FoxO3a as a Positive Prognostic Marker and a Therapeutic Target in Tamoxifen-Resistant Breast Cancer

 , and
, and 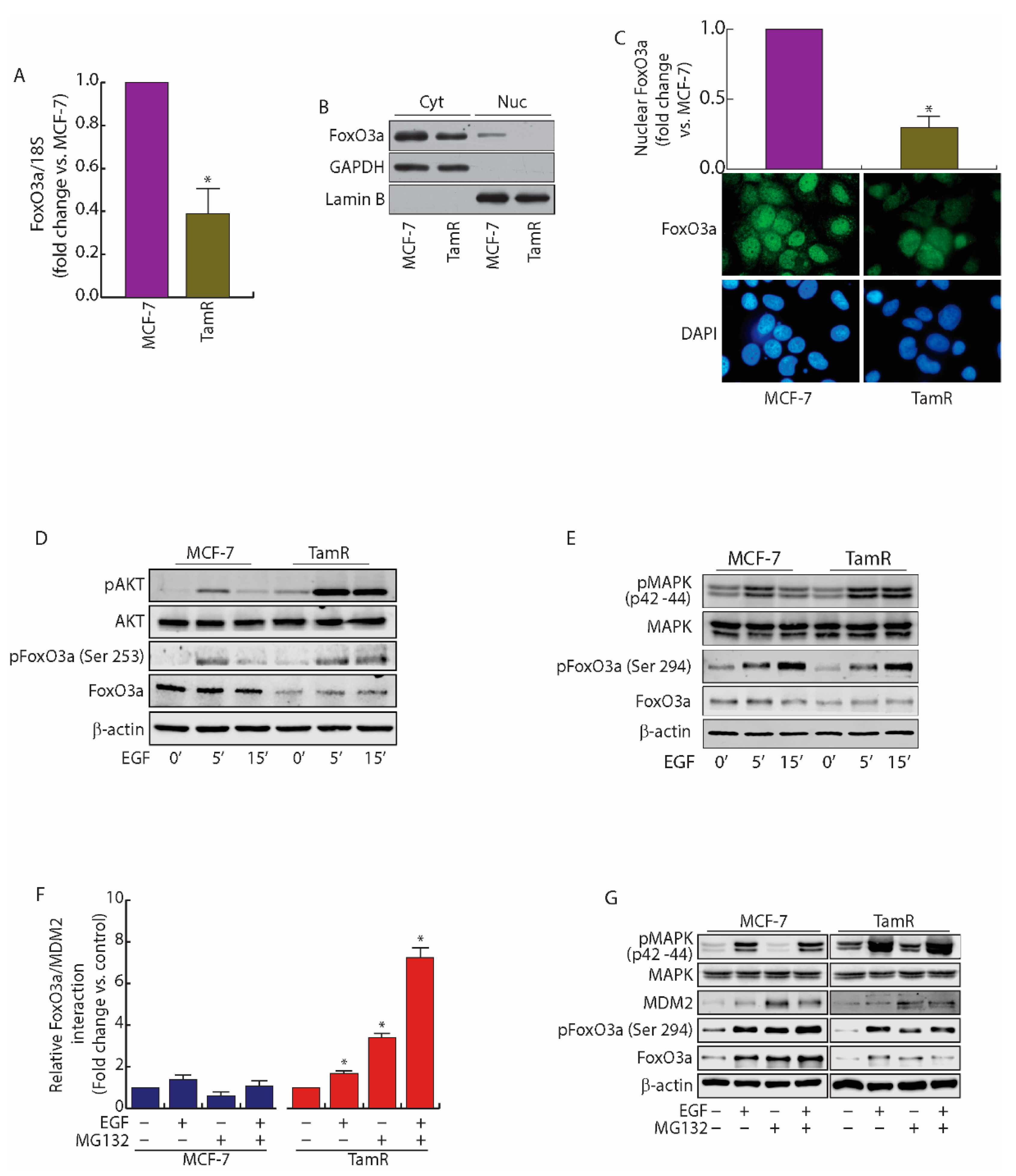{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. FoxO3a Is Downregulated in Tamoxifen Resistant (TamR) BCCs
2.2. MDM2 Mediates FoxO3a Protein Degradation in TamR Cells
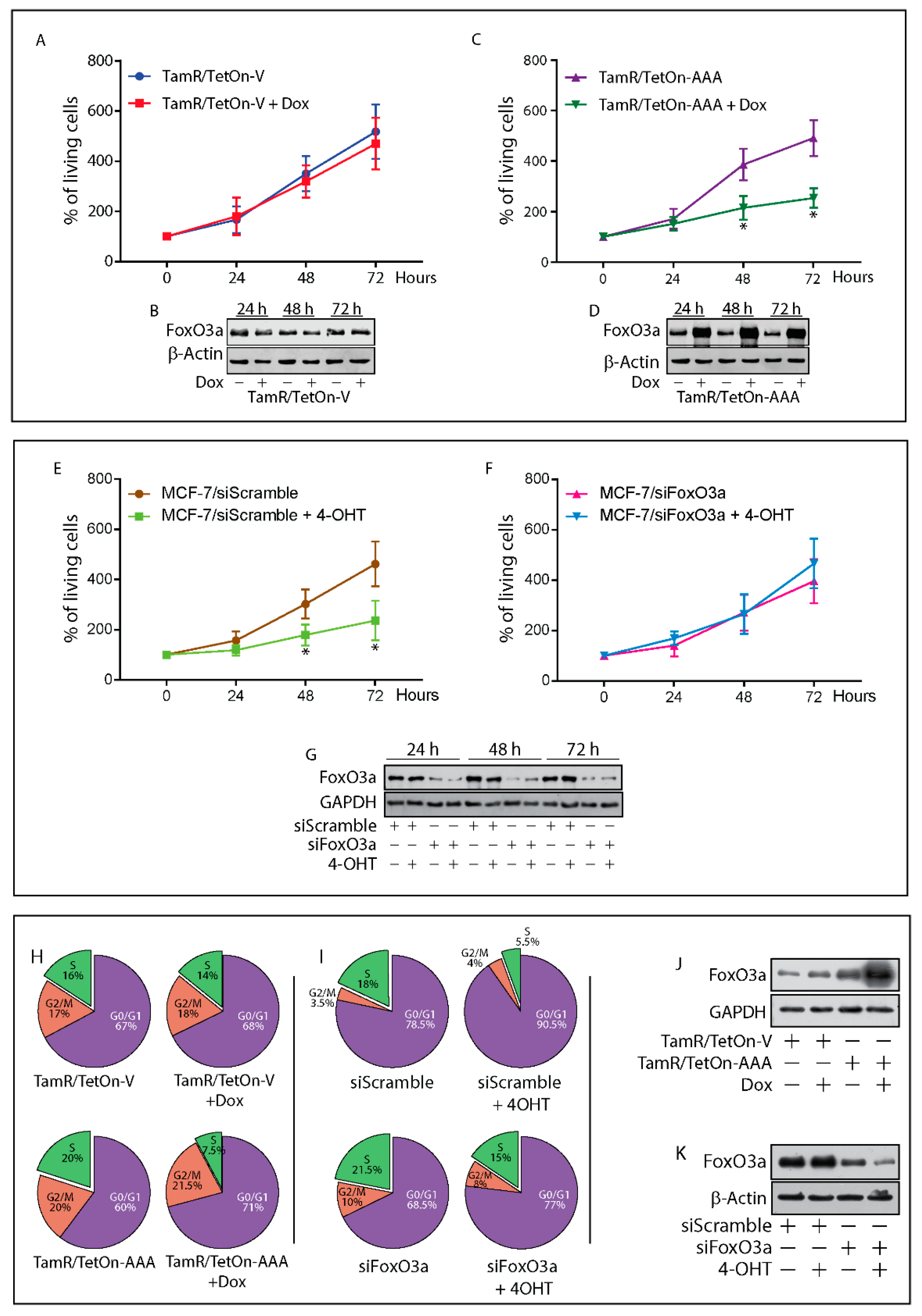
2.3. Re-Expression of Foxo3a in Tamr Cells Restores the Sensitivity to the Antiestrogen
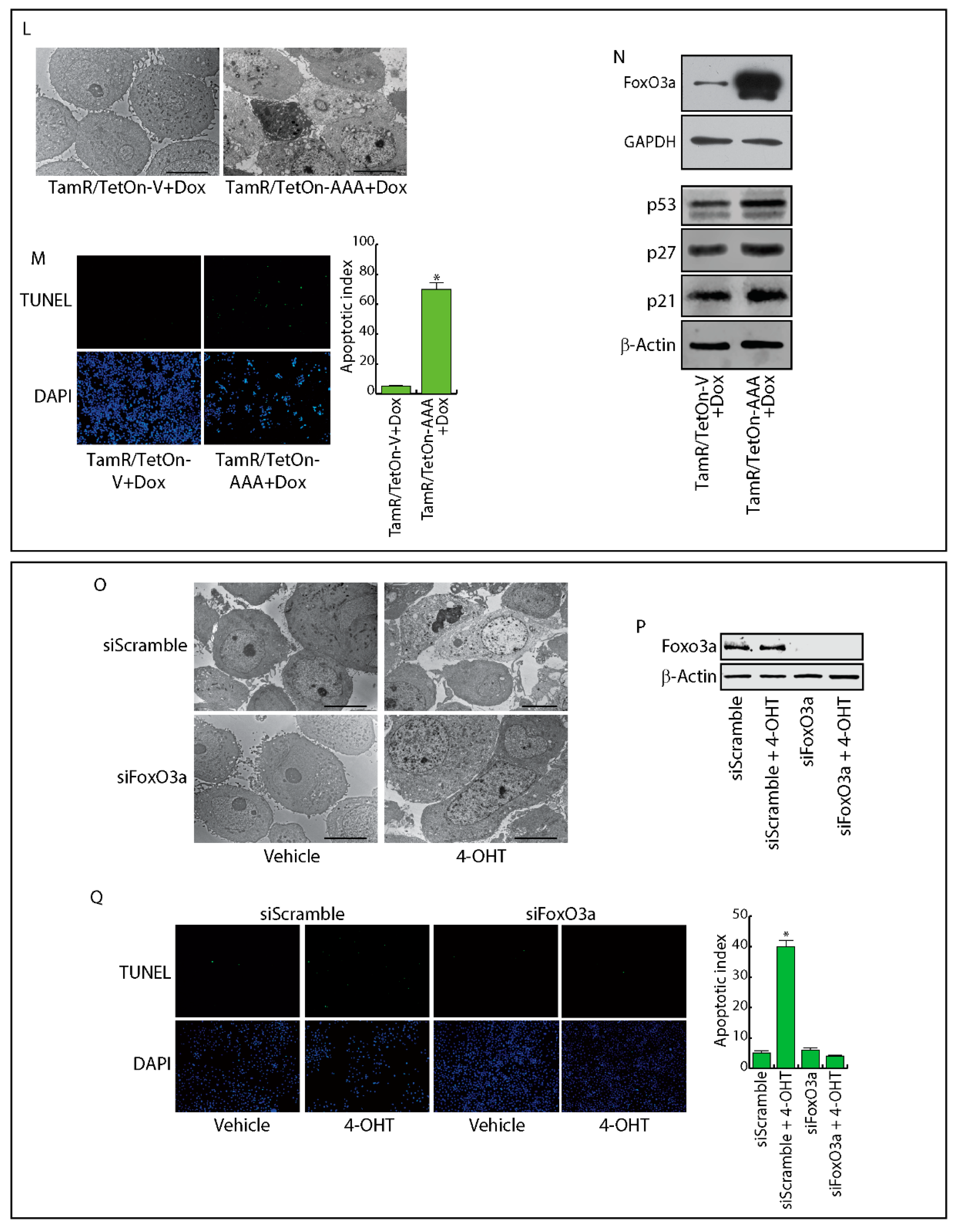
2.4. FoxO3a Restores the Apoptotic Response to Tamoxifen in TamR Cells
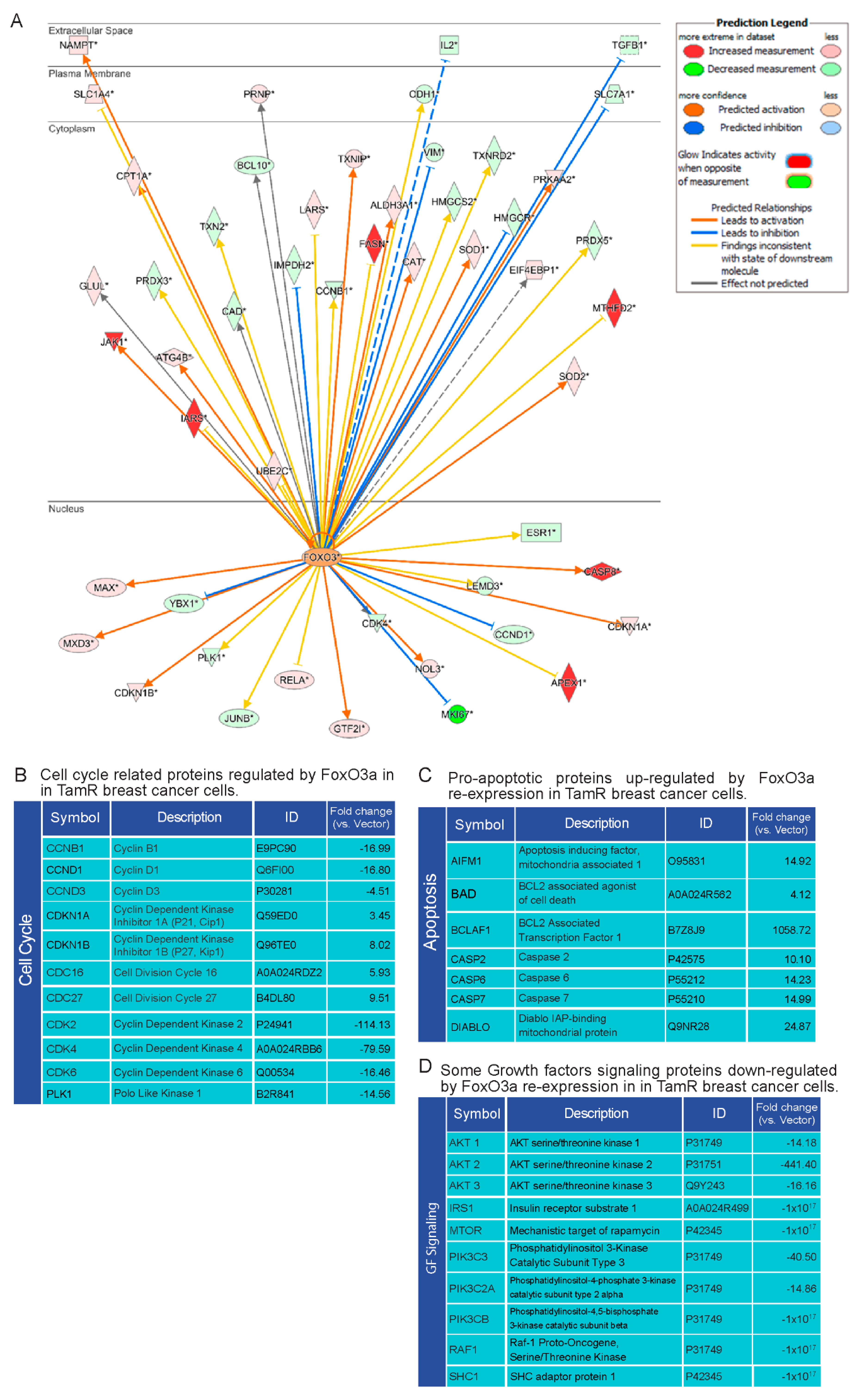
2.5. Proteomic Analysis of TamR Cells Expressing Active FoxO3a: Impact on Proteins Controlling G1/S Transition, Apoptosis and GF Signals
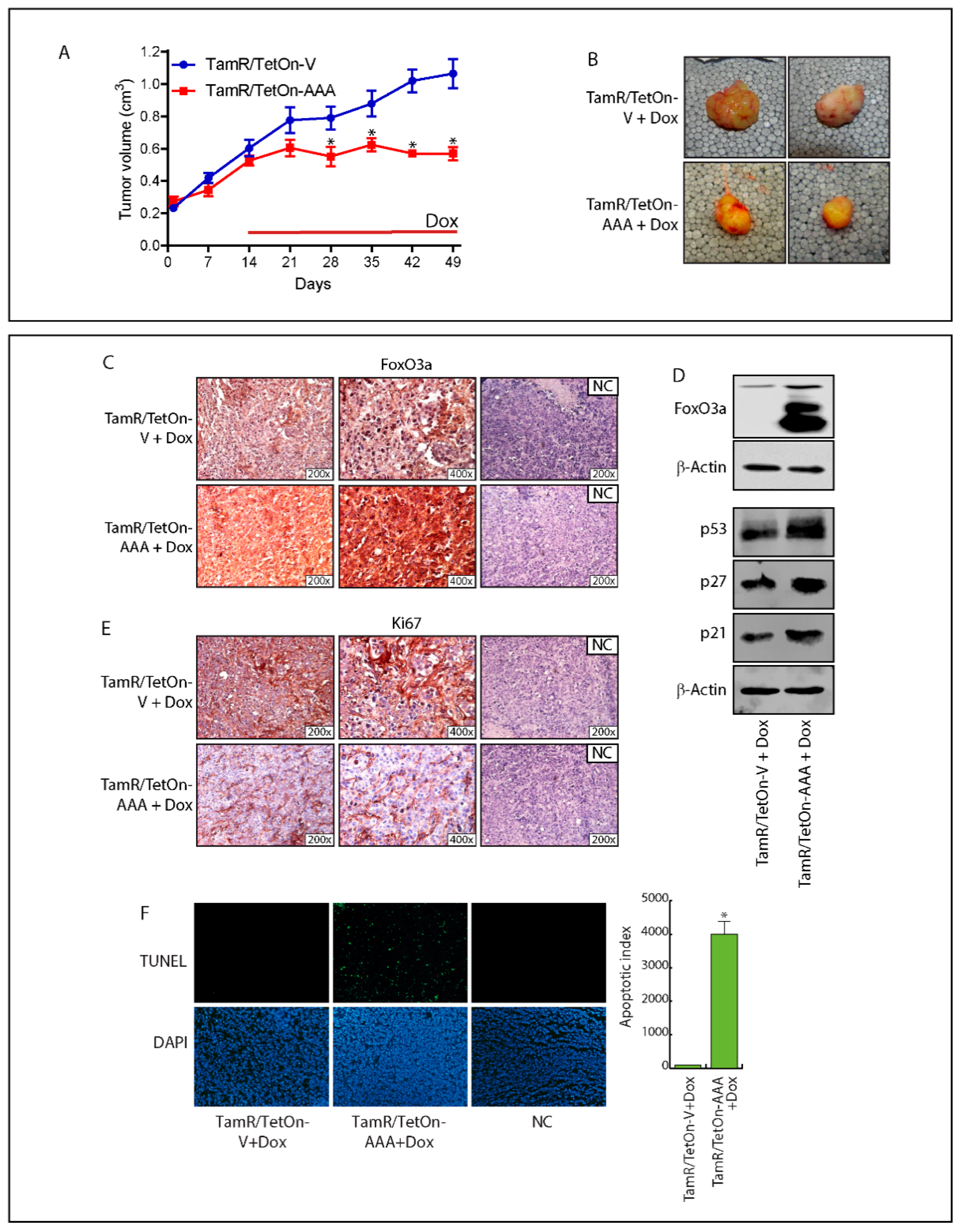
2.6. FoxO3a Over-Expression Inhibits TamR-Derived Tumor Growth
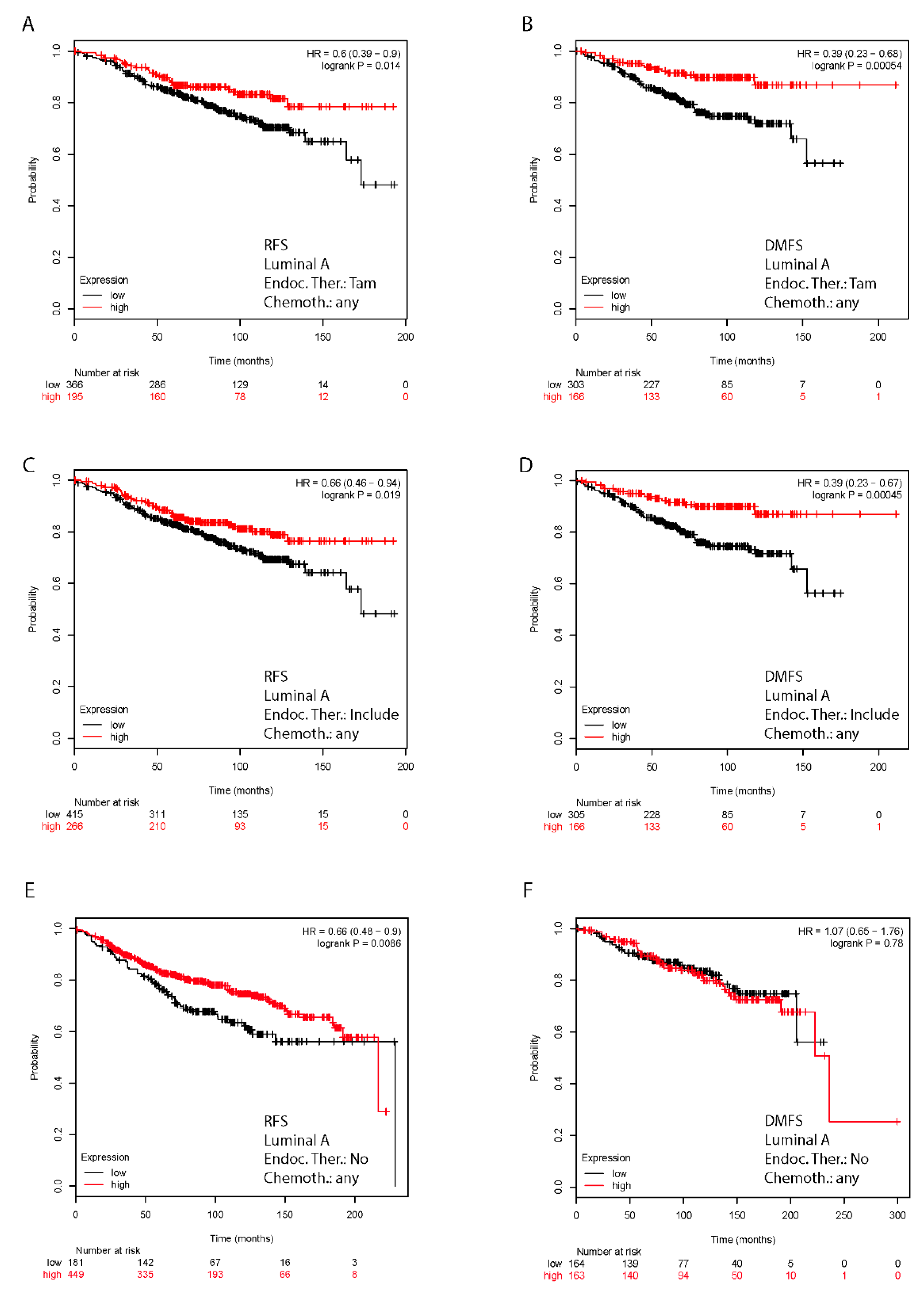
2.7. In Silico Validation of the Clinical Relevance of FoxO3a in Human BC Patients
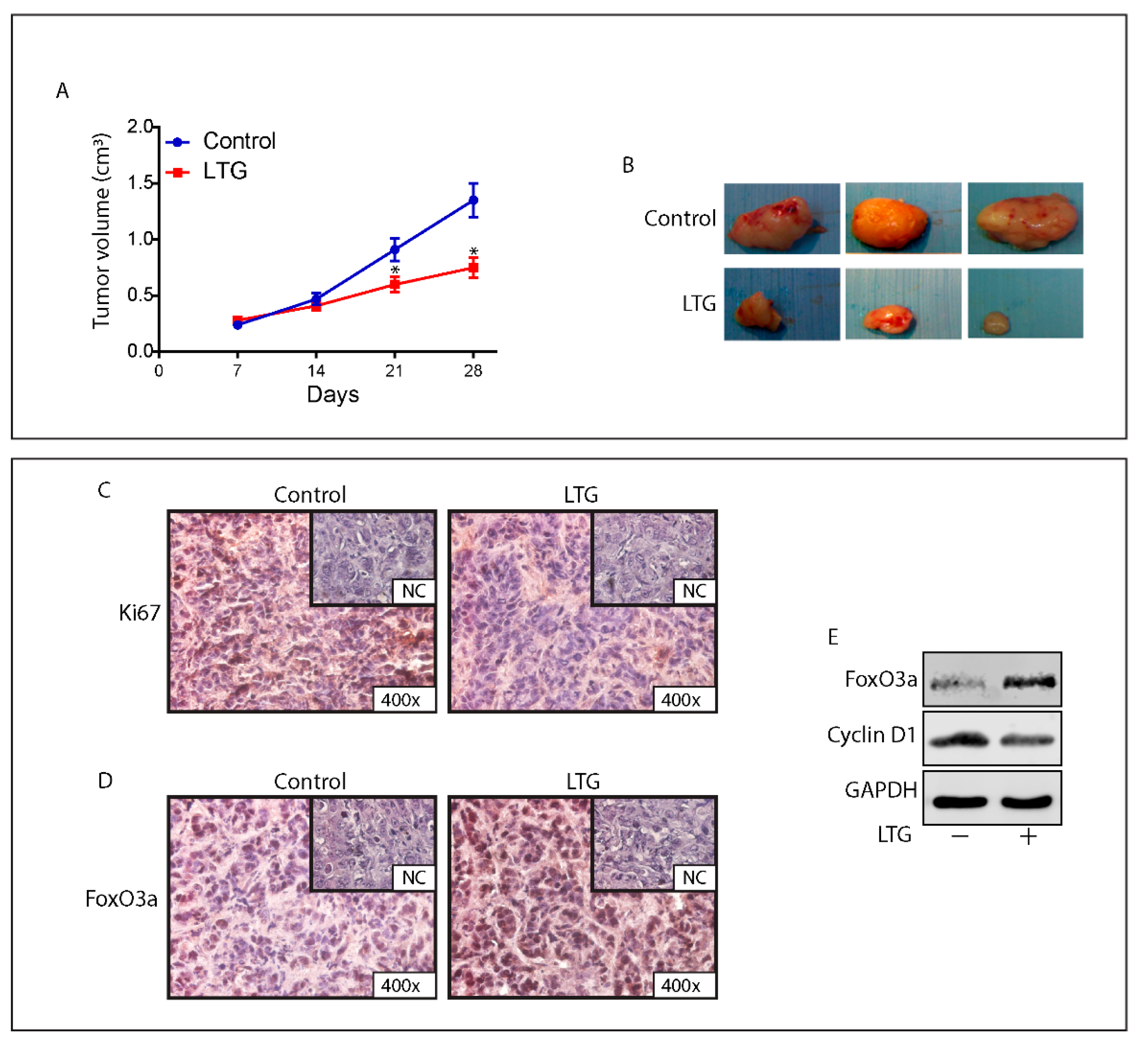
2.8. The AED Lamotrigine Restores the Sensitivity to Tamoxifen Treatment through FoxO3a Re-Expression in Tamoxifen Resistant Xenografts
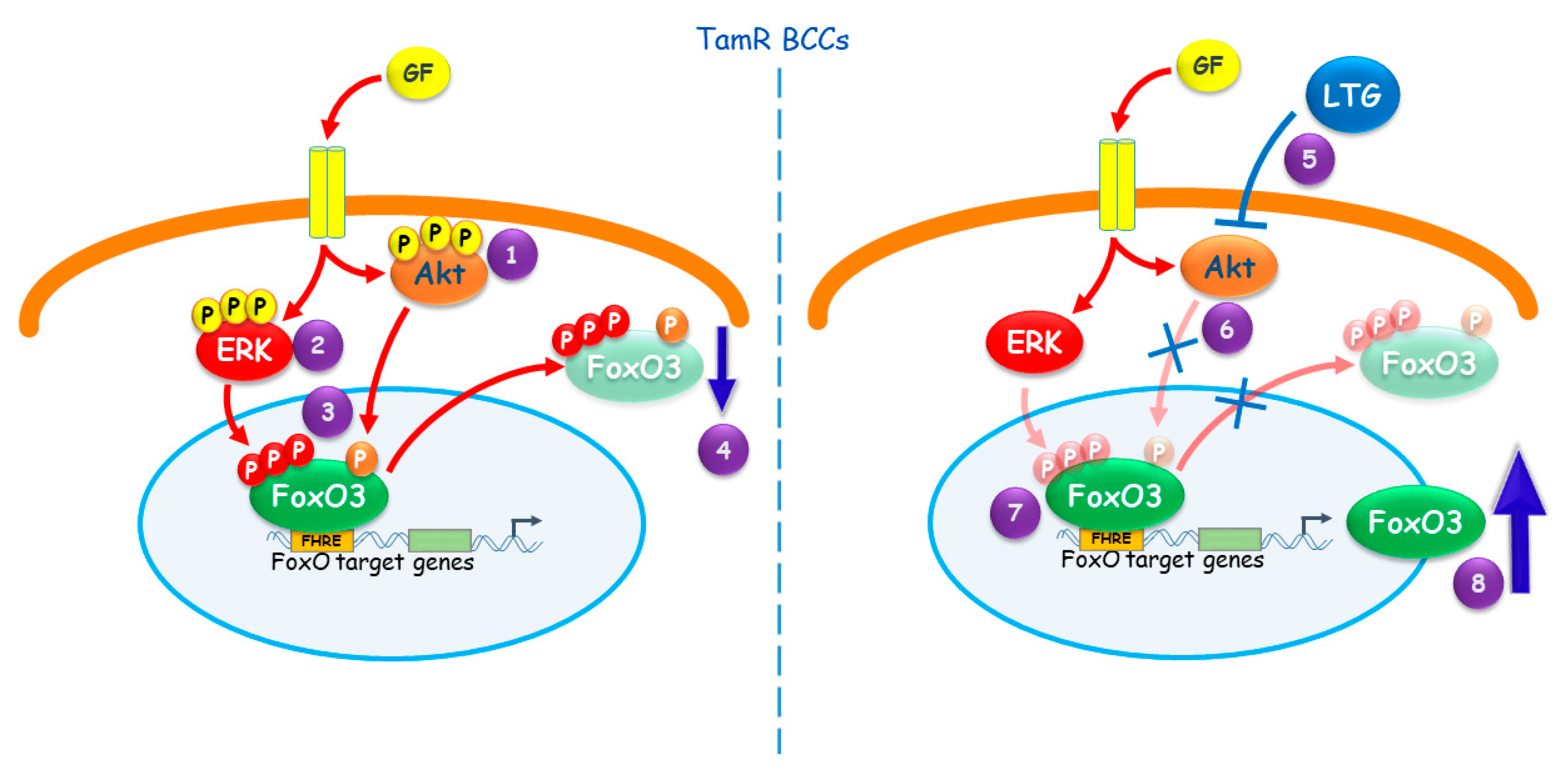
3. Discussion
4. Materials and Methods
4.1. Cell Cultures, Conditions, and Treatments
4.2. RNA Extraction, Reverse Transcription, and Real-Time (RT)-PCR
4.3. Western Blotting (WB) Assay
4.4. Immunostaining
4.5. Proximity Ligation Assay (PLA)
4.6. Generation of FoxO3a Inducible Stable Cell Lines
4.7. siRNA-Mediated RNA Interference
4.8. Growth Assay
4.9. Cell Cycle Analysis
4.10. Transmission Electron Microscopy (TEM)
4.11. TUNEL Assay
4.12. Label-Free Semi-Quantitative Proteomic Analysis
Ingenuity Pathway Analysis (IPA)
4.13. Kaplan–Meier Analysis
4.14. In Vivo Studies TamR/TetOn Derived Xenografts
4.15. In Vivo Studies TamR Derived Xenografts
Animal Care
4.16. Histological Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jordan, C. Historical perspective on hormonal therapy of advanced breast cancer. Clin. Ther. 2002, 24 (Suppl. A), A3–A16. [Google Scholar] [CrossRef]
- Cole, M.P.; Jones, C.T.; Todd, I.D. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br. J. Cancer 1971, 25, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 2016, 50, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.N.; Rutkovsky, A.C.; Giordano, A. Mechanisms of resistance in estrogen receptor positive breast cancer: Overcoming resistance to tamoxifen/aromatase inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, N.; Fang, J.; Huang, J.; Tian, F.; Li, C.; Xie, F. Role of PKC-ERK signaling in tamoxifen-induced apoptosis and tamoxifen resistance in human breast cancer cells. Oncol. Rep. 2012, 27, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Kohli, L.; Kaza, N.; Coric, T.; Byer, S.J.; Brossier, N.M.; Klocke, B.J.; Bjornsti, M.A.; Carroll, S.L.; Roth, K.A. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res. 2013, 73, 4395–4405. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-kappaB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Gee, J.M.; Robertson, J.F.; Ellis, I.O.; Nicholson, R.I. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int. J. Cancer 2001, 95, 247–254. [Google Scholar] [CrossRef]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 2005, 23, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Perez-Torres, M.; Narasanna, A.; Guix, M.; Stal, O.; Perez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009, 69, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.J.; Gee, J.M.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF-7 cells. Breast Cancer Res. Treat. 2004, 87, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M. FOXO factors and breast cancer: Outfoxing endocrine resistance. Endocr. Relat. Cancer 2016, 23, R113–R130. [Google Scholar] [CrossRef]
- Alvarez, B.; Martinez, A.C.; Burgering, B.M.; Carrera, A.C. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature 2001, 413, 744–747. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Dobson, M.; Ramakrishnan, G.; Ma, S.; Kaplun, L.; Balan, V.; Fridman, R.; Tzivion, G. Bimodal regulation of FoxO3 by AKT and 14-3-3. Biochim. Biophys. Acta 2011, 1813, 1453–1464. [Google Scholar] [CrossRef]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Sisci, D.; Maris, P.; Cesario, M.G.; Anselmo, W.; Coroniti, R.; Trombino, G.E.; Romeo, F.; Ferraro, A.; Lanzino, M.; Aquila, S.; et al. The estrogen receptor alpha is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle 2013, 12, 3405–3420. [Google Scholar] [CrossRef] [PubMed]
- Morelli, C.; Lanzino, M.; Garofalo, C.; Maris, P.; Brunelli, E.; Casaburi, I.; Catalano, S.; Bruno, R.; Sisci, D.; Ando, S. Akt2 inhibition enables the forkhead transcription factor FoxO3a to have a repressive role in estrogen receptor alpha transcriptional activity in breast cancer cells. Mol. Cell. Biol. 2010, 30, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Habashy, H.O.; Rakha, E.A.; Aleskandarany, M.; Ahmed, M.A.; Green, A.R.; Ellis, I.O.; Powe, D.G. FOXO3a nuclear localisation is associated with good prognosis in luminal-like breast cancer. Breast Cancer Res. Treat. 2011, 129, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.M.; Meussen-Elholm, E.T.; Roste, L.S.; Tauboll, E. Antiepileptic drugs inhibit cell growth in the human breast cancer cell line MCF7. Mol. Cell. Endocrinol. 2004, 213, 173–179. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef]
- Pellegrino, M.; Rizza, P.; Nigro, A.; Ceraldi, R.; Ricci, E.; Perrotta, I.; Aquila, S.; Lanzino, M.; Ando, S.; Morelli, C.; et al. FoxO3a Mediates the Inhibitory Effects of the Antiepileptic Drug Lamotrigine on Breast Cancer Growth. Mol. Cancer Res. 2018, 16, 923–934. [Google Scholar] [CrossRef]
- Peserico, A.; Chiacchiera, F.; Grossi, V.; Matrone, A.; Latorre, D.; Simonatto, M.; Fusella, A.; Ryall, J.G.; Finley, L.W.; Haigis, M.C.; et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell. Mol. Life Sci. 2013, 70, 2015–2029. [Google Scholar] [CrossRef]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Signorile, M.L.; Longo, G.; De Rasmo, D.; et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 2011, 1813, 1961–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inuzuka, H.; Fukushima, H.; Shaik, S.; Wei, W. Novel Insights into the Molecular Mechanisms Governing Mdm2 Ubiquitination and Destruction. Oncotarget 2010, 1, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris-Pages, M.; Smith, D.L.; Gyorffy, B.; Sotgia, F.; Lisanti, M.P. Proteomic identification of prognostic tumour biomarkers, using chemotherapy-induced cancer-associated fibroblasts. Aging 2015, 7, 816–838. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Hung, M.C. Deciphering the role of forkhead transcription factors in cancer therapy. Curr. Drug Targets 2011, 12, 1284–1290. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Yonezawa, A.; Matsubara, K.; Yano, I. Impact of P-glycoprotein and breast cancer resistance protein on the brain distribution of antiepileptic drugs in knockout mouse models. Eur. J. Pharmacol. 2013, 710, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Tsai, W.B.; Cheng, C.J.; Hsu, C.; Chung, Y.M.; Li, P.C.; Lin, S.H.; Hu, M.C. Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res. 2008, 10, R21. [Google Scholar] [CrossRef] [Green Version]
- Stone, A.; Valdes-Mora, F.; Gee, J.M.; Farrow, L.; McClelland, R.A.; Fiegl, H.; Dutkowski, C.; McCloy, R.A.; Sutherland, R.L.; Musgrove, E.A.; et al. Tamoxifen-induced epigenetic silencing of oestrogen-regulated genes in anti-hormone resistant breast cancer. PLoS ONE 2012, 7, e40466. [Google Scholar] [CrossRef]
- Lutzner, N.; Kalbacher, H.; Krones-Herzig, A.; Rosl, F. FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS ONE 2012, 7, e42166. [Google Scholar] [CrossRef] [Green Version]
- Burgering, B.M.; Medema, R.H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunekawa, S.; Demozay, D.; Briaud, I.; McCuaig, J.; Accili, D.; Stein, R.; Rhodes, C.J. FoxO feedback control of basal IRS-2 expression in pancreatic beta-cells is distinct from that in hepatocytes. Diabetes 2011, 60, 2883–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, K.; Tanaka, J.; Shuiqing, Y.; Welch, C.L.; DePinho, R.A.; Tabas, I.; Tall, A.R.; Goldberg, I.J.; Accili, D. FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 2012, 15, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Zekas, E.; Prossnitz, E.R. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer 2015, 15, 702. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zou, L.; Lu, W.Q.; Zhang, Y.; Shen, A.G. Foxo3a expression is a prognostic marker in breast cancer. PLoS ONE 2013, 8, e70746. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Kim, Y.; Kim, H.; Sim, J.; Ahn, H.; Chung, M.S.; Shin, S.J.; Jang, K. FOXO3a expression is associated with lymph node metastasis and poor disease-free survival in triple-negative breast cancer. J. Clin. Pathol. 2018, 71, 806–813. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Brodie, M.J. Lamotrigine. Lancet 1992, 339, 1397–1400. [Google Scholar] [CrossRef]
- Cacho-Diaz, B.; San-Juan, D.; Salmeron, K.; Boyzo, C.; Lorenzana-Mendoza, N. Choice of antiepileptic drugs affects the outcome in cancer patients with seizures. Clin. Transl. Oncol. 2018, 20, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.D.; Flynn, P.J.; Sloan, J.A.; Wong, G.Y.; Novotny, P.; Johnson, D.B.; Gross, H.M.; Renno, S.I.; Nashawaty, M.; Loprinzi, C.L. Efficacy of lamotrigine in the management of chemotherapy-induced peripheral neuropathy: A phase 3 randomized, double-blind, placebo-controlled trial, N01C3. Cancer 2008, 112, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Benit, C.P.; Vecht, C.J. Seizures and cancer: Drug interactions of anticonvulsants with chemotherapeutic agents, tyrosine kinase inhibitors and glucocorticoids. Neuro-Oncol. Pract. 2016, 3, 245–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tracy, T.S.; Remmel, R.P. Glucuronidation of dihydrotestosterone and trans-androsterone by recombinant UDP-glucuronosyltransferase (UGT) 1A4: Evidence for multiple UGT1A4 aglycone binding sites. Drug Metab. Dispos. 2010, 38, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Giordano, C.; Rizza, P.; Gu, G.; Barone, I.; Bonofiglio, D.; Giordano, F.; Malivindi, R.; Gaccione, D.; Lanzino, M.; et al. Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J. Cell. Physiol. 2009, 218, 490–500. [Google Scholar] [CrossRef]
- Morelli, C.; Maris, P.; Sisci, D.; Perrotta, E.; Brunelli, E.; Perrotta, I.; Panno, M.L.; Tagarelli, A.; Versace, C.; Casula, M.F.; et al. PEG-templated mesoporous silica nanoparticles exclusively target cancer cells. Nanoscale 2011, 3, 3198–3207. [Google Scholar] [CrossRef]
- Lamb, R.; Fiorillo, M.; Chadwick, A.; Ozsvari, B.; Reeves, K.J.; Smith, D.L.; Clarke, R.B.; Howell, S.J.; Cappello, A.R.; Martinez-Outschoorn, U.E.; et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget 2015, 6, 14005–14025. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.Y.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, M.; Rizza, P.; Donà, A.; Nigro, A.; Ricci, E.; Fiorillo, M.; Perrotta, I.; Lanzino, M.; Giordano, C.; Bonofiglio, D.; et al. FoxO3a as a Positive Prognostic Marker and a Therapeutic Target in Tamoxifen-Resistant Breast Cancer. Cancers 2019, 11, 1858. https://doi.org/10.3390/cancers11121858
Pellegrino M, Rizza P, Donà A, Nigro A, Ricci E, Fiorillo M, Perrotta I, Lanzino M, Giordano C, Bonofiglio D, et al. FoxO3a as a Positive Prognostic Marker and a Therapeutic Target in Tamoxifen-Resistant Breast Cancer. Cancers. 2019; 11(12):1858. https://doi.org/10.3390/cancers11121858
Chicago/Turabian StylePellegrino, Michele, Pietro Rizza, Ada Donà, Alessandra Nigro, Elena Ricci, Marco Fiorillo, Ida Perrotta, Marilena Lanzino, Cinzia Giordano, Daniela Bonofiglio, and et al. 2019. "FoxO3a as a Positive Prognostic Marker and a Therapeutic Target in Tamoxifen-Resistant Breast Cancer" Cancers 11, no. 12: 1858. https://doi.org/10.3390/cancers11121858