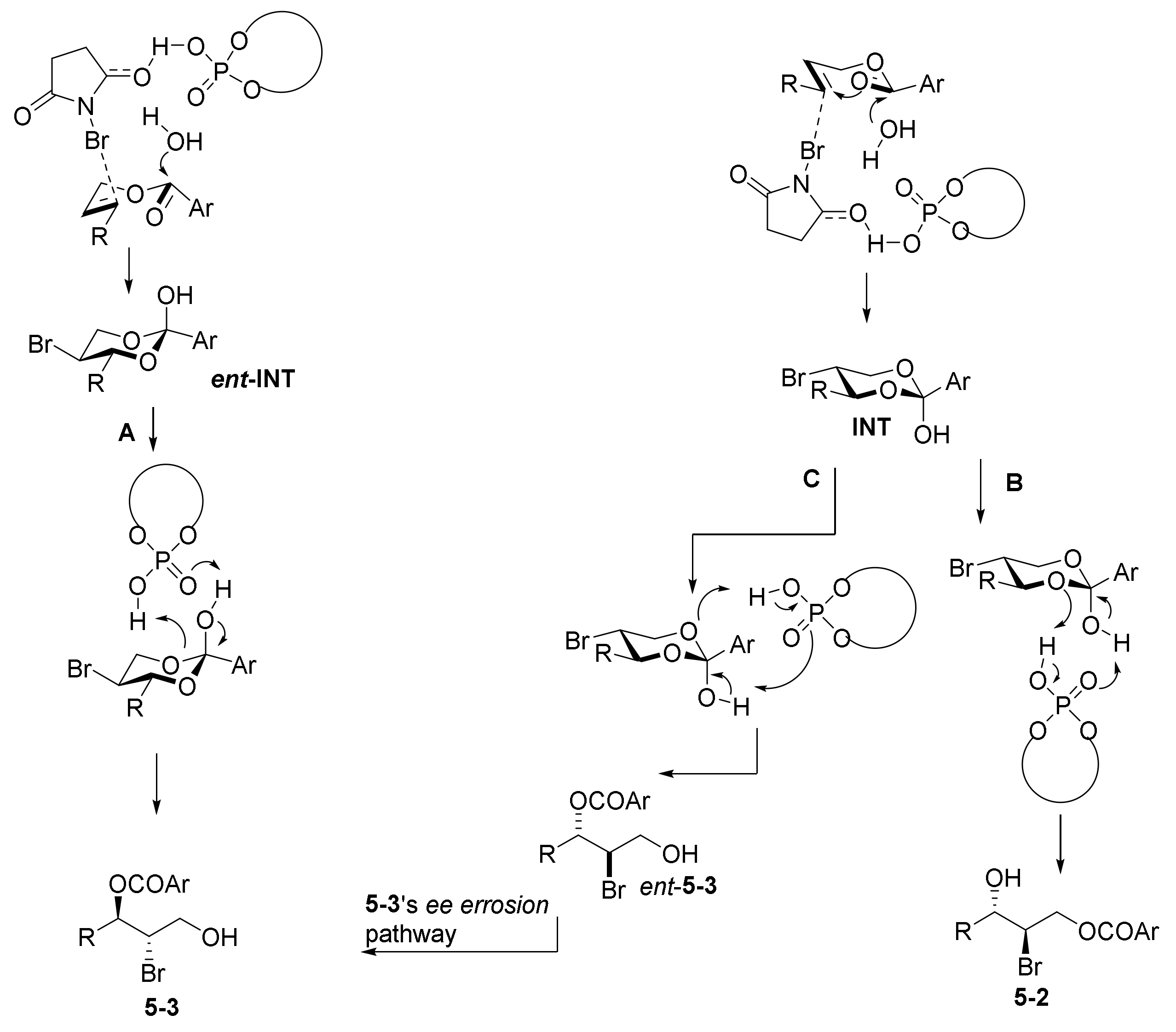
- Cao, Y.-M.; Lentz, D.; Christmann, M. Synthesis of Enantioenriched Bromohydrins via Divergent Reactions of Racemic Intermediates from Anchimeric Oxygen Borrowing. J. Am. Chem. Soc. 2018, 140, 10677–10681. [Google Scholar] [CrossRef]
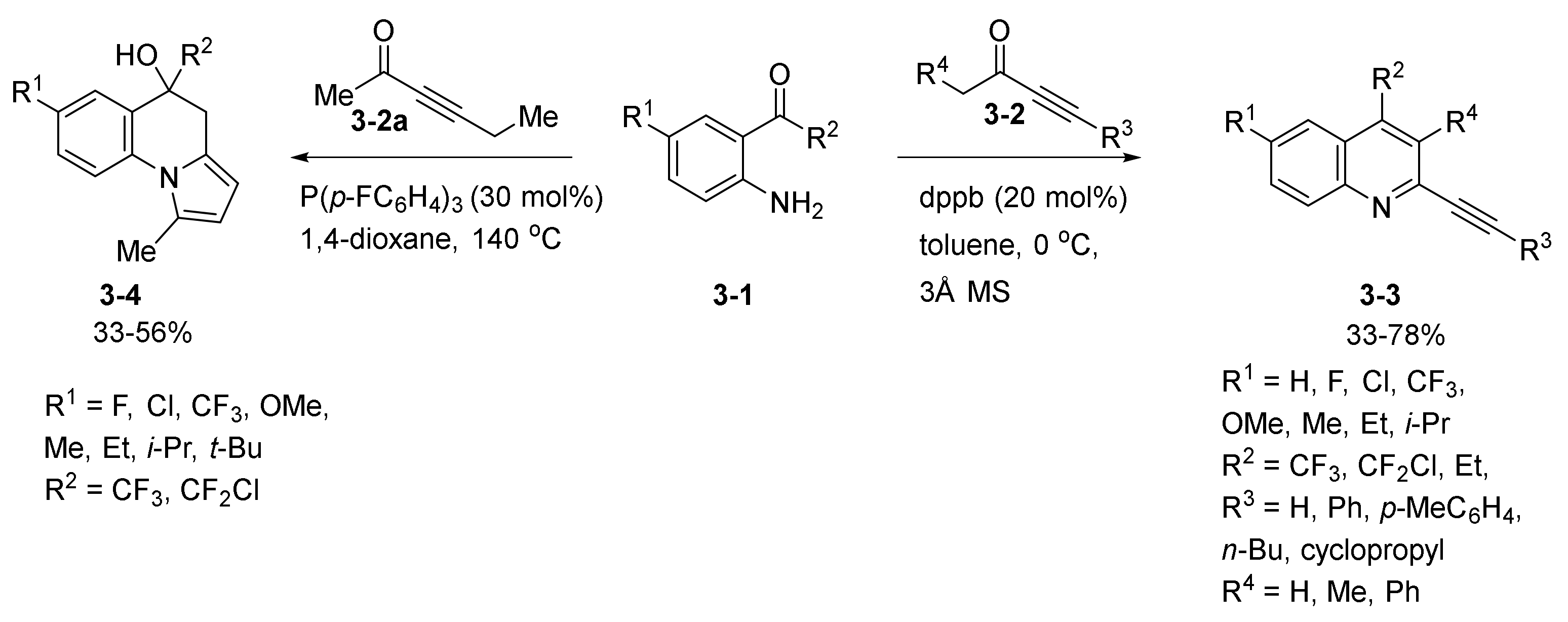
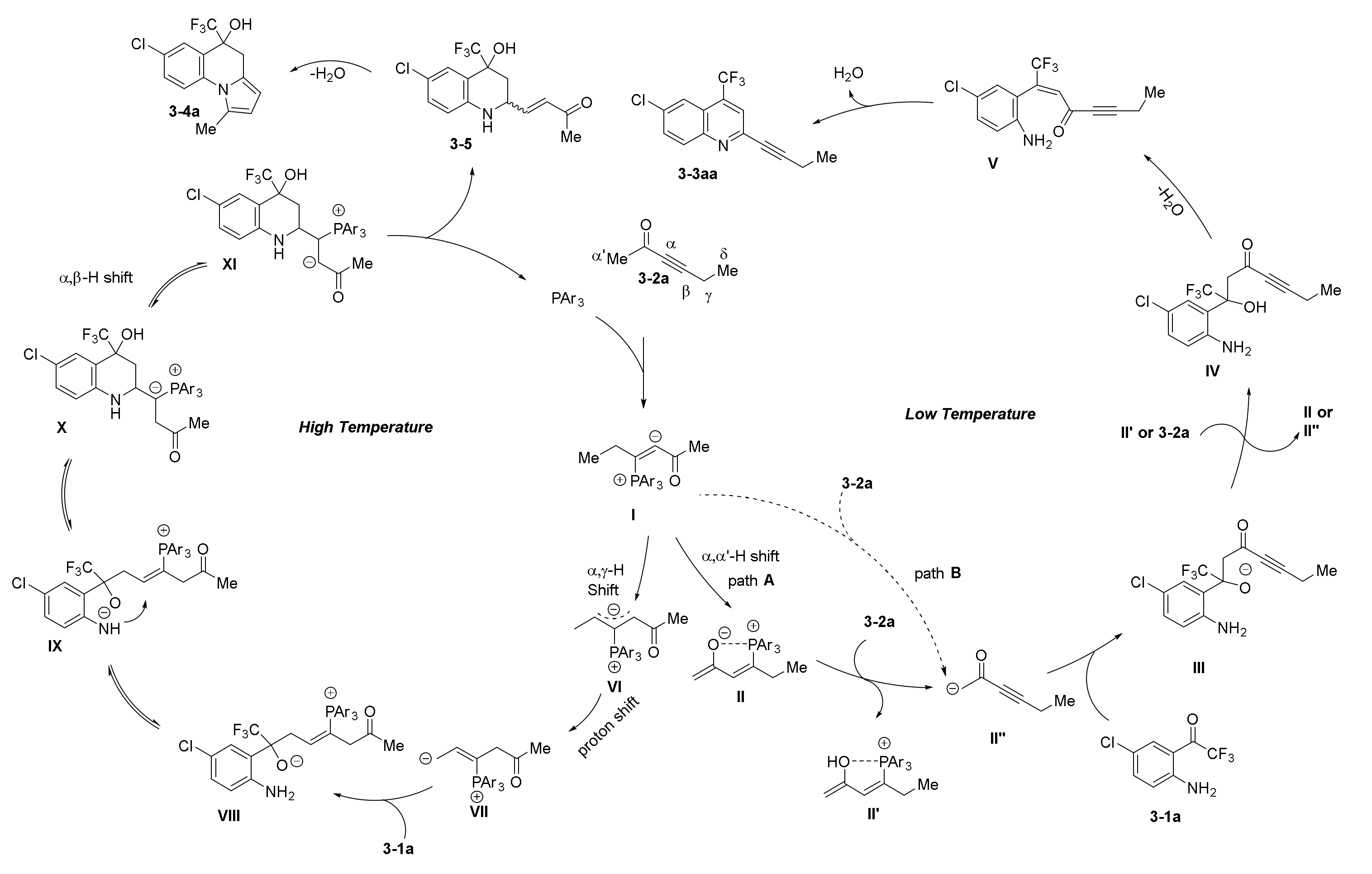
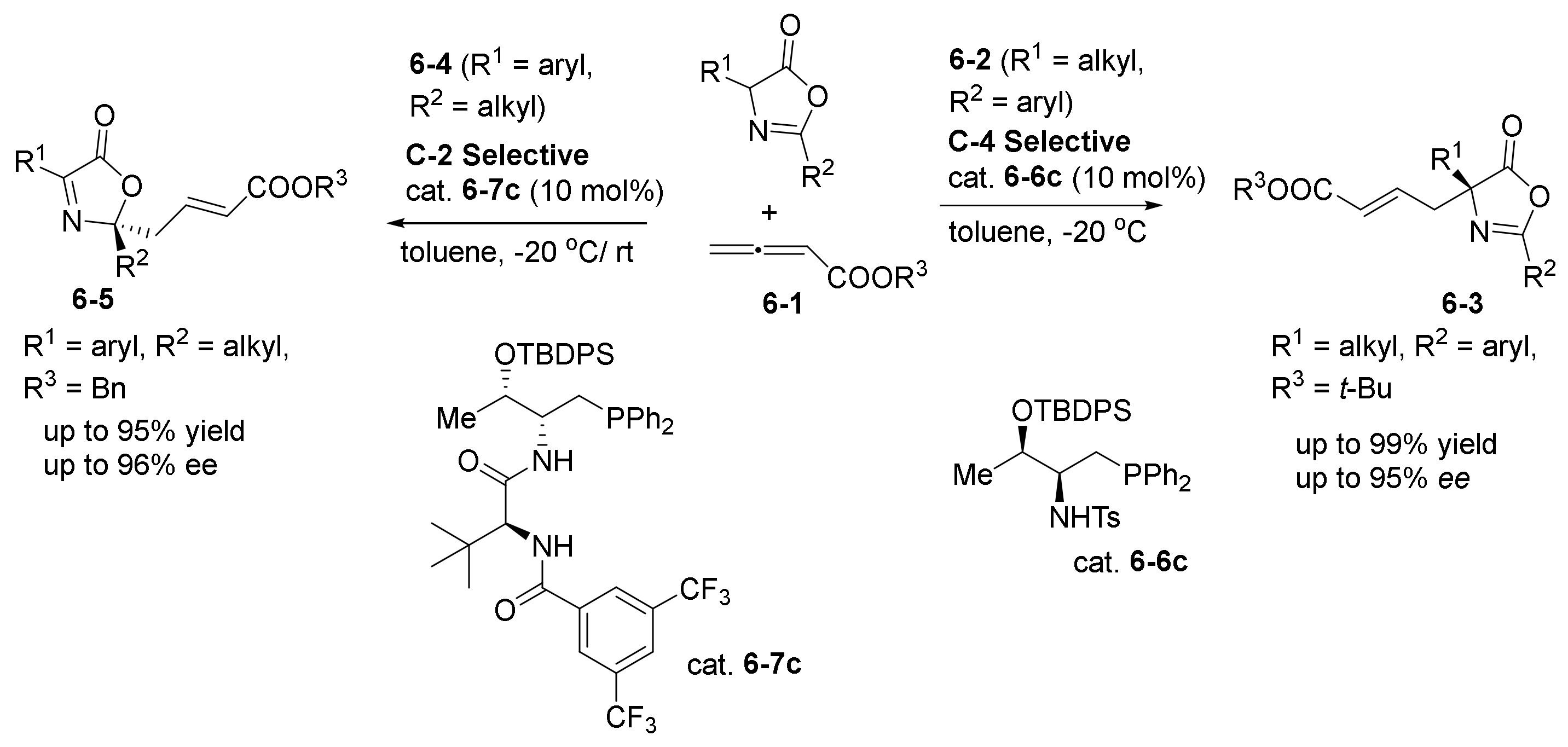
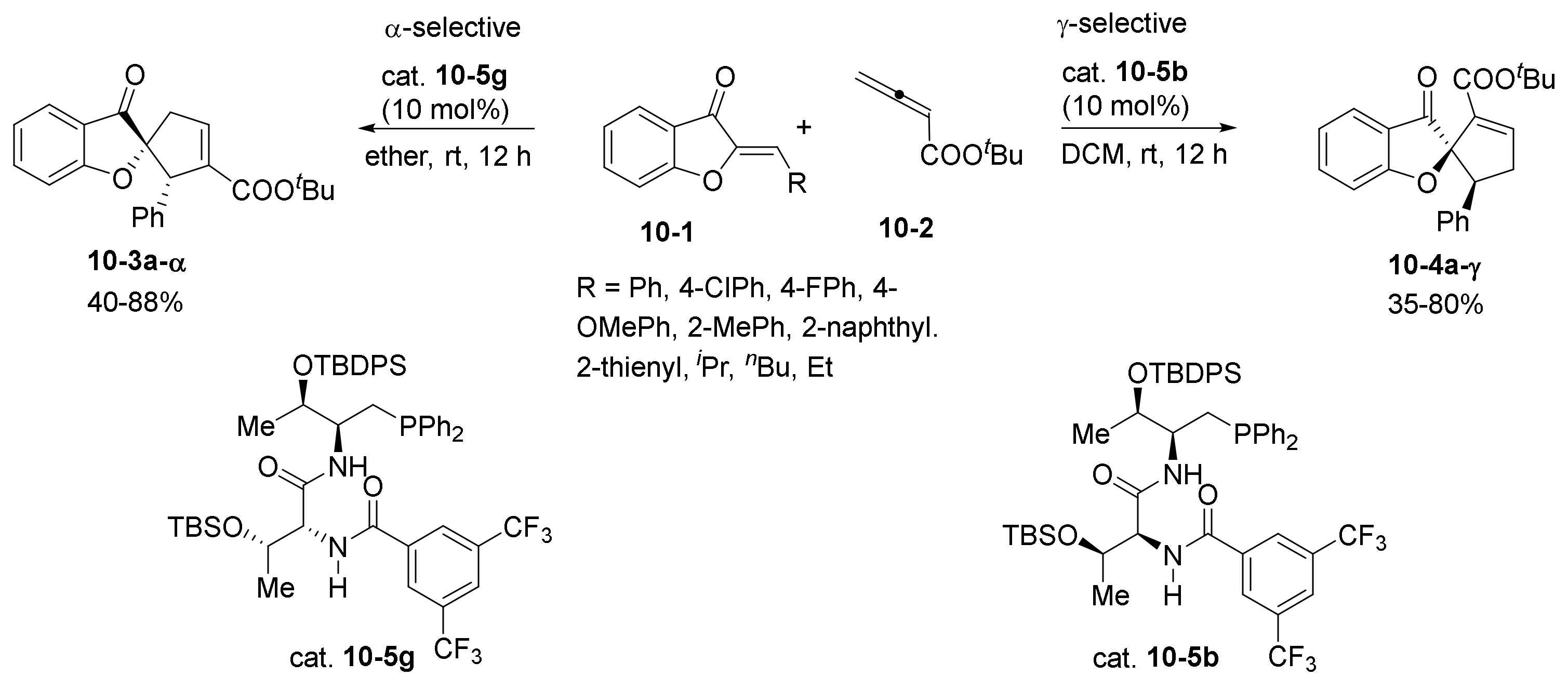
- Wang, T.; Yu, Z.; Hoon, D.L.; Phee, C.Y.; Lan, Y.; Lu, Y. Regiodivergent Enantioselective γ-Additions of Oxazolones to 2,3-Butadienoates Catalyzed by Phosphines: Synthesis of α,α-Disubstituted α-Amino Acids and N,O-Acetal Derivatives. J. Am. Chem. Soc. 2016, 138, 265–271. [Google Scholar] [CrossRef]
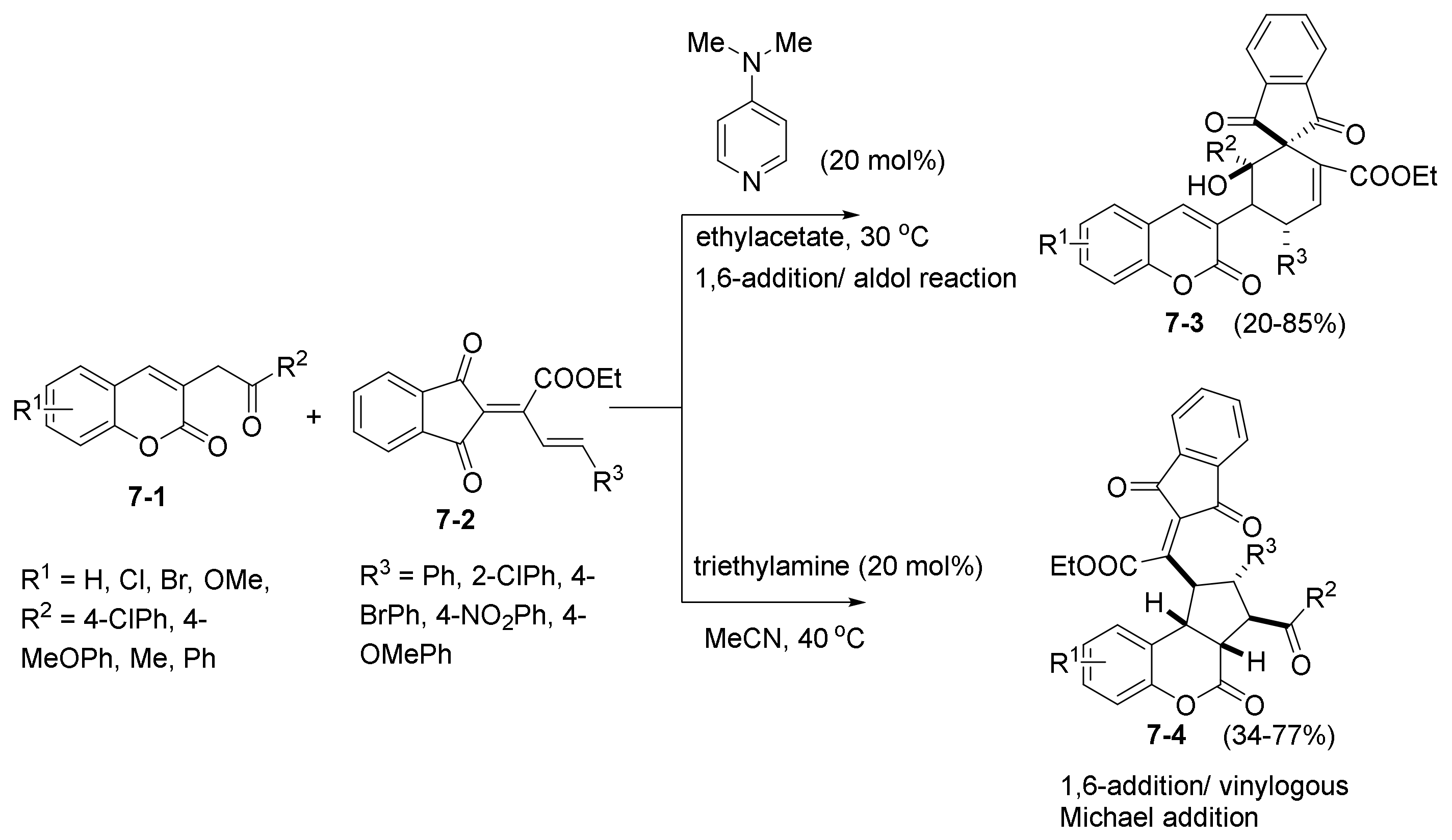
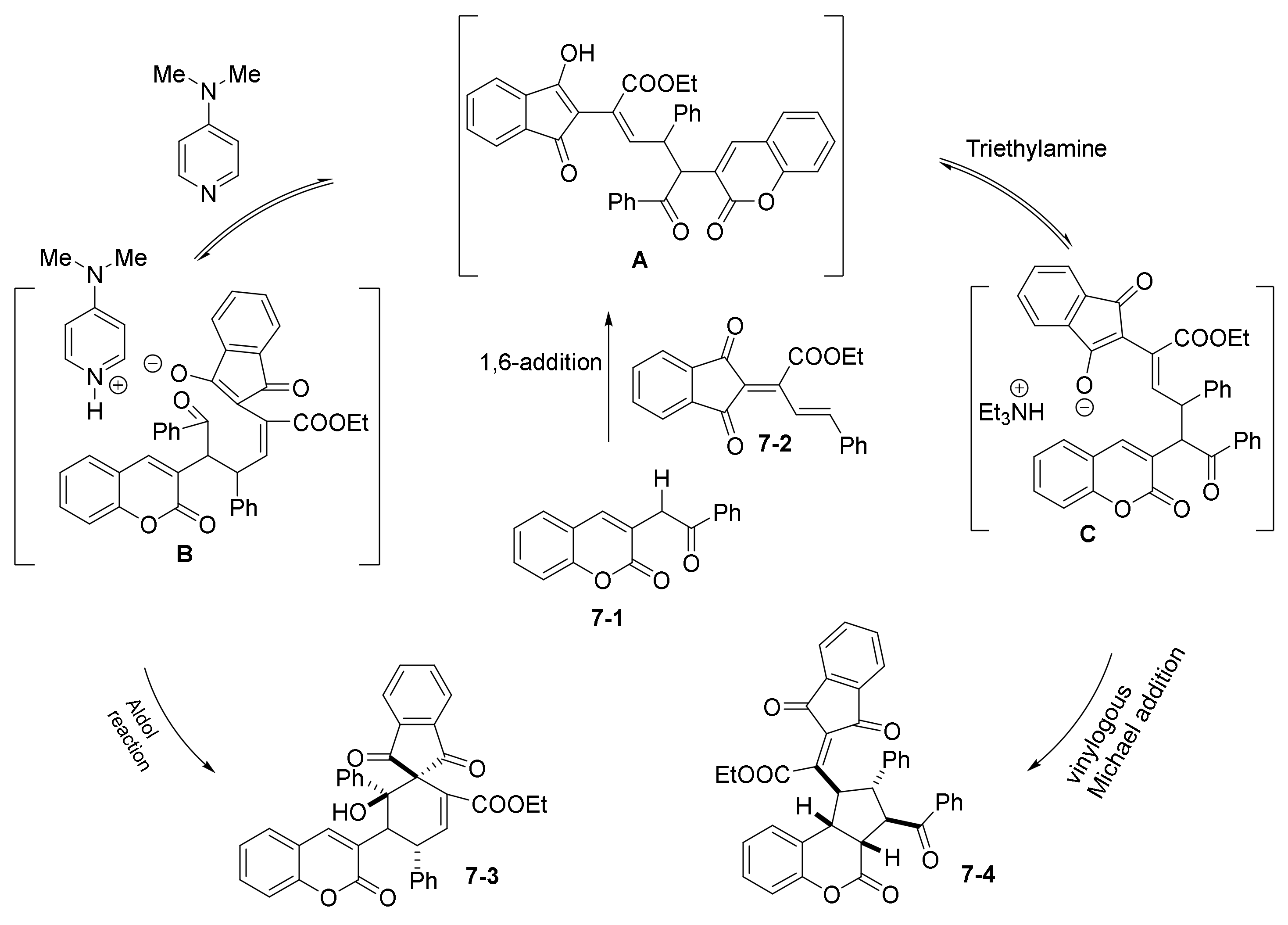
- Wang, M.; Tseng, P.-Y.; Chi, W.-J.; Suresh, S.; Edukondalu, A.; Chen, Y.-R.; Lin, W. Diversity-Oriented Synthesis of Spirocyclohexene Indane-1,3-diones and Coumarin-Fused Cyclopentanes via an Organobase-Controlled Cascade Reaction. Adv. Synth. Catal. 2020, 362, 3407–3415. [Google Scholar] [CrossRef]
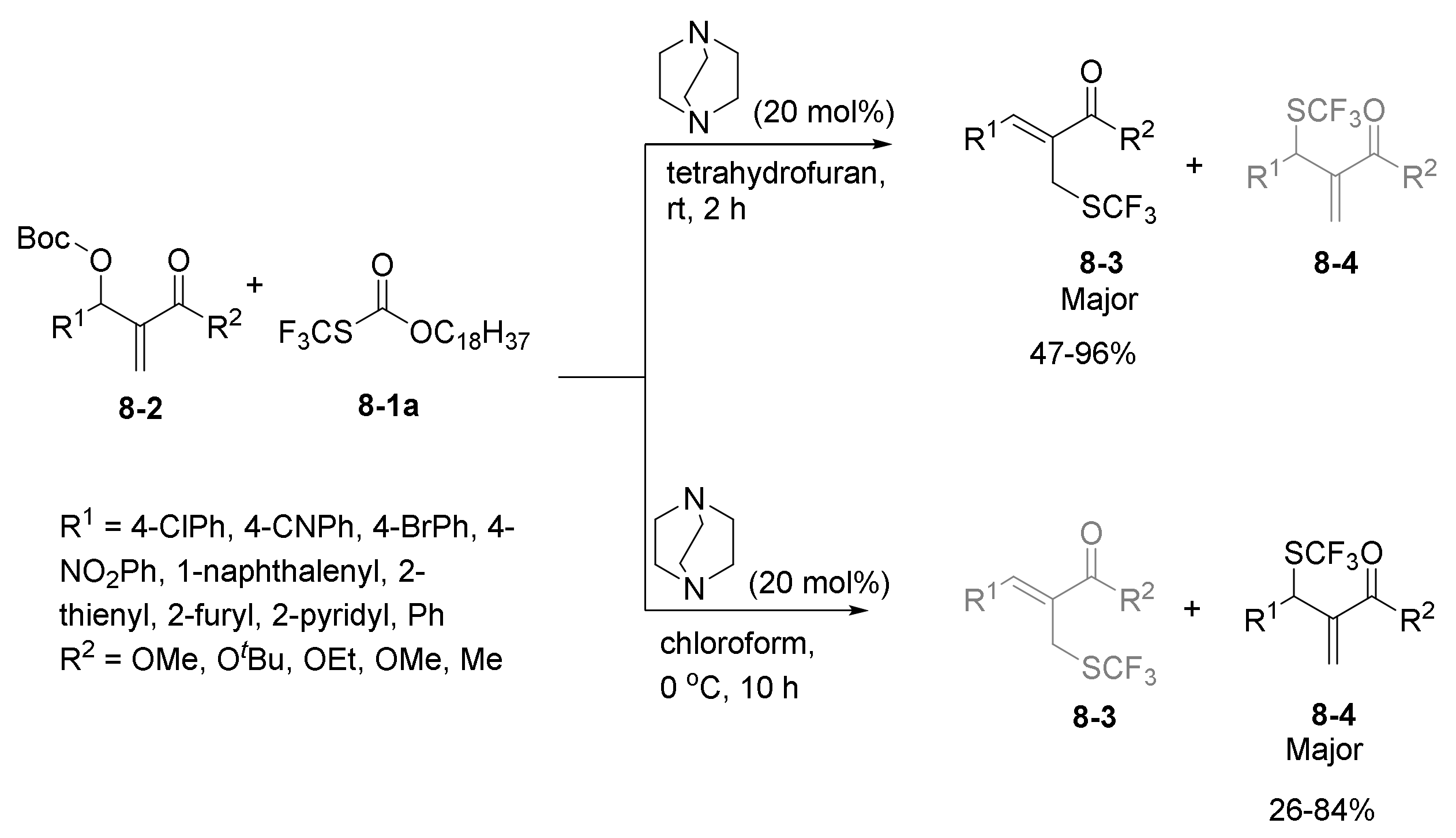
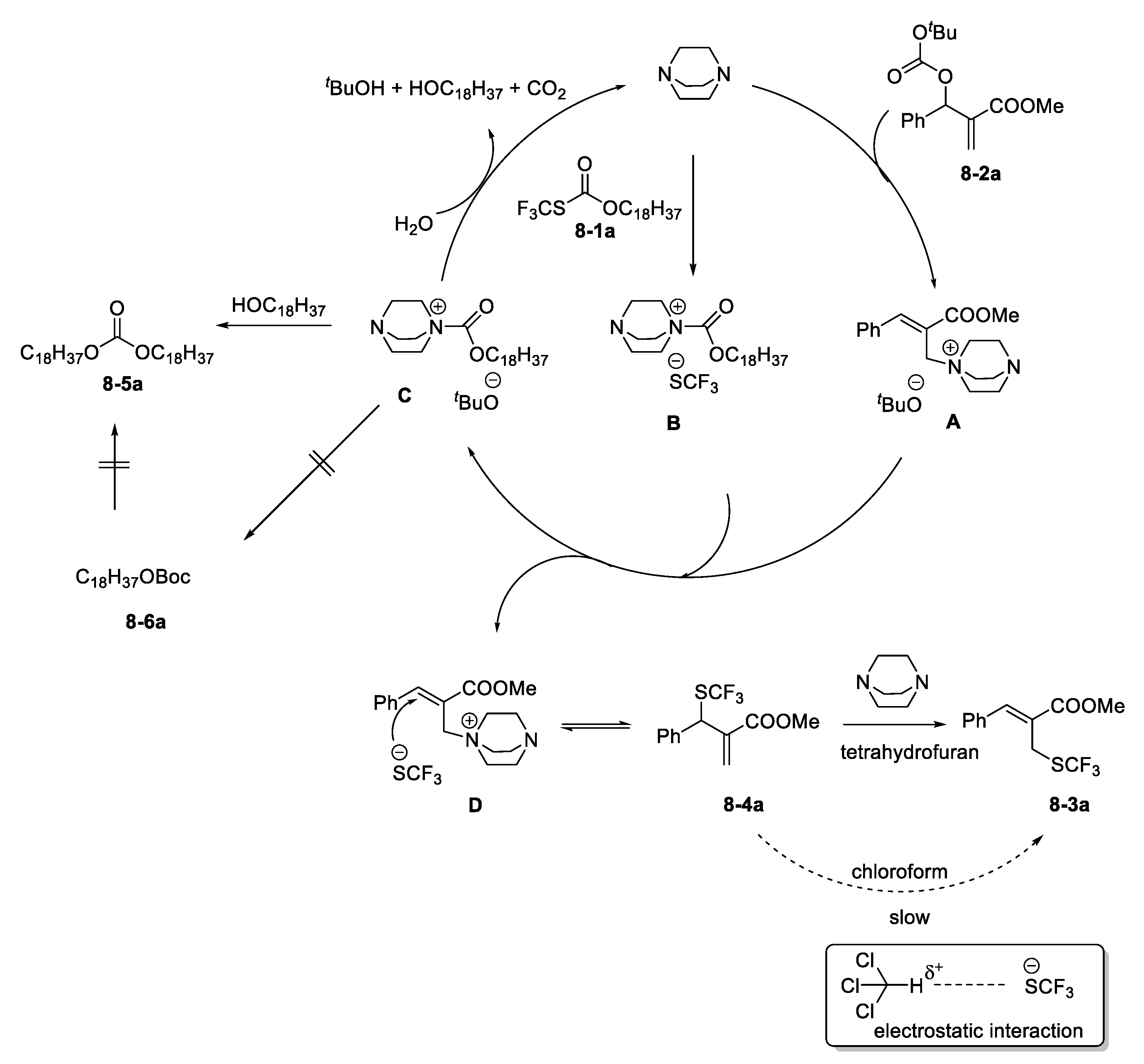
- Yang, H.-B.; Fan, X.; Wei, Y.; Shi, M. Solvent-controlled nucleophilic trifluoromethylthiolation of Morita–Baylis–Hillman carbonates: Dual roles of DABCO in activating the Zard’s trifluoromethylthiolation reagent and the MBH carbonates. Org. Chem. Front. 2015, 2, 1088–1093. [Google Scholar] [CrossRef]
- Luo, H.-Y.; Dong, J.-W.; Xie, Y.-Y.; Song, X.-F.; Zhu, D.; Ding, T.; Liu, Y.; Chen, Z.-M. Lewis Base/Brønsted Acid Co-Catalyzed Asymmetric Thiolation of Alkenes with Acid-Controlled Divergent Regioselectivity. Chem. Eur. J. 2019, 25, 15411–15418. [Google Scholar] [CrossRef]
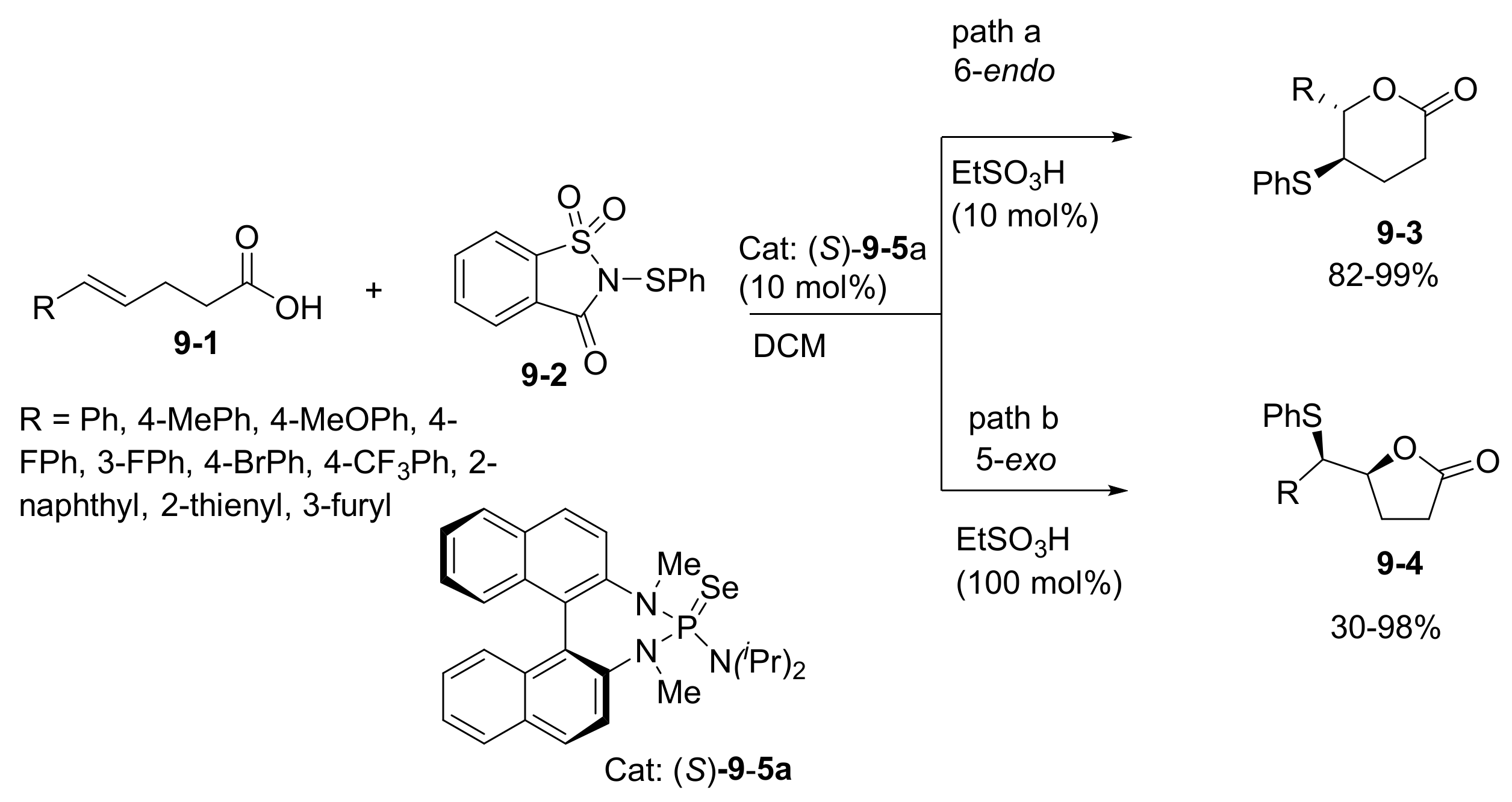
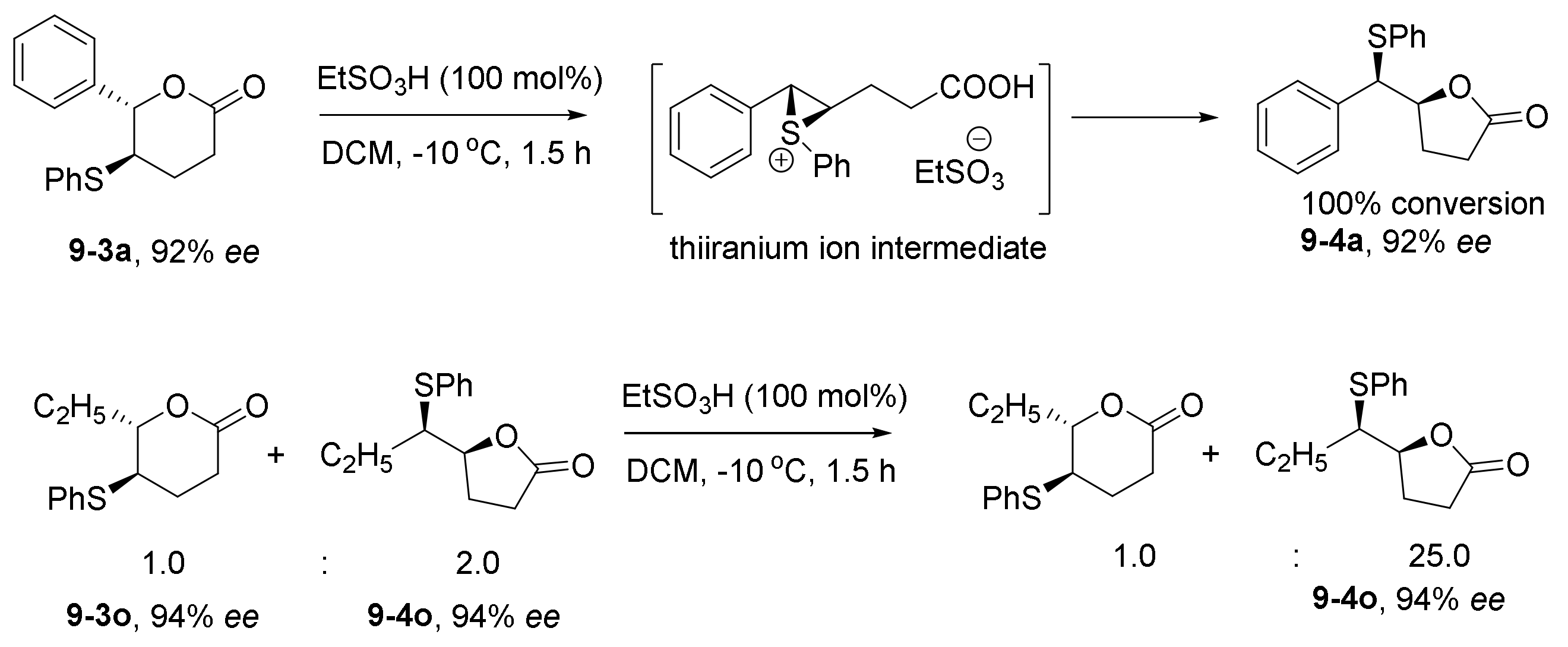
- Ni, H.; Yu, Z.; Yao, W.; Lan, Y.; Ullah, N.; Lu, Y. Catalyst-controlled regioselectivity in phosphine catalysis: The synthesis of spirocyclic benzofuranones via regiodivergent [3+2] annulations of aurones and an allenoate. Chem. Sci. 2017, 8, 5699–5704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Cahard, D. Regio- and Stereocontrolled Nucleophilic Trifluoromethylthiolation of Morita–Baylis–Hillman Carbonates. Synlett 2015, 26, 40–44. [Google Scholar]
- Chen, X.-Y.; Lin, R.-C.; Ye, S. Catalytic [2+2] and [3+2] cycloaddition reactions of allenoates with cyclic ketimines. Chem. Commun. 2012, 48, 1317–1319. [Google Scholar] [CrossRef]
- Luo, W.; Shao, B.; Li, J.; Song, D.; Yi, X.; Ling, F.; Zhong, W. Divergent synthesis of spirocyclopentene-pyrazolones and pyrano[2,3-c]-pyrazoles via Lewis base controlled annulation reactions. Tetrahedron Lett. 2019, 60, 151206. [Google Scholar] [CrossRef]
- Sun, F.; Yin, T.; Feng, A.; Hu, Y.; Yu, C.; Li, T.; Yao, C. Base-promoted regiodivergent allylation of N-acylhydrazones with Morita–Baylis–Hillman carbonates by tuning the catalyst. Org. Biomol. Chem. 2019, 17, 5283–5293. [Google Scholar] [CrossRef]
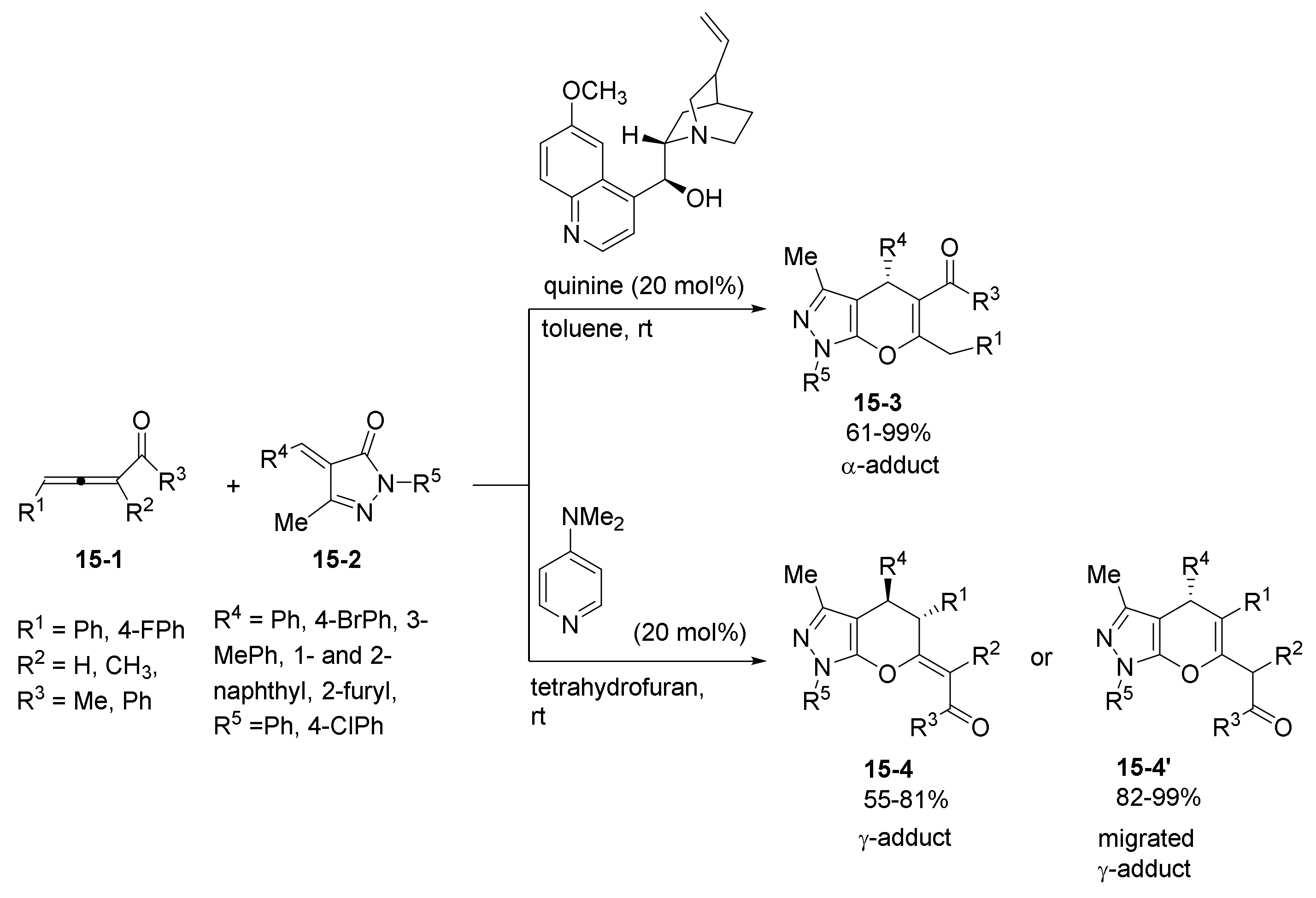
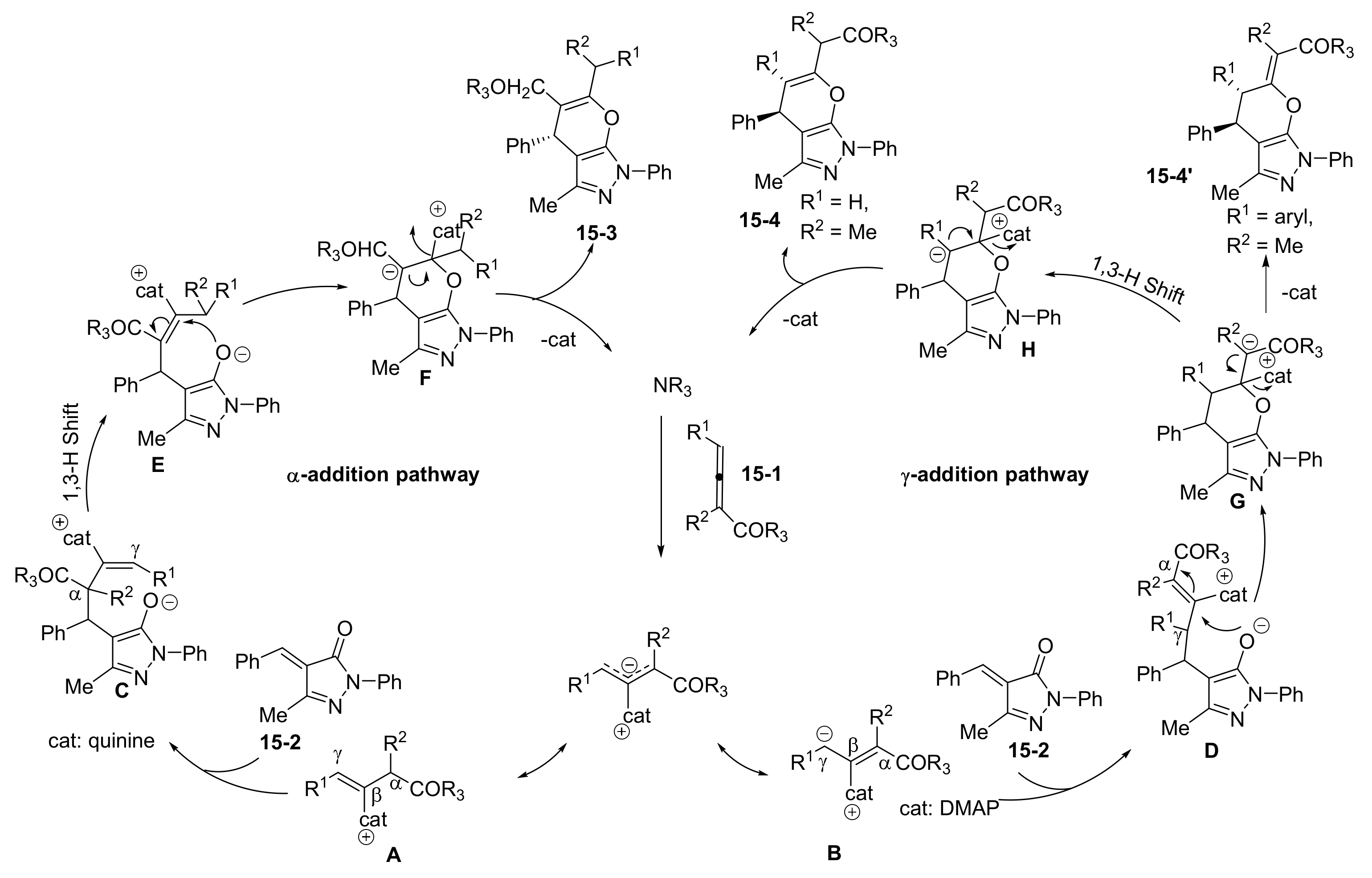
- Zhang, W.; Wei, S.; Wang, W.; Qu, J.; Wang, B. Catalytic asymmetric construction of C-4 alkenyl substituted pyrazolone derivatives bearing multiple stereoelements. Chem. Commun. 2021, 57, 6550–6553. [Google Scholar] [CrossRef] [PubMed]
- Majdecki, M.; Grodek, P.; Jurczak, J. Stereoselective α-Chlorination of β-Keto Esters in the Presence of Hybrid Amide-Based Cinchona Alkaloids as Catalysts. J. Org. Chem. 2021, 86, 995–1001. [Google Scholar] [CrossRef]
- Anif Pasha, M.; Peraka, S.; Ramachary, D.B. Catalytic Asymmetric Synthesis of Benzobicyclo[3.2.1]octanes. Chem. Eur. J. 2021, 27, 10563–10568. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yan, R.-J.; Du, W.; Chen, Y.-C. Asymmetric Dearomative Cascade Multiple Functionalizations of Activated N-Alkylpyridinium and N-Alkylquinolinium Salts. Org. Lett. 2020, 22, 7617–7621. [Google Scholar] [CrossRef]
- Krishna, A.V.; Reddy, G.S.; Gorachand, B.; Ramachary, D.B. Organocatalytic Asymmetric Formal [3+3]-Cycloaddition to Access 2,3-Diazaspiro[4.5]deca-3,6-dien-1-ones. Eur. J. Org. Chem. 2020, 2020, 6623–6628. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Song, S.-G.; Ren, L.; Li, Y.-G.; Wu, X. Enantioselective γ-Alkylation of α,β-Unsaturated Aldehydes Using New Cinchona-Based Primary Amine Catalyst. Eur. J. Org. Chem. 2019, 2019, 6838–6841. [Google Scholar] [CrossRef]
- Dočekal, V.; Formánek, B.; Císařová, I.; Veselý, J. A formal [4+2] cycloaddition of sulfur-containing alkylidene heterocycles with allenic compounds. Org. Chem. Front. 2019, 6, 3259–3263. [Google Scholar] [CrossRef]
- Deng, Y.-H.; Chu, W.-D.; Zhang, X.-Z.; Yan, X.; Yu, K.-Y.; Yang, L.-L.; Huang, H.; Fan, C.-A. Cinchona Alkaloid Catalyzed Enantioselective [4+2] Annulation of Allenic Esters and in Situ Generated ortho-Quinone Methides: Asymmetric Synthesis of Functionalized Chromans. J. Org. Chem. 2017, 82, 5433–5440. [Google Scholar] [CrossRef]
- Gui, Y.-Y.; Yang, J.; Qi, L.-W.; Wang, X.; Tian, F.; Li, X.-N.; Peng, L.; Wang, L.-X. A cinchona alkaloid catalyzed enantioselective sulfa-Michael/aldol cascade reaction of isoindigos: Construction of chiral bispirooxindole tetrahydrothiophenes with vicinal quaternary spirocenters. Org. Biomol. Chem. 2015, 13, 6371–6379. [Google Scholar] [CrossRef]
- Marcelli, T.; Hiemstra, H. Cinchona Alkaloids in Asymmetric Organocatalysis. Synthesis 2010, 2010, 1229–1279. [Google Scholar] [CrossRef]
- Cui, X.-Y.; Duan, H.-X.; Zhang, Y.; Wang, Y.-Q. Asymmetric Mannich Reaction of Aryl Methyl Ketones with Cyclic Imines Benzo[e][1,2,3]oxathiazine 2,2-Dioxides Catalyzed by Cinchona Alkaloid-based Primary Amines. Chem. Asian J. 2016, 11, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, D.; Zhang, X.R. Direct Asymmetric anti-Mannich-Type Reactions Catalyzed by Cinchona Alkaloid Derivatives. Chirality 2014, 26, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Kong, S.; Cai, Y.; Wu, G.; Miao, Z. Diastereo- and enantioselective nitro-Mannich reaction of α-substituted nitroacetates to N-phosphoryl imines catalyzed by cinchona alkaloid thiourea organocatalysts. Org. Biomol. Chem. 2013, 11, 3223–3229. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K.; Kumar, R.; Singh, S.; Jain, S.; Vanka, K.; Singh, R.P. The Vinylogous Michael Addition of 3-Alkylidene-2-oxindoles to β,γ-Unsaturated α-Keto Esters by Bifunctional Cinchona Alkaloids. Eur. J. Org. Chem. 2020, 2020, 5690–5694. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, M.; Wang, Z.; Tang, L.; Chen, W.; Ban, S.; Li, Q. Highly enantioselective Michael addition of pyrazolin-5-ones to nitroolefins catalyzed by cinchona alkaloid derived 4-methylbenzoylthioureas. Chirality 2018, 30, 1096–1104. [Google Scholar] [CrossRef]
- Cabanillas, A.; Davies, C.D.; Male, L.; Simpkins, N.S. Highly enantioselective access to diketopiperazines via cinchona alkaloid catalyzed Michael additions. Chem. Sci. 2015, 6, 1350–1354. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.D.; He, X.Y.; Han, D.D.; Yang, X.R.; Yao, H.B.; Cheng, D.J. Asymmetric Aza-Henry Reaction of Indolenines Mediated by a Cinchona-Alkaloid-Thiourea Organocatalyst. Asian J. Org. Chem. 2019, 8, 2023–2026. [Google Scholar] [CrossRef]
- Lu, N.; Fang, Y.; Gao, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Bifunctional Thiourea-Ammonium Salt Catalysts Derived from Cinchona Alkaloids: Cooperative Phase-Transfer Catalysts in the Enantioselective Aza-Henry Reaction of Ketimines. J. Org. Chem. 2018, 83, 1486–1492. [Google Scholar] [CrossRef]
- Majdecki, M.; Tyszka-Gumkowska, A.; Jurczak, J. Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones Using Amide-Based Cinchona Alkaloids as Hybrid Phase-Transfer Catalysts. Org. Lett. 2020, 22, 8687–8691. [Google Scholar] [CrossRef]
- Berkessel, A.; Guixà, M.; Schmidt, F.; Neudörfl, J.M.; Lex, J. Highly enantioselective epoxidation of 2-methylnaphthoquinone (vitamin K3) mediated by new cinchona alkaloid phase-transfer catalysts. Chem. Eur. J. 2007, 13, 4483–4498. [Google Scholar] [CrossRef]
- Cheng, C.; Sun, X.; Wu, Z.; Liu, Q.; Xiong, L.; Miao, Z. Lewis base catalyzed regioselective cyclization of allene ketones or α-methyl allene ketones with unsaturated pyrazolones. Org. Biomol. Chem. 2019, 17, 3232–3238. [Google Scholar] [CrossRef] [PubMed]
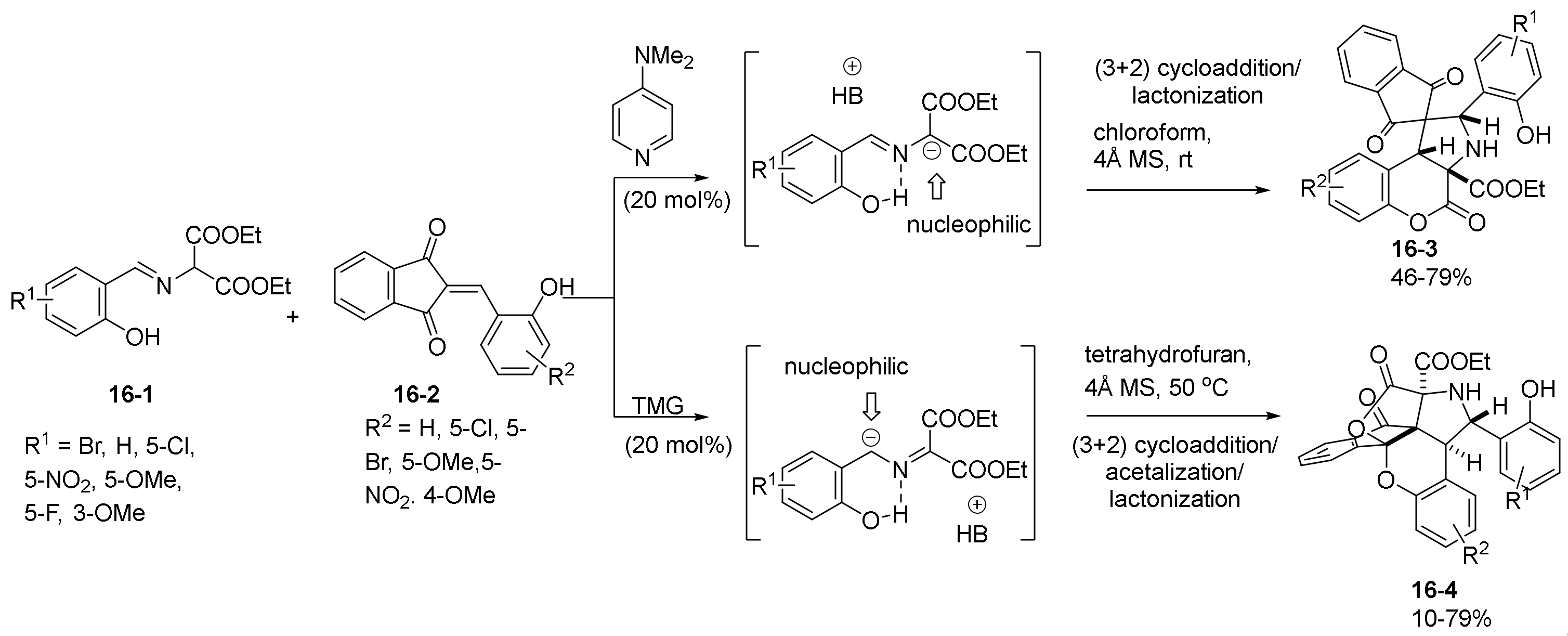
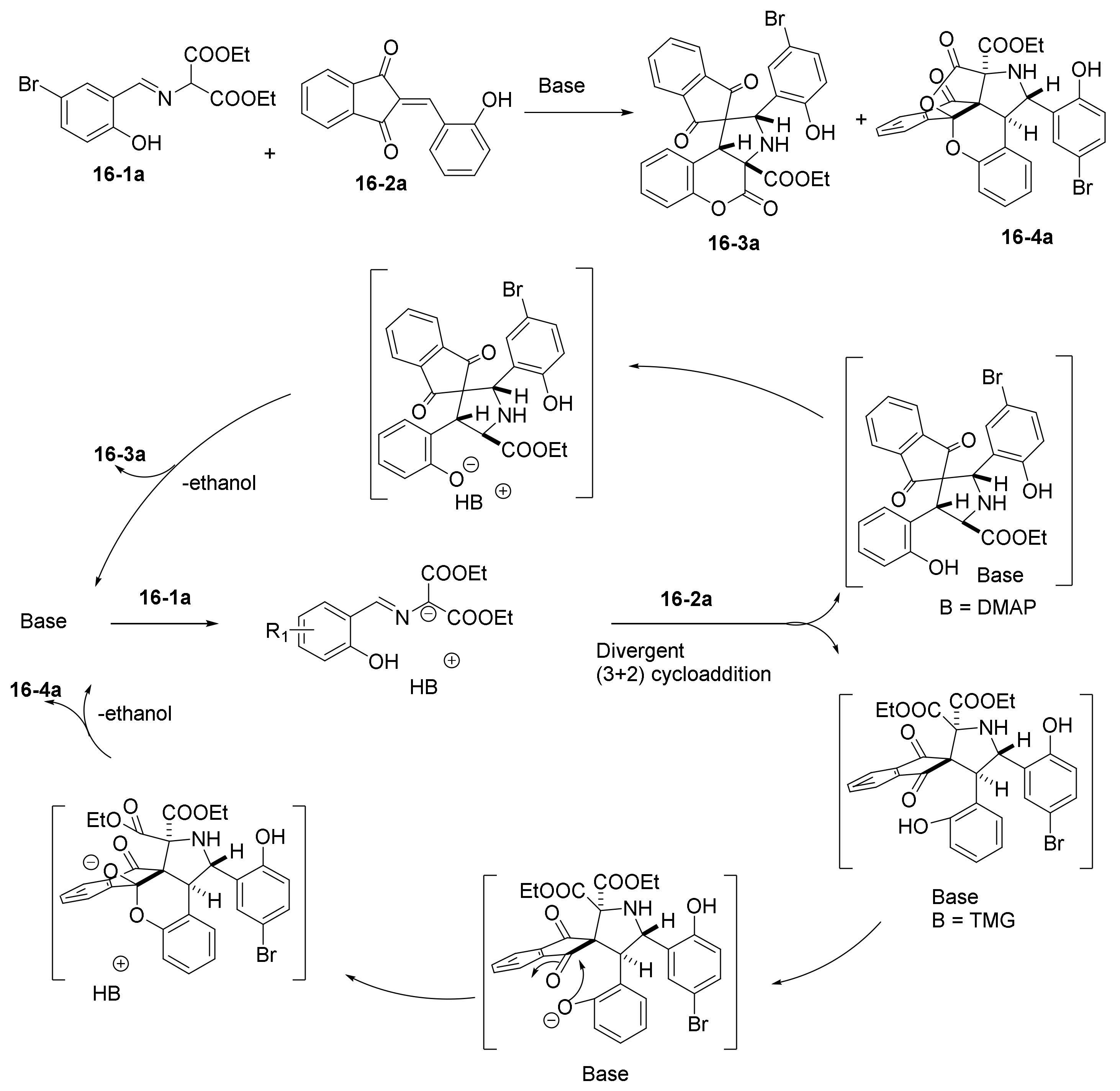
- Yu, J.-K.; Chien, H.-W.; Lin, Y.-J.; Karanam, P.; Chen, Y.-H.; Lin, W. Diversity-oriented synthesis of chromenopyrrolidines from azomethine ylides and 2-hydroxybenzylidene indandiones via base-controlled regiodivergent (3+2) cycloaddition. Chem. Commun. 2018, 54, 9921–9924. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-B.; Wan, D.-H. C–C Bond Acylation of Oxime Ethers via NHC Catalysis. Org. Lett. 2021, 23, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Suresh, S. N-Heterocyclic Carbene (NHC)-Catalyzed Intramolecular Stetter Reaction to Access Dibenzo-fused Seven-membered Heterocycles. Asian J. Org. Chem. 2021, 10, 1406–1409. [Google Scholar] [CrossRef]
- Wang, C.; Liu, L. NHC-catalyzed oxindole synthesis via single electron transfer. Org. Chem. Front. 2021, 8, 1454–1460. [Google Scholar] [CrossRef]
- Suresh, P.; Thamotharan, S.; Selva Ganesan, S. NHC Organocatalysis in D2O for the Highly Diastereoselective Synthesis of Deuterated Spiropyran Analogues. ChemistrySelect 2021, 6, 2036–2040. [Google Scholar] [CrossRef]
- Song, R.; Jin, Z.; Chi, Y.R. NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions. Chem. Sci. 2021, 12, 5037–5043. [Google Scholar] [CrossRef]
- Singh, A.; Narula, A.K. N-Heterocyclic carbene (NHC) catalyzed amidation of aldehydes with amines via the tandem N-hydroxysuccinimide ester formation. New J. Chem. 2021, 45, 7486–7490. [Google Scholar] [CrossRef]
- Satyam, K.; Ramarao, J.; Suresh, S. N-Heterocyclic carbene (NHC)-catalyzed intramolecular benzoin condensation–oxidation. Org. Biomol. Chem. 2021, 19, 1488–1492. [Google Scholar] [CrossRef]
- Leng, H.; Zhao, Q.; Mao, Q.; Liu, S.; Luo, M.; Qin, R.; Huang, W.; Zhan, G. NHC-catalysed retro-aldol/aldol cascade reaction enabling solvent-controlled stereodivergent synthesis of spirooxindoles. Chin. Chem. Lett. 2021, in press. [Google Scholar] [CrossRef]
- Barman, D.; Ghosh, T.; Show, K.; Debnath, S.; Ghosh, T.; Maiti, D.K. NHC-Mediated Stetter-Aldol and Imino-Stetter-Aldol Domino Cyclization to Naphthalen-1(2H)-ones and Isoquinolines. Org. Lett. 2021, 23, 2178–2182. [Google Scholar] [CrossRef]
- Barik, S.; Biju, A.T. N-Heterocyclic carbene (NHC) organocatalysis using aliphatic aldehydes. Chem. Commun. 2020, 56, 15484–15495. [Google Scholar] [CrossRef]
- Wu, Q.; Li, C.; Wang, W.; Wang, H.; Pan, D.; Zheng, P. NHC-catalyzed enantioselective synthesis of dihydropyran-4-carbonitriles bearing all-carbon quaternary centers. Org. Chem. Front. 2017, 4, 2323–2326. [Google Scholar] [CrossRef]
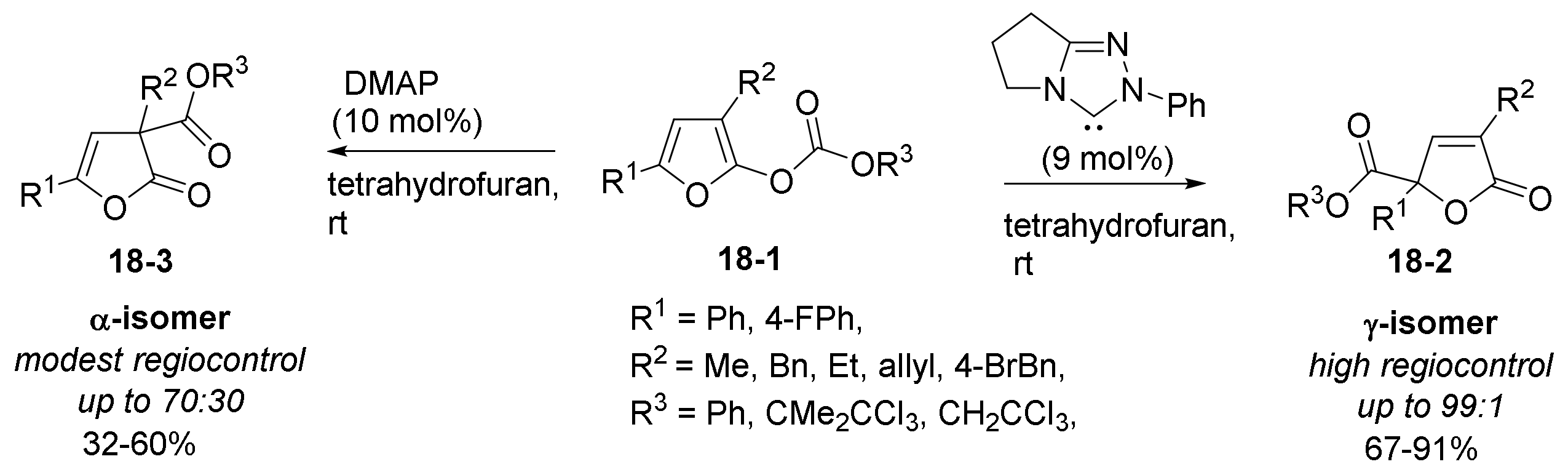
- Gould, E.; Walden, D.M.; Kasten, K.; Johnston, R.C.; Wu, J.; Slawin, A.M.Z.; Mustard, T.J.L.; Johnston, B.; Davies, T.; Ha-Yeon Cheong, P.; et al. Catalyst selective and regiodivergent O- to C- or N-carboxyl transfer of pyrazolyl carbonates: Synthetic and computational studies. Chem. Sci. 2014, 5, 3651–3658. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.D.; Joannesse, C.; Morrill, L.C.; Philp, D.; Smith, A.D. Regiodivergent Lewis base-promoted O- to C-carboxyl transfer of furanyl carbonates. Org. Biomol. Chem. 2015, 13, 2895–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
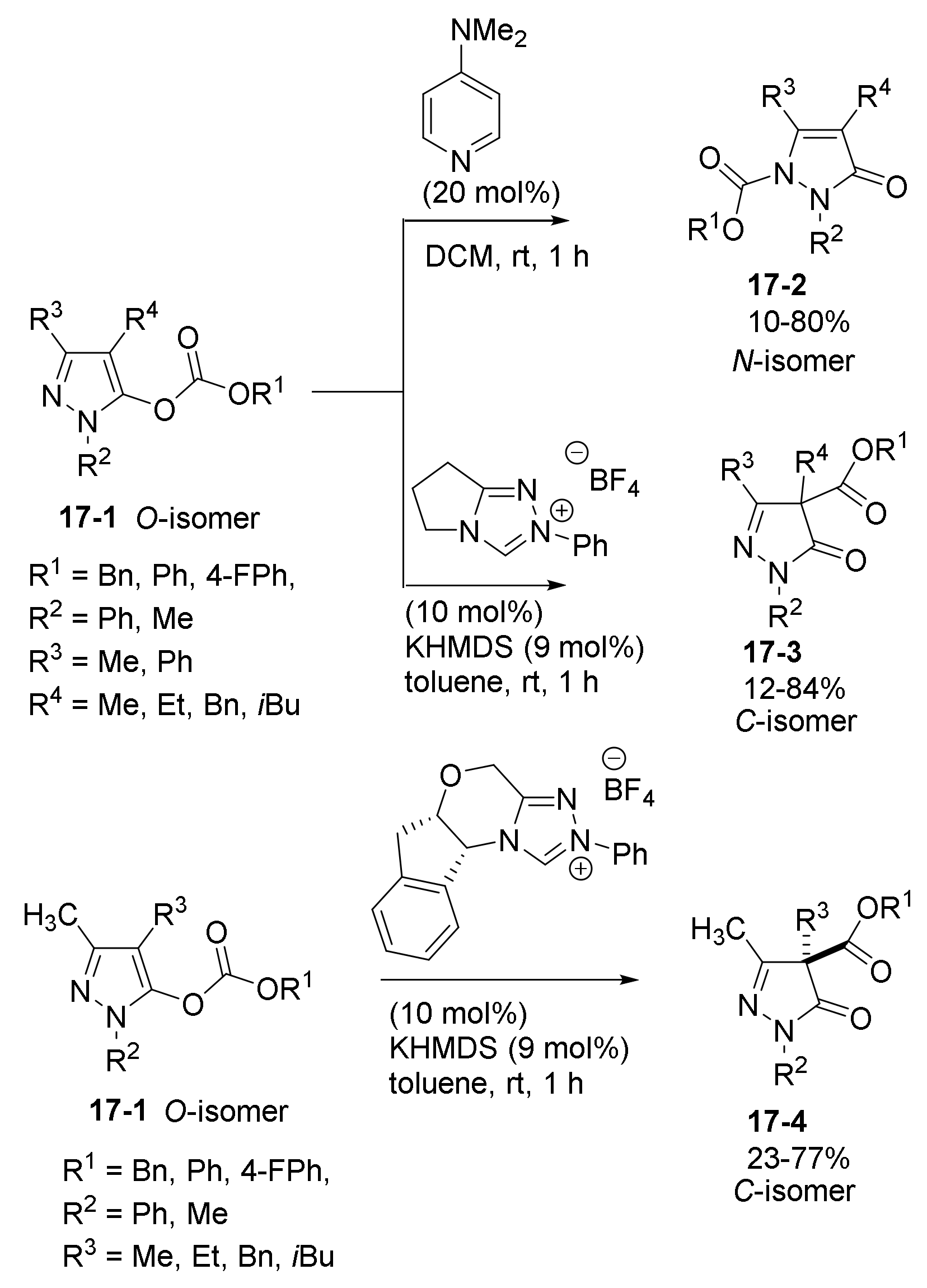
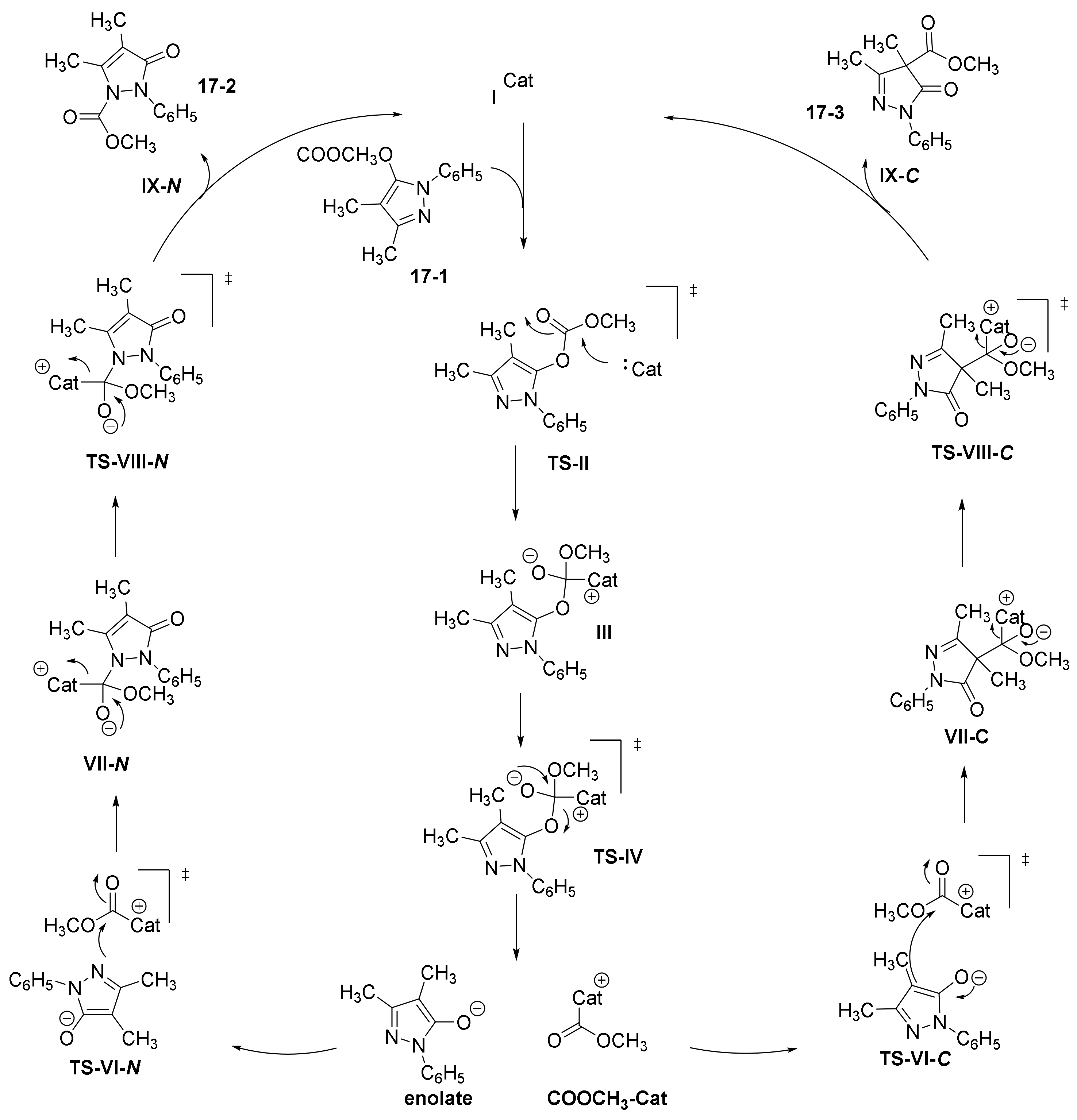
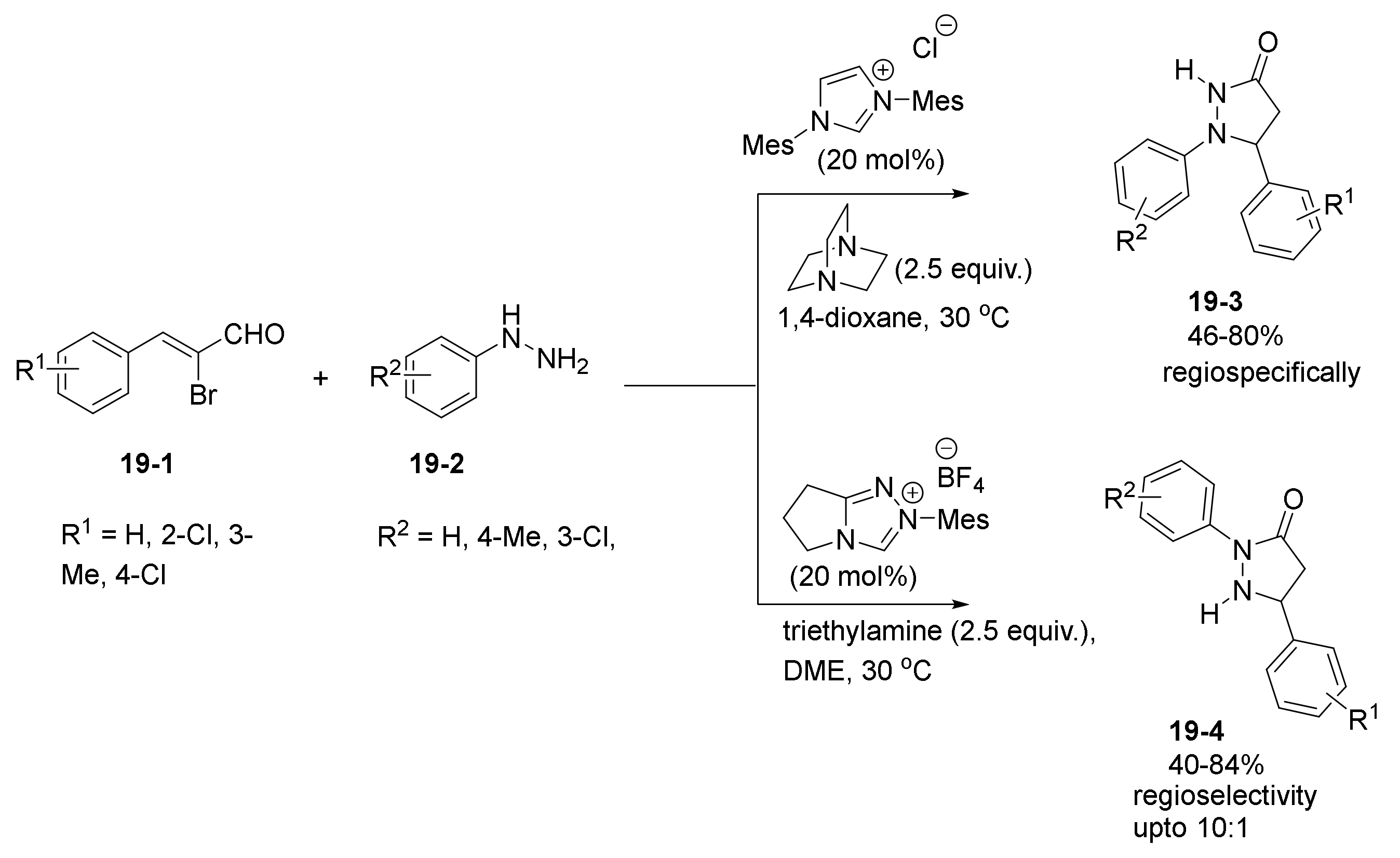
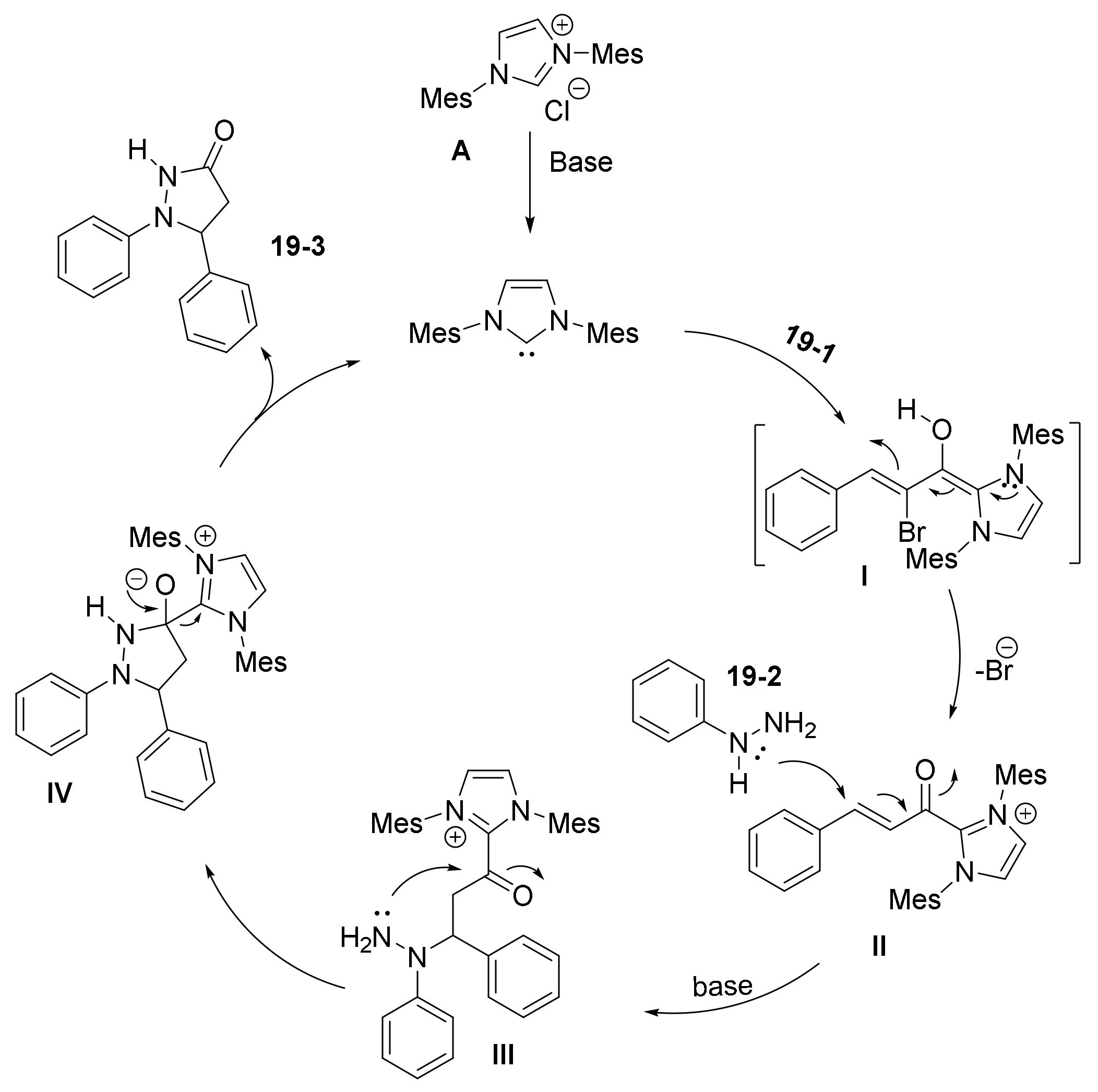
- Yu, C.; Shen, S.; Jiang, L.; Li, J.; Lu, Y.; Li, T.; Yao, C. NHC-catalyzed regiodivergent syntheses of difunctionalized 3-pyrazolidinones from α-bromoenal and monosubstituted hydrazine. Org. Biomol. Chem. 2017, 15, 9149–9155. [Google Scholar] [CrossRef] [PubMed]
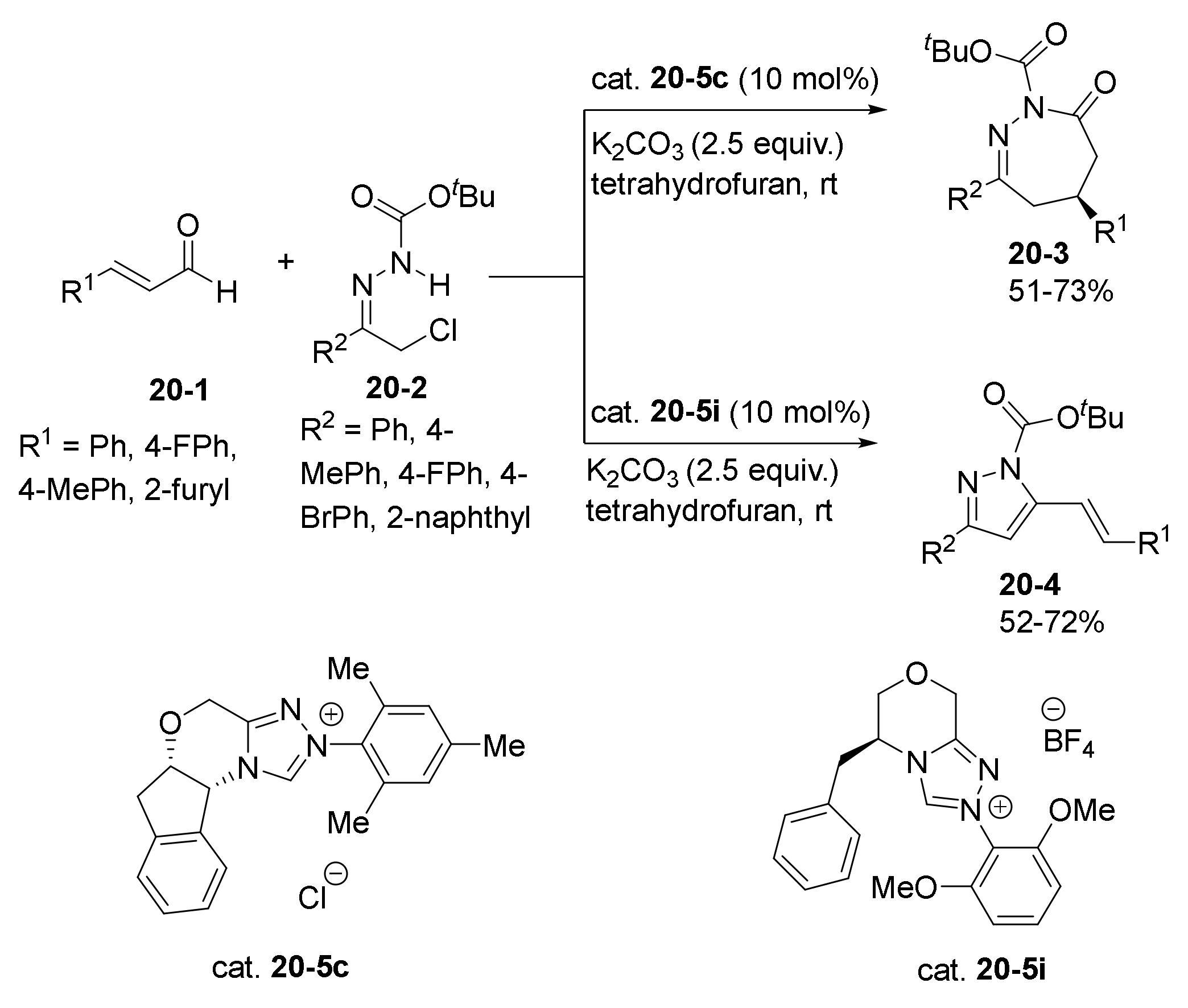
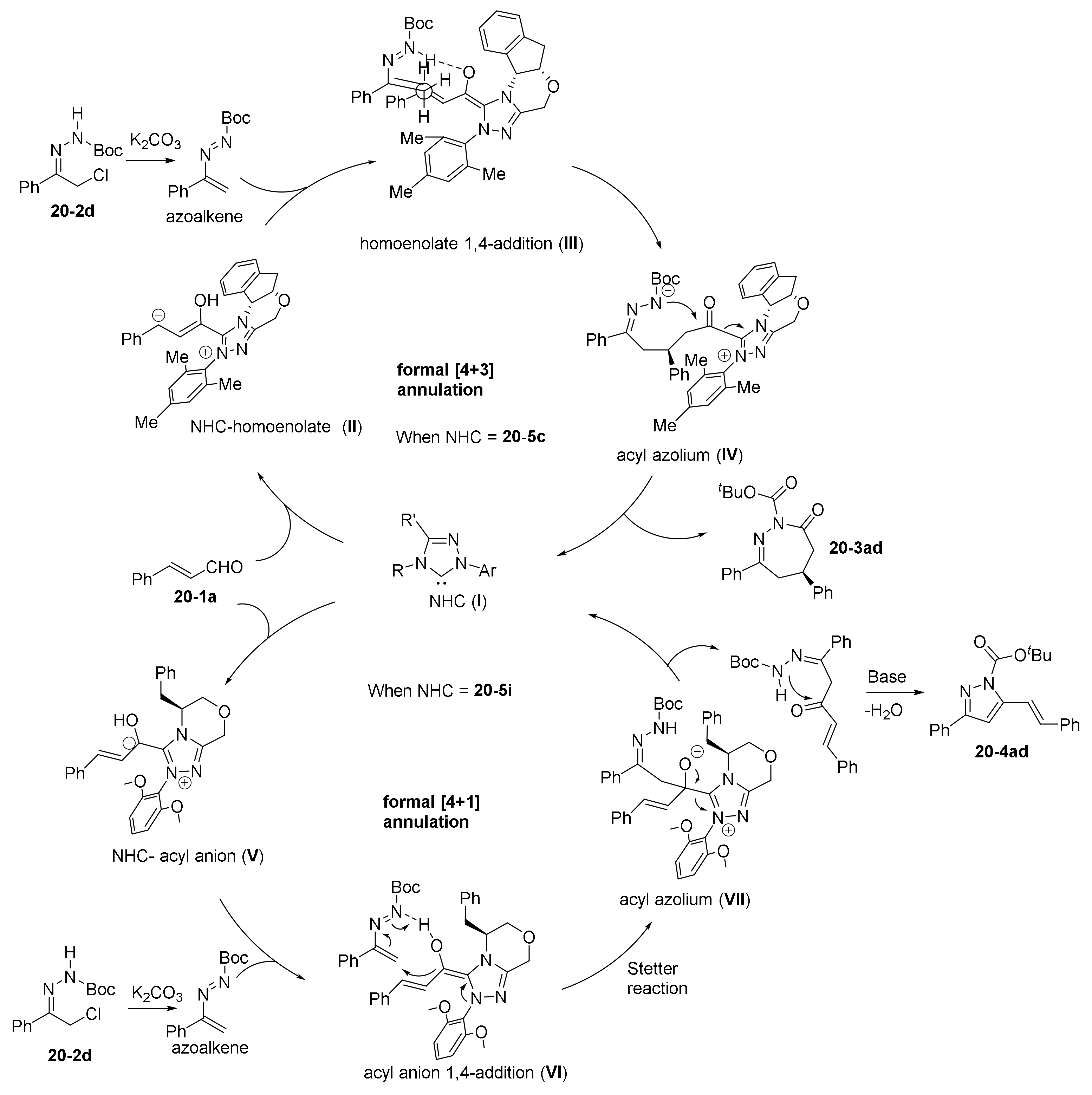
- Guo, C.; Sahoo, B.; Daniliuc, C.G.; Glorius, F. N-Heterocyclic Carbene Catalyzed Switchable Reactions of Enals with Azoalkenes: Formal [4+3] and [4+1] Annulations for the Synthesis of 1,2-Diazepines and Pyrazoles. J. Am. Chem. Soc. 2014, 136, 17402–17405. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C.G.; Glorius, F. Switchable selectivity in an NHC-catalysed dearomatizing annulation reaction. Nat. Chem. 2015, 7, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zha, T.; Shao, Z. Enantiodivergent synthesis of tricyclic chromans: Remote nucleophilic groups switch selectivity in catalytic asymmetric cascade reactions of trifunctional substrates. Green Synth. Catal. 2021, 2, 241–245. [Google Scholar] [CrossRef]
- Chen, Z.-C.; Du, W.; Chen, Y.-C. New Amines and Activation Modes in Asymmetric Aminocatalysis. Chin. J. Chem. 2021, 39, 1775–1786. [Google Scholar] [CrossRef]
- Da Silva, A.F.; Leonarczyk, I.A.; Ferreira, M.A.B.; Jurberg, I.D. Diastereodivergent aminocatalyzed spirocyclization strategies using 4-alkylideneisoxazol-5-ones and methyl vinyl ketones. Org. Chem. Front. 2020, 7, 3599–3607. [Google Scholar] [CrossRef]
- Arimitsu, S.; Gima, E. Improvement of primary-amine-catalyzed asymmetric α-benzoyloxylation of α-branched enals by a synergistic effect of water and sulfonic acids. Tetrahedron Lett. 2020, 61, 152032. [Google Scholar] [CrossRef]
- Przydacz, A.; Skrzyńska, A.; Albrecht, Ł. Breaking Aromaticity with Aminocatalysis: A Convenient Strategy for Asymmetric Synthesis. Angew. Chem. Int. Ed. 2019, 58, 63–73. [Google Scholar] [CrossRef]
- Maity, S.; Sar, S.; Ghorai, P. Primary Aminothiourea-Catalyzed Enantioselective Synthesis of Rauhut–Currier Adducts of 3-Arylcyclohexenone with a Tethered Enone on the Aryl Moiety at the Ortho-Position. Org. Lett. 2018, 20, 1707–1711. [Google Scholar] [CrossRef]
- Cui, L.; You, Y.E.; Mi, X.; Luo, S. Asymmetric Fluorination of α-Branched Aldehydes by Chiral Primary Amine Catalysis: Reagent-Controlled Enantioselectivity Switch. J. Org. Chem. 2018, 83, 4250–4256. [Google Scholar] [CrossRef]
- Vicario, J.L. Aminocatalytic Enantioselective Cycloadditions. Synlett 2016, 27, 1006–1021. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Vesely, J.; Rios, R.; Córdova, A. Proline and Lewis base co-catalyzed addition of α,β-unsaturated aldehydes to nitrostyrenes. Tetrahedron Lett. 2008, 49, 1137–1140. [Google Scholar] [CrossRef]
- Córdova, A.; Notz, W.; Barbas, C.F. Proline-Catalyzed One-Step Asymmetric Synthesis of 5-Hydroxy-(2E)-hexenal from Acetaldehyde. J. Org. Chem. 2002, 67, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Ramachary, D.B.; Barbas, C.F. Direct Amino Acid-Catalyzed Asymmetric Desymmetrization of meso-Compounds: Tandem Aminoxylation/O−N Bond Heterolysis Reactions. Org. Lett. 2005, 7, 1577–1580. [Google Scholar] [CrossRef]
- Watanabe, S.-I.; Córdova, A.; Tanaka, F.; Barbas, C.F. One-Pot Asymmetric Synthesis of β-Cyanohydroxymethyl α-Amino Acid Derivatives: Formation of Three Contiguous Stereogenic Centers. Org. Lett. 2002, 4, 4519–4522. [Google Scholar] [CrossRef] [PubMed]
- Juaristi, E. Recent developments in next generation (S)-proline-derived chiral organocatalysts. Tetrahedron 2021, 88, 132143. [Google Scholar] [CrossRef]
- Jessen, N.I.; Bertuzzi, G.; Bura, M.; Skipper, M.L.; Jørgensen, K.A. Enantioselective Construction of the Cycl[3.2.2]azine Core via Organocatalytic [12+2] Cycloadditions. J. Am. Chem. Soc. 2021, 143, 6140–6151. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, G.; Alamillo-Ferrer, C.; Burés, J. Mechanistically Guided Design of an Efficient and Enantioselective Aminocatalytic α-Chlorination of Aldehydes. J. Am. Chem. Soc. 2021, 143, 6805–6809. [Google Scholar] [CrossRef]
- Vachan, B.S.; Karuppasamy, M.; Vinoth, P.; Vivek Kumar, S.; Perumal, S.; Sridharan, V.; Menéndez, J.C. Proline and its Derivatives as Organocatalysts for Multi- Component Reactions in Aqueous Media: Synergic Pathways to the Green Synthesis of Heterocycles. Adv. Synth. Catal. 2020, 362, 87–110. [Google Scholar] [CrossRef]
- Vega-Peñaloza, A.; Paria, S.; Bonchio, M.; Dell’Amico, L.; Companyó, X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 9, 6058–6072. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Yan, R.-J.; Xiao, B.-X.; Du, W.; Albrecht, Ł.; Chen, Y.-C. Asymmetric Formal Vinylogous Iminium Ion Activation for Vinyl-Substituted Heteroaryl and Aryl Aldehydes. Org. Lett. 2019, 21, 9628–9632. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, B.M. Proline-Catalyzed Asymmetric α-Amination in the Synthesis of Bioactive Molecules. Synlett 2018, 29, 1944–1956. [Google Scholar] [CrossRef]
- Klier, L.; Tur, F.; Poulsen, P.H.; Jørgensen, K.A. Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers. Chem. Soc. Rev. 2017, 46, 1080–1102. [Google Scholar] [CrossRef]
- Donslund, B.S.; Johansen, T.K.; Poulsen, P.H.; Halskov, K.S.; Jørgensen, K.A. The Diarylprolinol Silyl Ethers: Ten Years After. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Chandler, C.; Stadler, M.; Kampen, D.; List, B. Proline-catalysed Mannich reactions of acetaldehyde. Nature 2008, 452, 453–455. [Google Scholar] [CrossRef] [PubMed]
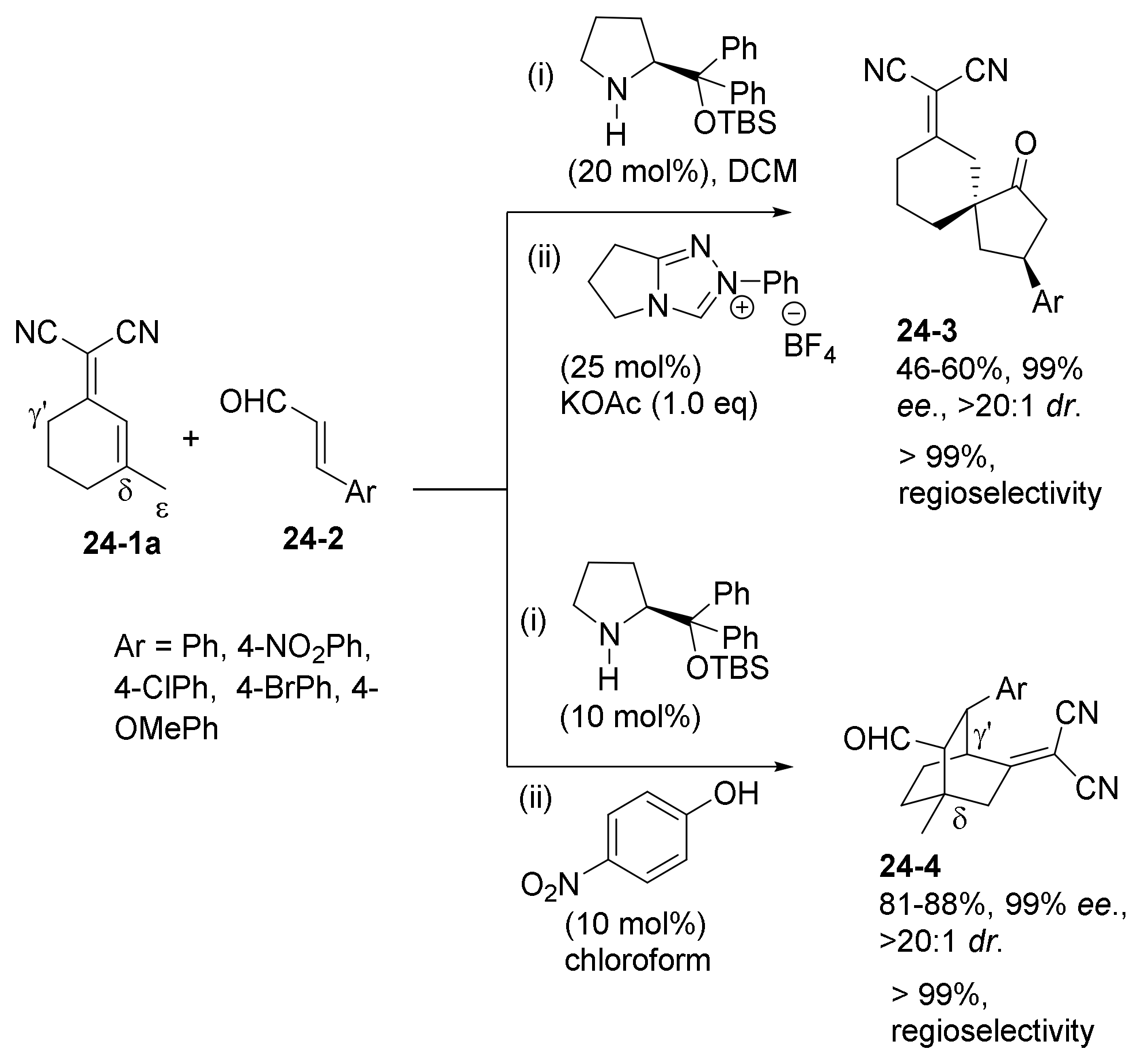
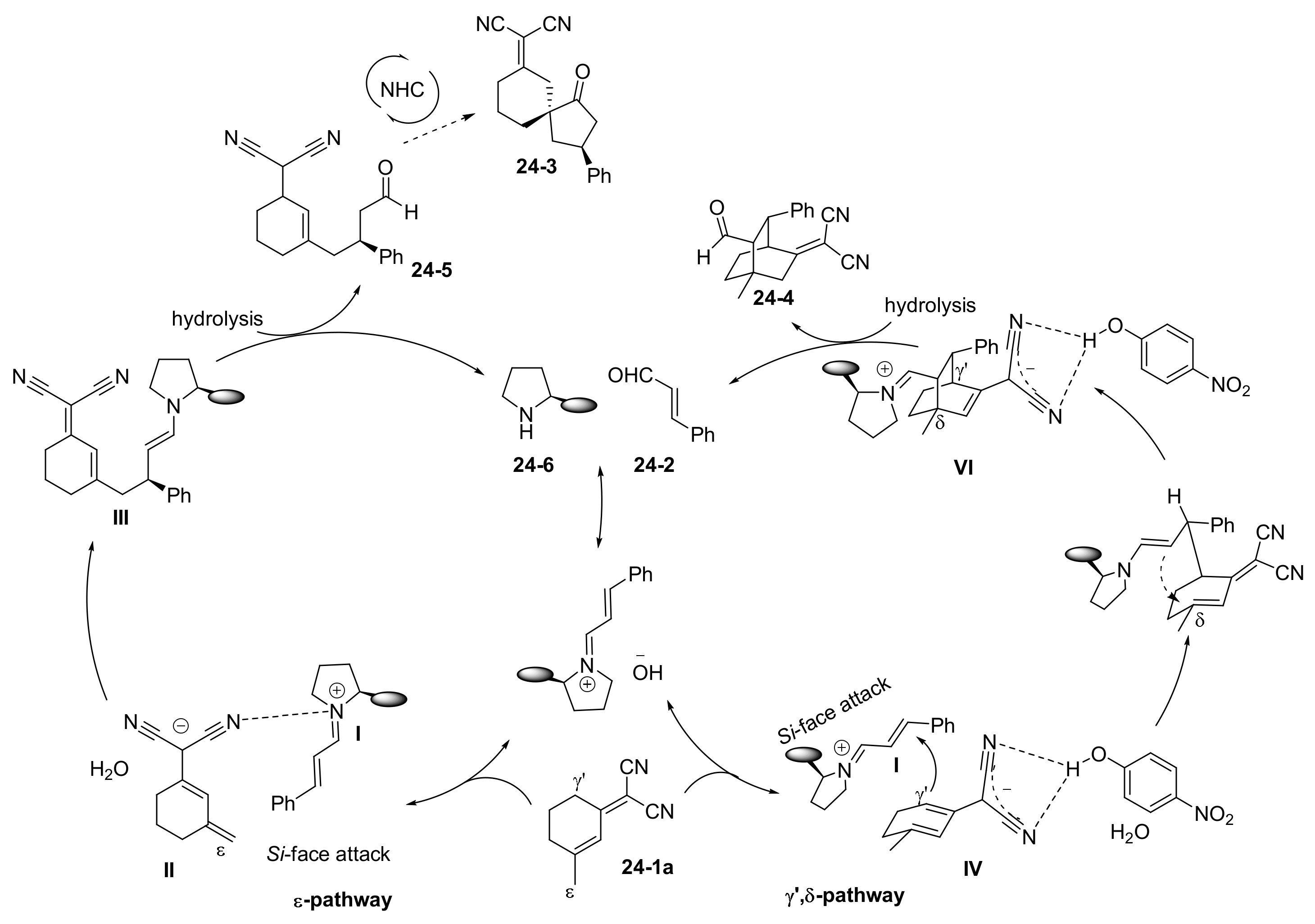
- Zhou, Z.; Wang, Z.-X.; Zhou, Y.-C.; Xiao, W.; Ouyang, Q.; Du, W.; Chen, Y.-C. Switchable regioselectivity in amine-catalysed asymmetric cycloadditions. Nat. Chem. 2017, 9, 590–594. [Google Scholar] [CrossRef] [PubMed]
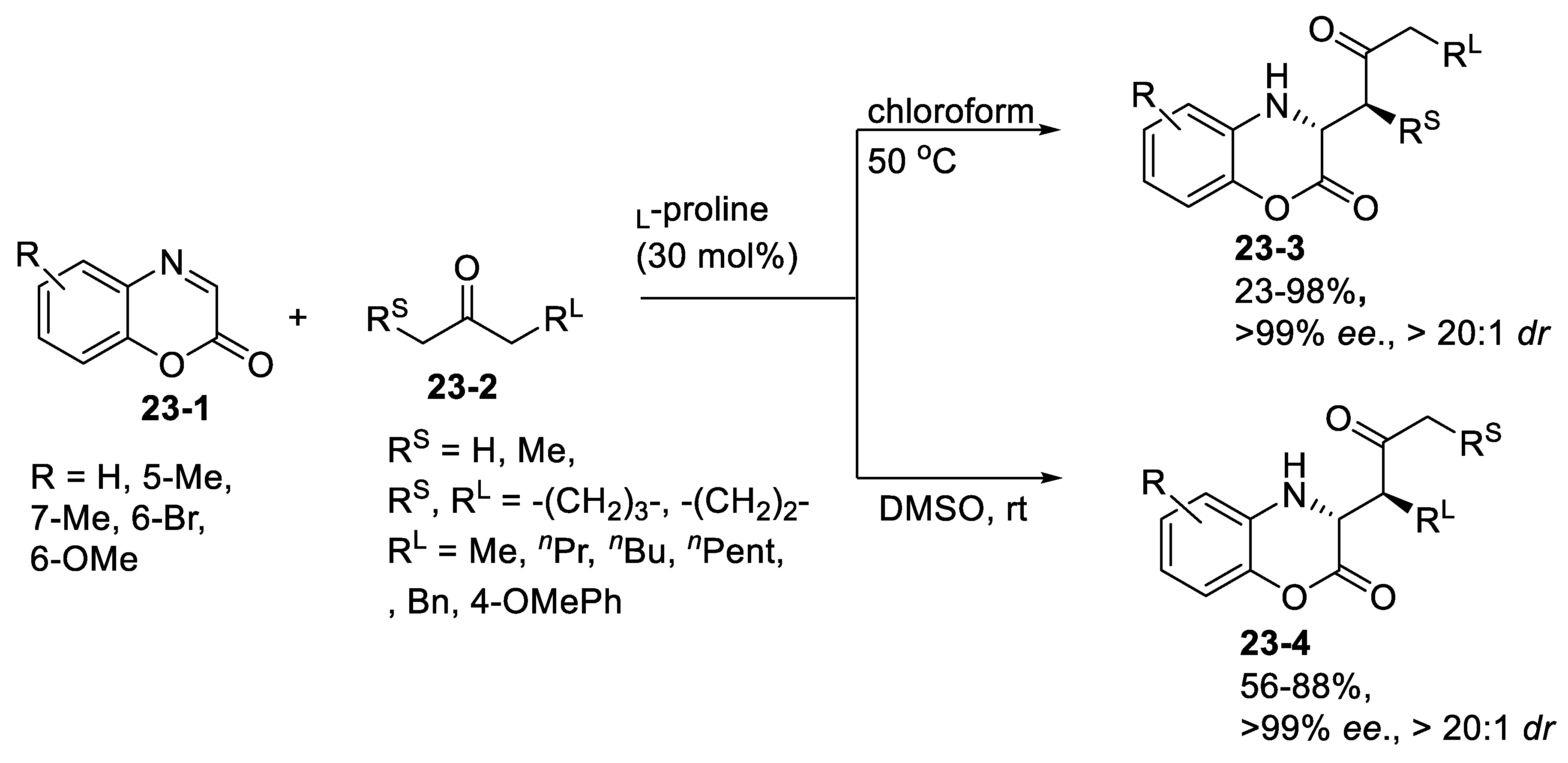
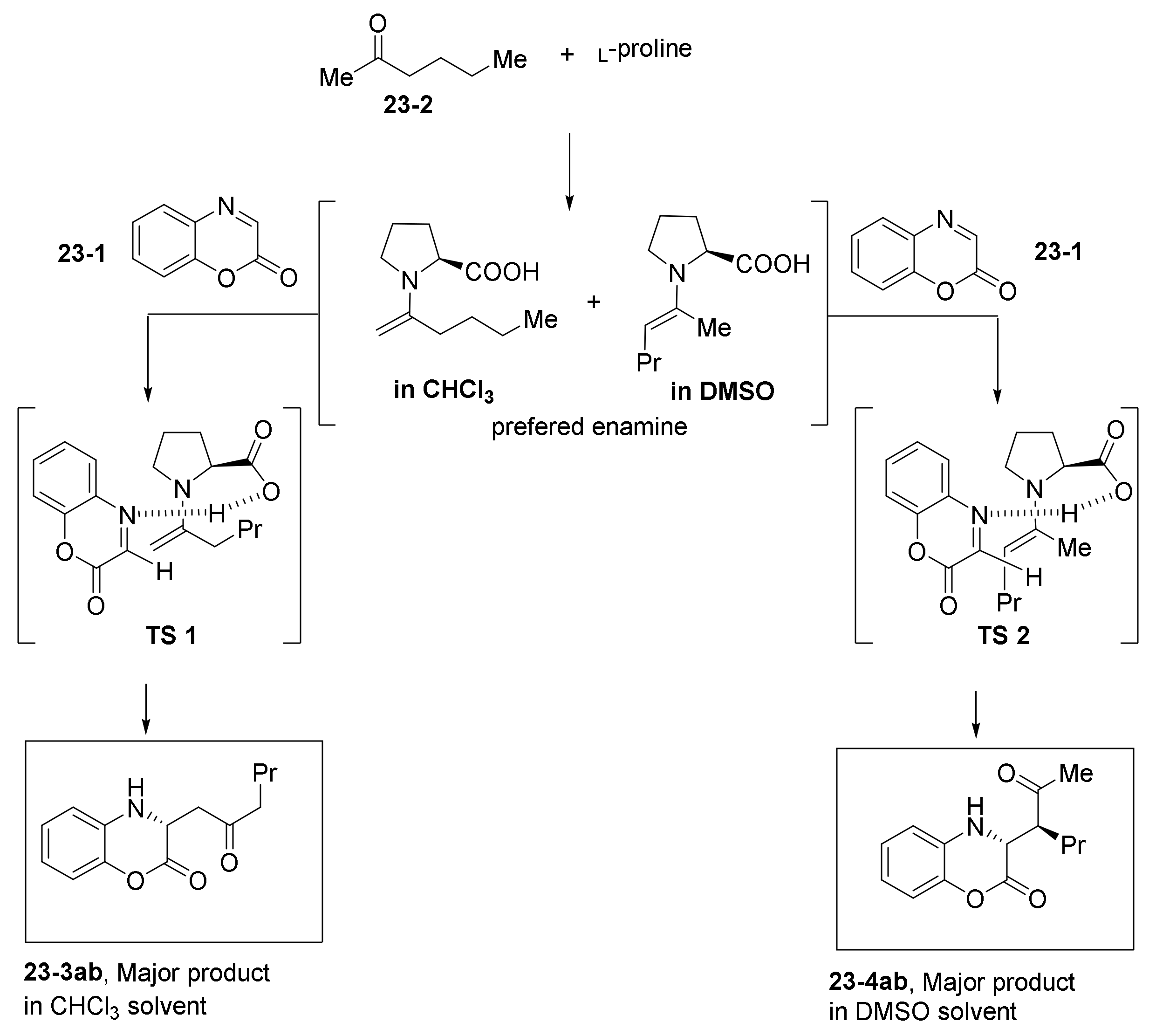
- Viji, M.; Sim, J.; Li, S.; Lee, H.; Oh, K.; Jung, J.-K. Organocatalytic and Regiodivergent Mannich Reaction of Ketones with Benzoxazinones. Adv. Synth. Catal. 2018, 360, 4464–4469. [Google Scholar] [CrossRef]
- Dell’Amico, L.; Rassu, G.; Zambrano, V.; Sartori, A.; Curti, C.; Battistini, L.; Pelosi, G.; Casiraghi, G.; Zanardi, F. Exploring the Vinylogous Reactivity of Cyclohexenylidene Malononitriles: Switchable Regioselectivity in the Organocatalytic Asymmetric Addition to Enals Giving Highly Enantioenriched Carbabicyclic Structures. J. Am. Chem. Soc. 2014, 136, 11107–11114. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, M.; Huang, M.; Liu, H.; Yan, Y.; Zhang, X. Enantioselective [3+2] annulation of 3-hydroxymaleimides with quinone monoimines. Org. Chem. Front. 2021, 8, 2268–2273. [Google Scholar] [CrossRef]
- Varlet, T.; Masson, G. Enamides and dienamides in phosphoric acid-catalysed enantioselective cycloadditions for the synthesis of chiral amines. Chem. Commun. 2021, 57, 4089–4105. [Google Scholar] [CrossRef]
- Liu, H.; Yan, Y.; Li, M.; Zhang, X. An enantioselective aza-Friedel–Crafts reaction of 5-aminoisoxazoles with isatin-derived N-Boc ketimines. Org. Biomol. Chem. 2021, 19, 3820–3824. [Google Scholar] [CrossRef]
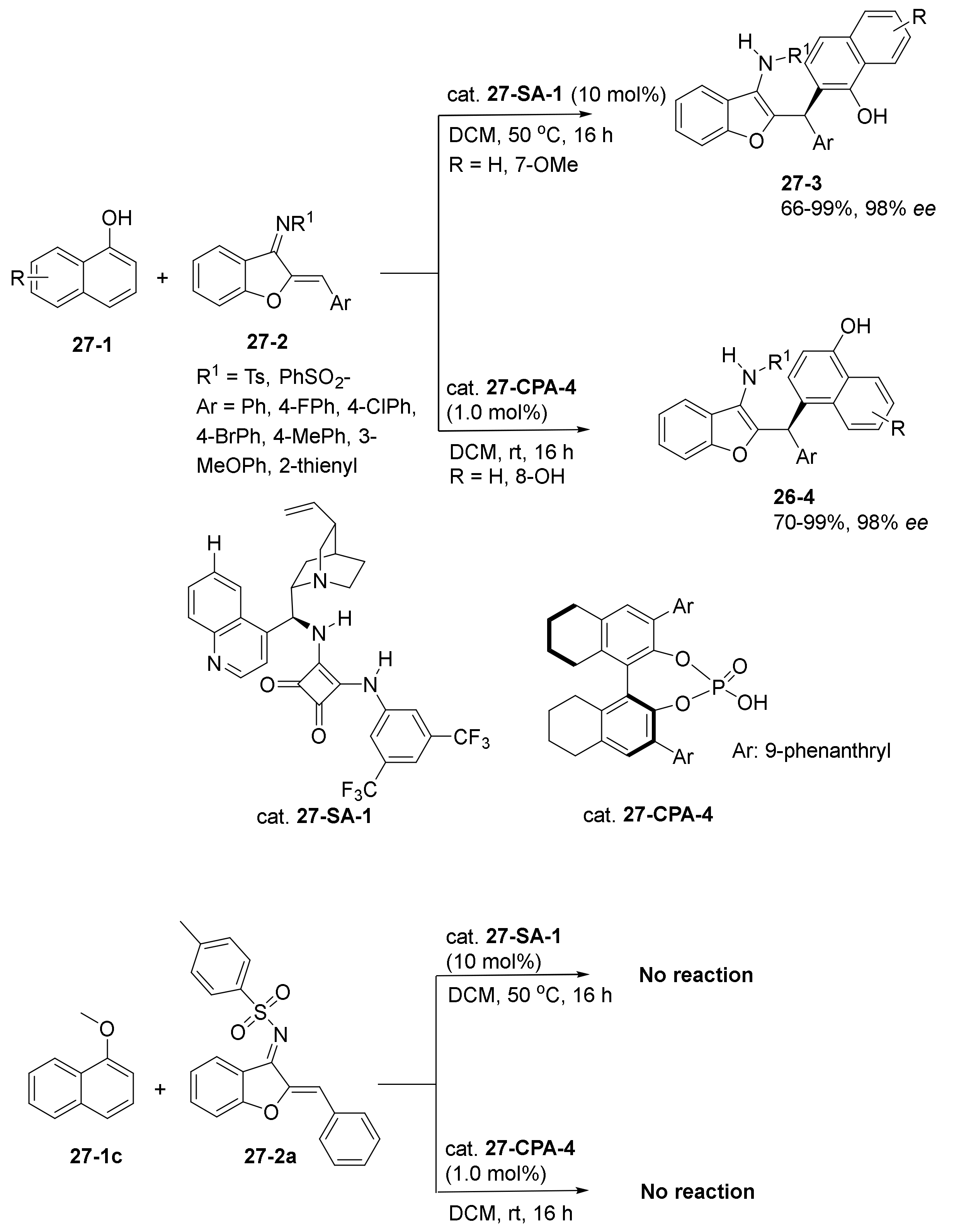
- Koay, W.L.; Mei, G.-J.; Lu, Y. Facile access to benzofuran-fused tetrahydropyridines via catalytic asymmetric [4 + 2] cycloaddition of aurone-derived 1-azadienes with 3-vinylindoles. Org. Chem. Front. 2021, 8, 968–974. [Google Scholar] [CrossRef]
- Gao, Y.-Q.; Hou, Y.; Chen, J.; Zhen, Y.; Xu, D.; Zhang, H.; Wei, H.; Xie, W. Asymmetric synthesis of 9-alkyl tetrahydroxanthenones via tandem asymmetric Michael/cyclization promoted by chiral phosphoric acid. Org. Biomol. Chem. 2021, 19, 348–354. [Google Scholar] [CrossRef]
- Wu, S.-F.; Tu, M.-S.; Hang, Q.-Q.; Zhang, S.; Ding, H.; Zhang, Y.-C.; Shi, F. Construction of chiral chroman scaffolds via catalytic asymmetric (4 + 2) cyclizations of para-quinone methide derivatives with 3-vinylindoles. Org. Biomol. Chem. 2020, 18, 5388–5399. [Google Scholar] [CrossRef]
- Varlet, T.; Gelis, C.; Retailleau, P.; Bernadat, G.; Neuville, L.; Masson, G. Enantioselective Redox-Divergent Chiral Phosphoric Acid Catalyzed Quinone Diels–Alder Reactions. Angew. Chem. Int. Ed. 2020, 59, 8491–8496. [Google Scholar] [CrossRef]
- Liu, H.; Yan, Y.; Zhang, J.; Liu, M.; Cheng, S.; Wang, Z.; Zhang, X. Enantioselective dearomative [3+2] annulation of 5-amino-isoxazoles with quinone monoimines. Chem. Commun. 2020, 56, 13591–13594. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, C.; Cheng, Y.; Jia, Q.; Li, W.; Liu, K.; Li, P. Enantioselective Construction of Vicinal Sulfur-functionalized Quaternary and Tertiary Stereocenters via Organocatalytic Michael Addition of 5H-Thiazol-4-ones to 1-Azadienes. Asian J. Org. Chem. 2020, 9, 1183–1186. [Google Scholar] [CrossRef]
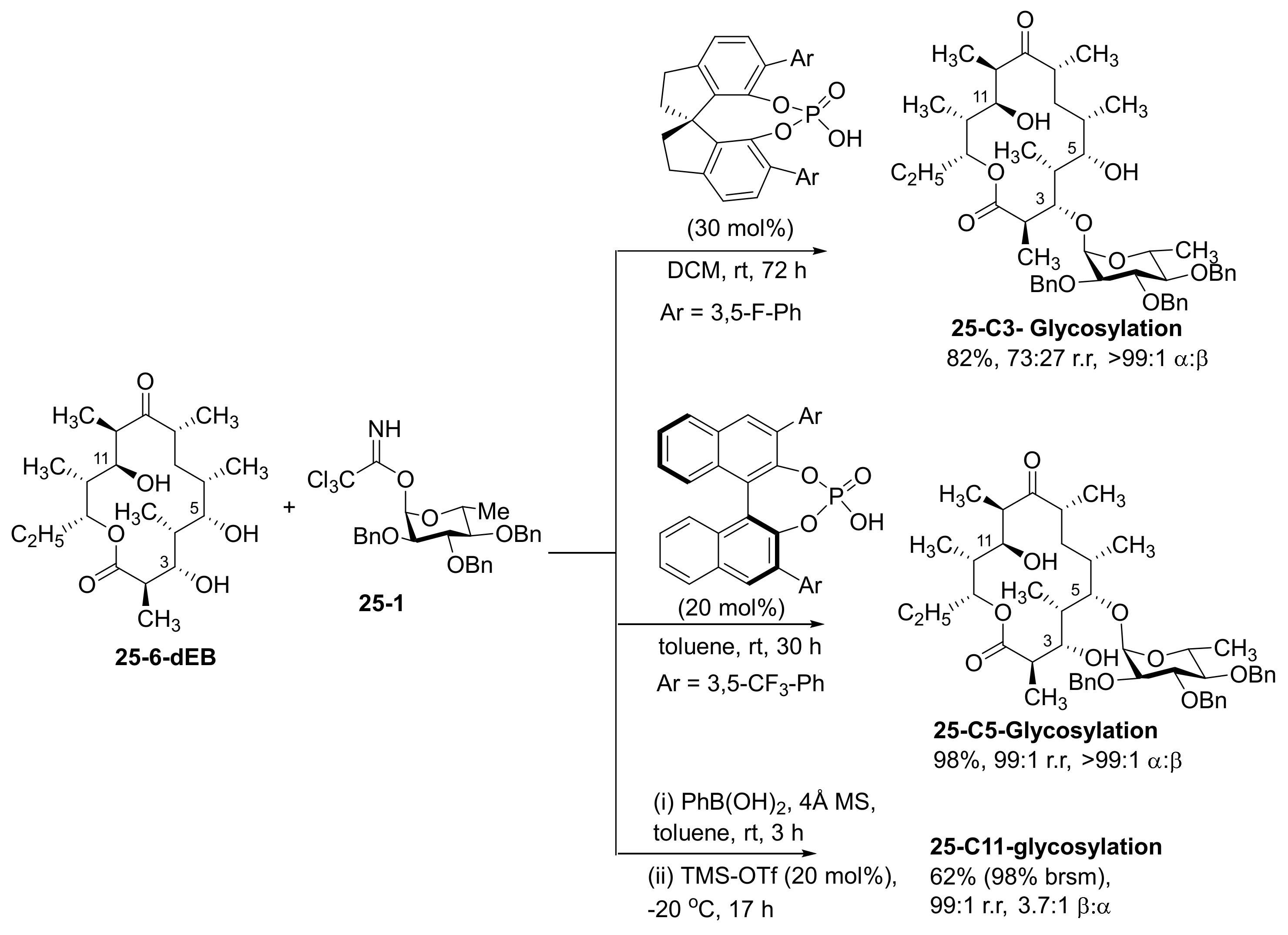
- Tay, J.-H.; Argüelles, A.J.; DeMars, M.D.; Zimmerman, P.M.; Sherman, D.H.; Nagorny, P. Regiodivergent Glycosylations of 6-Deoxy-erythronolide B and Oleandomycin-Derived Macrolactones Enabled by Chiral Acid Catalysis. J. Am. Chem. Soc. 2017, 139, 8570–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Sun, Z.; Fernando, E.H.N.; Nesterov, V.N.; Cundari, T.R.; Wang, H. Formal oxo- and aza-[3+2] reactions of α-enaminones and quinones: A double divergent process and the roles of chiral phosphoric acid and molecular sieves. Chem. Sci. 2020, 11, 9386–9394. [Google Scholar] [CrossRef]
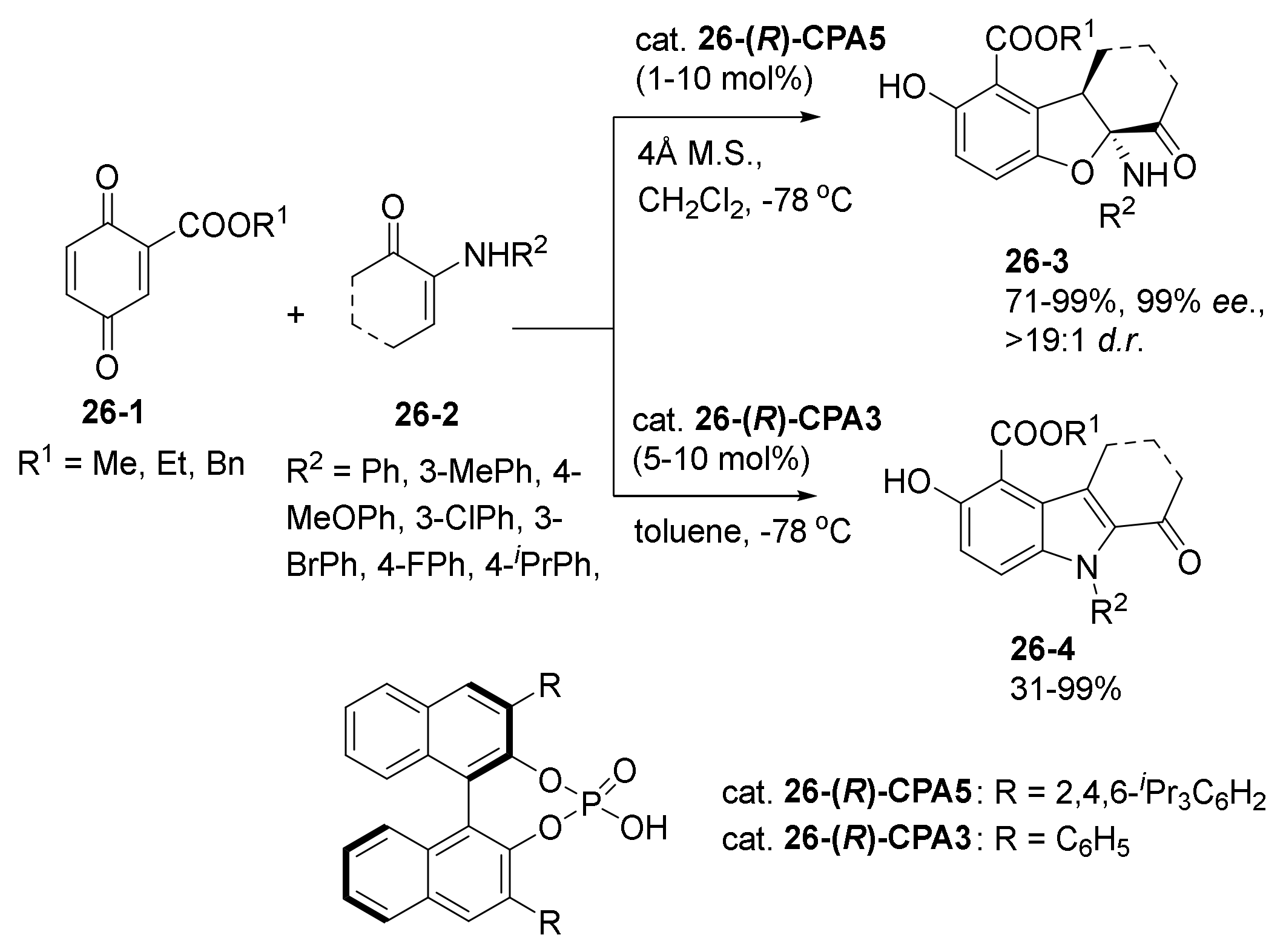
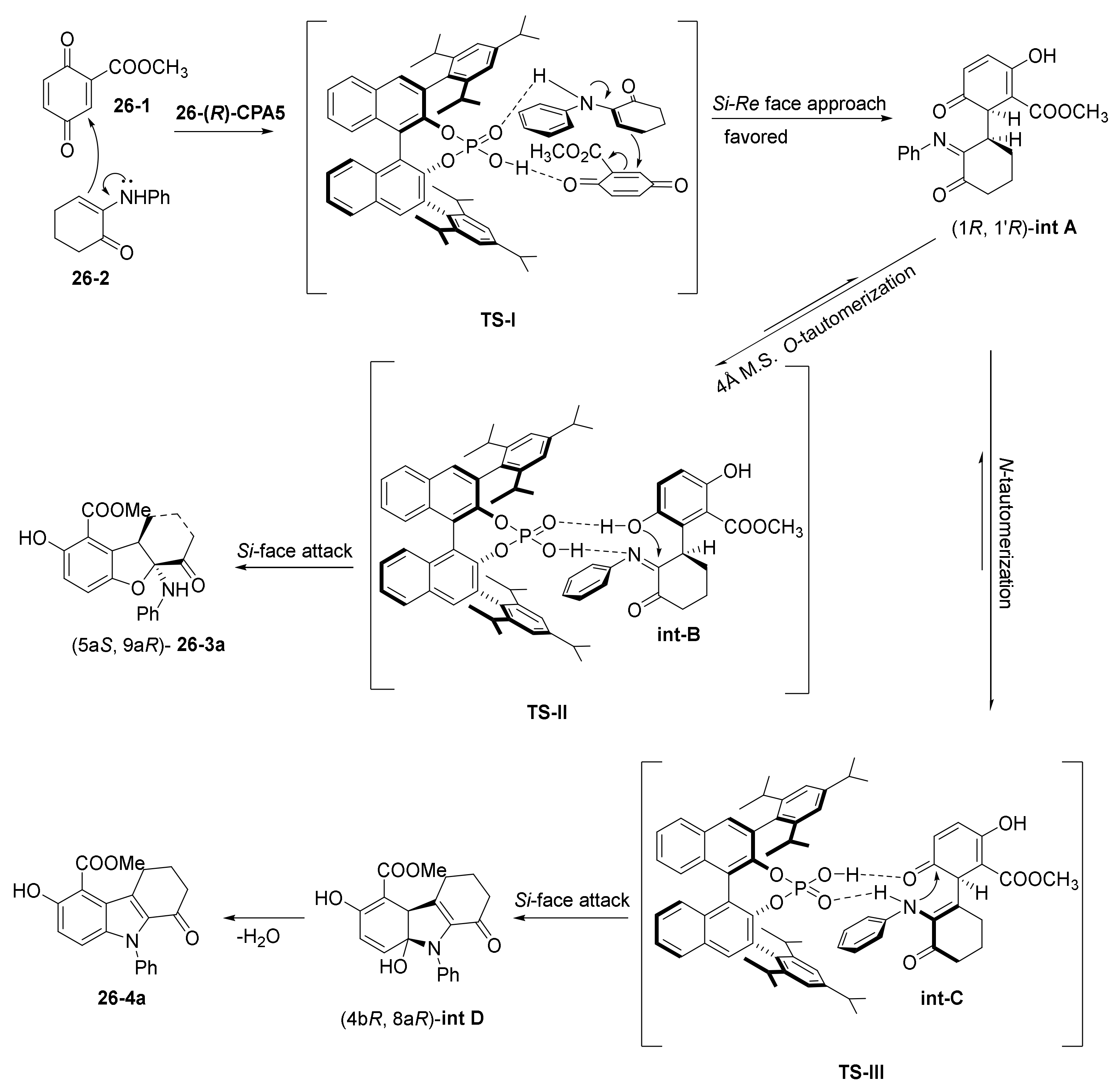
- Zhang, C.; Cheng, Y.; Li, F.; Luan, Y.; Li, P.; Li, W. Organocatalytic Enantioselective Regiodivergent C−H Bond Functionalization of 1-Naphthols with 1-Azadienes. Adv. Synth. Catal. 2020, 362, 1286–1291. [Google Scholar] [CrossRef]
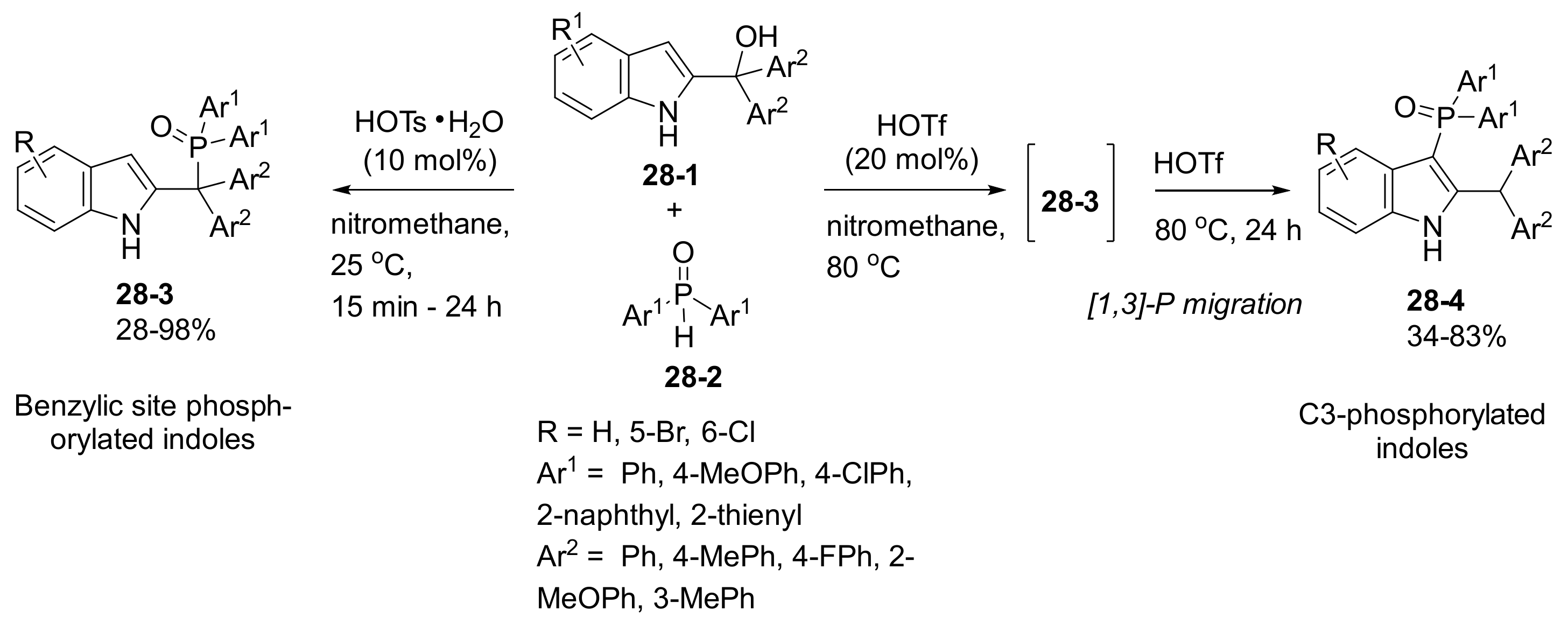
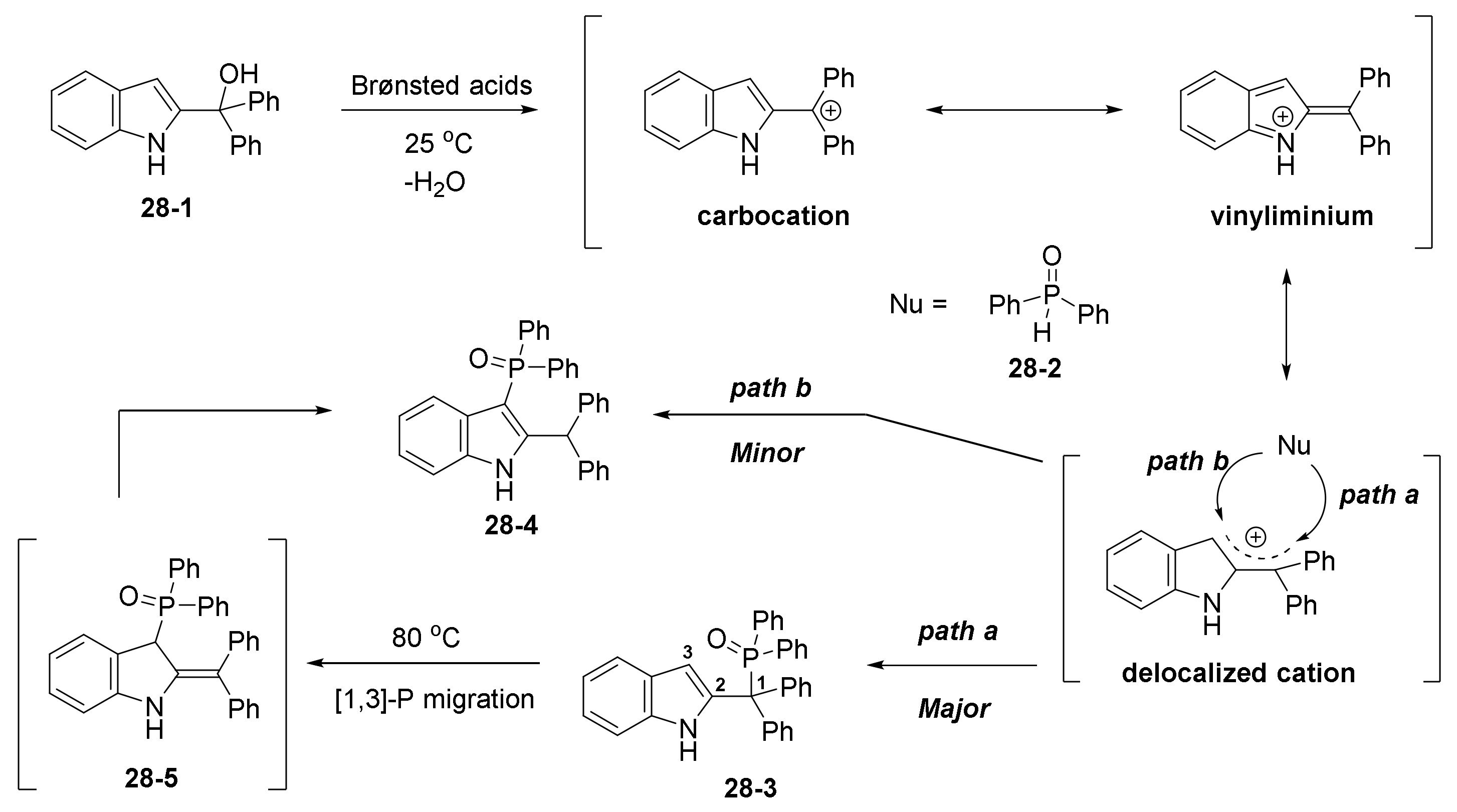
- Chen, L.; Zou, Y.-X.; Fang, X.-Y.; Wu, J.; Sun, X.-H. Brønsted acid-catalysed regiodivergent phosphorylation of 2-indolylmethanols to synthesize benzylic site or C3-phosphorylated indole derivatives. Org. Biomol. Chem. 2018, 16, 7417–7424. [Google Scholar] [CrossRef]
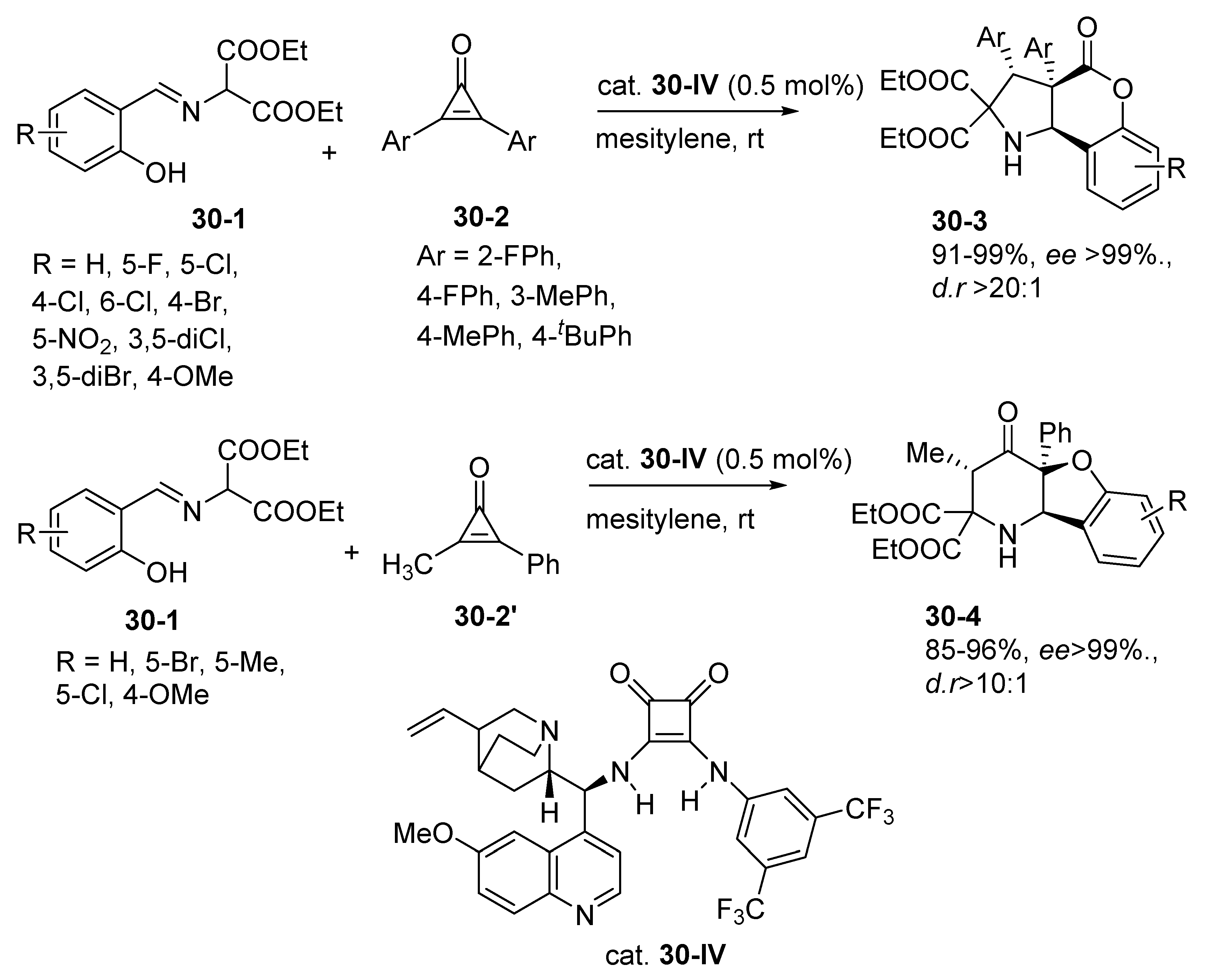
- Marcantonio, E.; Curti, C.; Battistini, L.; Sartori, A.; Cardinale, L.; Pelosi, G.; Zanardi, F. Direct, Asymmetric Synthesis of Carbocycle-Fused Uracils via [4+2] Cycloadditions: A Noncovalent Organocatalysis Approach. Adv. Synth. Catal. 2021, 363, 2625–2633. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, T.; Sun, Y.; Wang, L.; Jin, Y. Organocatalytic enantioselective aza-Friedel–Crafts alkylation of β-naphthols and isatin-derived ketimines via a Takemoto-type catalyst. New J. Chem. 2021, 45, 10481–10487. [Google Scholar] [CrossRef]
- Sun, B.-B.; Chen, J.-B.; Zhang, J.-Q.; Yang, X.-P.; Lv, H.-P.; Wang, Z.; Wang, X.-W. Organo-catalyzed asymmetric cascade annulation reaction for the construction of bi-spirocyclic pyrazolone and oxindole derivatives. Org. Chem. Front. 2020, 7, 796–809. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, M.; Maestro, A.; Andrés, J.M.; Pedrosa, R. Supported Bifunctional Chiral Thioureas as Catalysts in the Synthesis of 3-Amino-2-Oxindoles through Enantioselective aza-Friedel-Crafts Reaction: Application in Continuous Flow Processes. Adv. Synth. Catal. 2020, 362, 2744–2754. [Google Scholar] [CrossRef]
- Da Silva, T.L.; Rambo, R.S.; Jacoby, C.G.; Schneider, P.H. Asymmetric Michael reaction promoted by chiral thiazolidine-thiourea catalyst. Tetrahedron 2020, 76, 130874. [Google Scholar] [CrossRef]
- Yang, X.; Zhou, Y.-H.; Yang, H.; Wang, S.-S.; Ouyang, Q.; Luo, Q.-L.; Guo, Q.-X. Asymmetric Diels–Alder Reaction of 3-Vinylindoles and Nitroolefins Promoted by Multiple Hydrogen Bonds. Org. Lett. 2019, 21, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ferrer, P.; Sanz-Novo, M.; Maestro, A.; Andrés, J.M.; Pedrosa, R. Synthesis of Enantioenriched 3-Amino-3-Substituted Oxindoles by Stereoselective Mannich Reaction Catalyzed by Supported Bifunctional Thioureas. Adv. Synth. Catal. 2019, 361, 3645–3655. [Google Scholar] [CrossRef]
- Andrés, J.M.; Maestro, A.; Valle, M.; Pedrosa, R. Chiral Bifunctional Thioureas and Squaramides and Their Copolymers as Recoverable Organocatalysts. Stereoselective Synthesis of 2-Substituted 4-Amino-3-nitrobenzopyrans and 3-Functionalized 3,4-Diamino-4H-Chromenes. J. Org. Chem. 2018, 83, 5546–5557. [Google Scholar] [CrossRef] [Green Version]
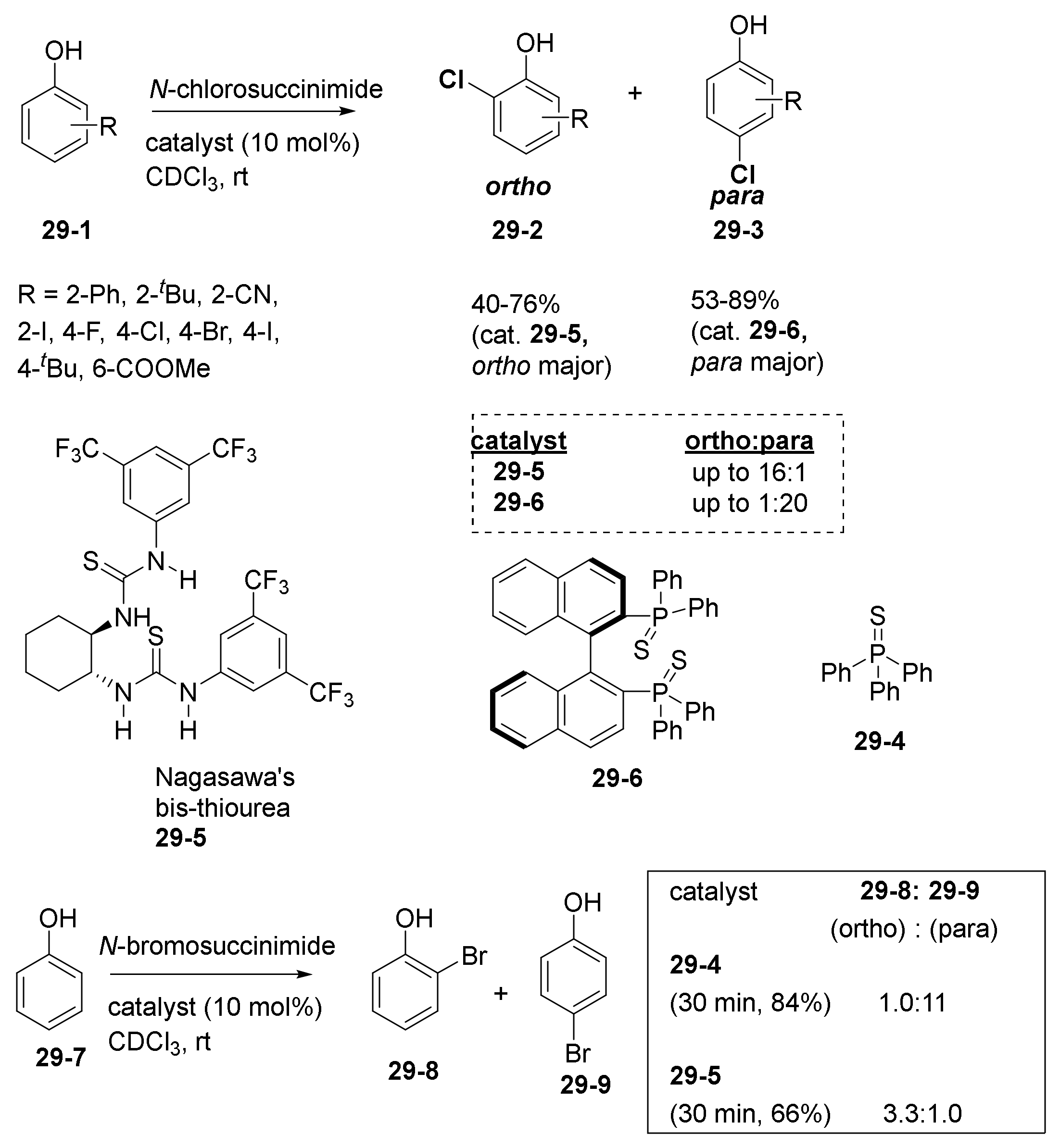
- Maddox, S.M.; Dinh, A.N.; Armenta, F.; Um, J.; Gustafson, J.L. The Catalyst-Controlled Regiodivergent Chlorination of Phenols. Org. Lett. 2016, 18, 5476–5479. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Lei, C.-W.; Zhao, J.-Q.; Wang, Z.-H.; You, Y. Organocatalytic Asymmetric Cyclopropanation of 3-Acylcoumarins with 3-Halooxindoles: Access to Spirooxindole-cyclopropa[c]coumarin Compounds. J. Org. Chem. 2021, 86, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Huang, Q.-W.; Gou, C.; Li, Q.-Z.; Dai, Q.-S.; Leng, H.-J.; Li, J.-L. Organocatalytic Enantioselective Synthesis of Tetrahydro-Furanyl Spirooxindoles via [3+2] Annulations of 3-Hydroxyoxindoles and Cyclic Ketolactams. Adv. Synth. Catal. 2021, 363, 2177–2182. [Google Scholar] [CrossRef]
- Biswas, A.; Ghosh, A.; Shankhdhar, R.; Chatterjee, I. Squaramide Catalyzed Asymmetric Synthesis of Five- and Six-Membered Rings. Asian J. Org. Chem. 2021, 10, 1345–1376. [Google Scholar] [CrossRef]
- Xu, K.; Chen, W.; Chen, X.; Wang, B.; Huang, J.; Tian, X. Organocatalytic asymmetric Friedel–Crafts alkylation/hemiketalization/lactonization cascade reactions: Highly enantioselective synthesis of furo[2,3-[b]]benzofuranones. Org. Chem. Front. 2020, 7, 1679–1684. [Google Scholar] [CrossRef]
- Wen, J.-B.; Du, D.-M. Squaramide-catalysed asymmetric cascade reactions of 2,3-dioxopyrrolidines with 3-chlorooxindoles. Org. Biomol. Chem. 2020, 18, 1647–1656. [Google Scholar] [CrossRef]
- Wang, Y.; Du, D.-M. Highly Diastereo- and Enantioselective Synthesis of Isoxazolone-Spirooxindoles via Squaramide-Catalyzed Cascade Michael/Michael Addition Reactions. J. Org. Chem. 2020, 85, 15325–15336. [Google Scholar] [CrossRef]
- Hou, X.-Q.; Du, D.-M. Recent Advances in Squaramide-Catalyzed Asymmetric Mannich Reactions. Adv. Synth. Catal. 2020, 362, 4487–4512. [Google Scholar] [CrossRef]
- Zhao, X.; Xiong, J.; An, J.; Yu, J.; Zhu, L.; Feng, X.; Jiang, X. Diastereodivergent construction of bispiro[oxindole-bi-pyrrolidine]s with four consecutive stereocenters via asymmetric [3+2] cycloaddition of 2,3-dioxopyrrolidines using identical catalysts. Org. Chem. Front. 2019, 6, 1989–1995. [Google Scholar] [CrossRef]
- Zhao, B.-L.; Li, J.-H.; Du, D.-M. Squaramide-Catalyzed Asymmetric Reactions. Chem. Rec. 2017, 17, 994–1018. [Google Scholar] [CrossRef] [PubMed]
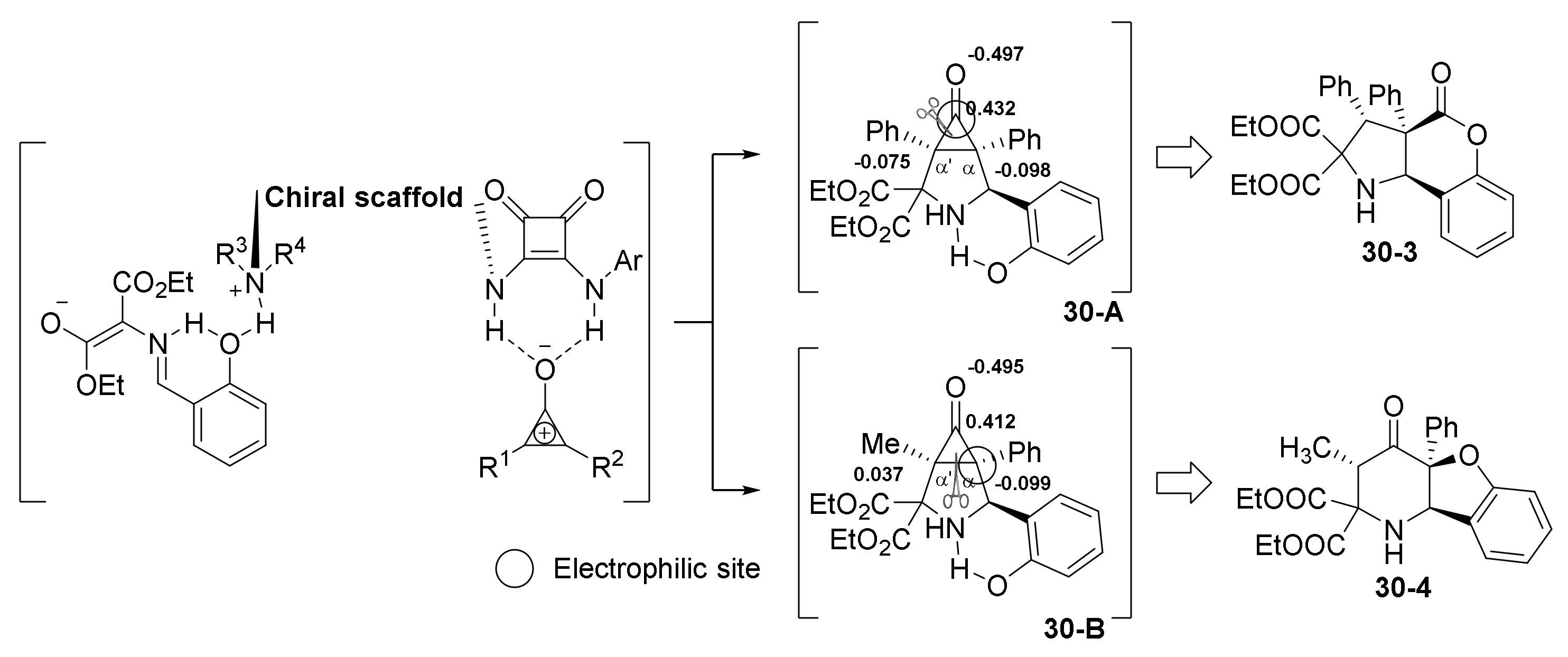
- Cao, J.; Fang, R.; Liu, J.-Y.; Lu, H.; Luo, Y.-C.; Xu, P.-F. Organocatalytic Regiodivergent C−C Bond Cleavage of Cyclopropenones: A Highly Efficient Cascade Approach to Enantiopure Heterocyclic Frameworks. Chem. Eur. J. 2018, 24, 18863–18867. [Google Scholar] [CrossRef] [PubMed]

























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viji, M.; Lanka, S.; Sim, J.; Jung, C.; Lee, H.; Vishwanath, M.; Jung, J.-K. Regiodivergent Organocatalytic Reactions. Catalysts 2021, 11, 1013. https://doi.org/10.3390/catal11081013
Viji M, Lanka S, Sim J, Jung C, Lee H, Vishwanath M, Jung J-K. Regiodivergent Organocatalytic Reactions. Catalysts. 2021; 11(8):1013. https://doi.org/10.3390/catal11081013
Chicago/Turabian StyleViji, Mayavan, Srinu Lanka, Jaeuk Sim, Chanhyun Jung, Heesoon Lee, Manjunatha Vishwanath, and Jae-Kyung Jung. 2021. "Regiodivergent Organocatalytic Reactions" Catalysts 11, no. 8: 1013. https://doi.org/10.3390/catal11081013