RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy



Abstract
:1. Introduction
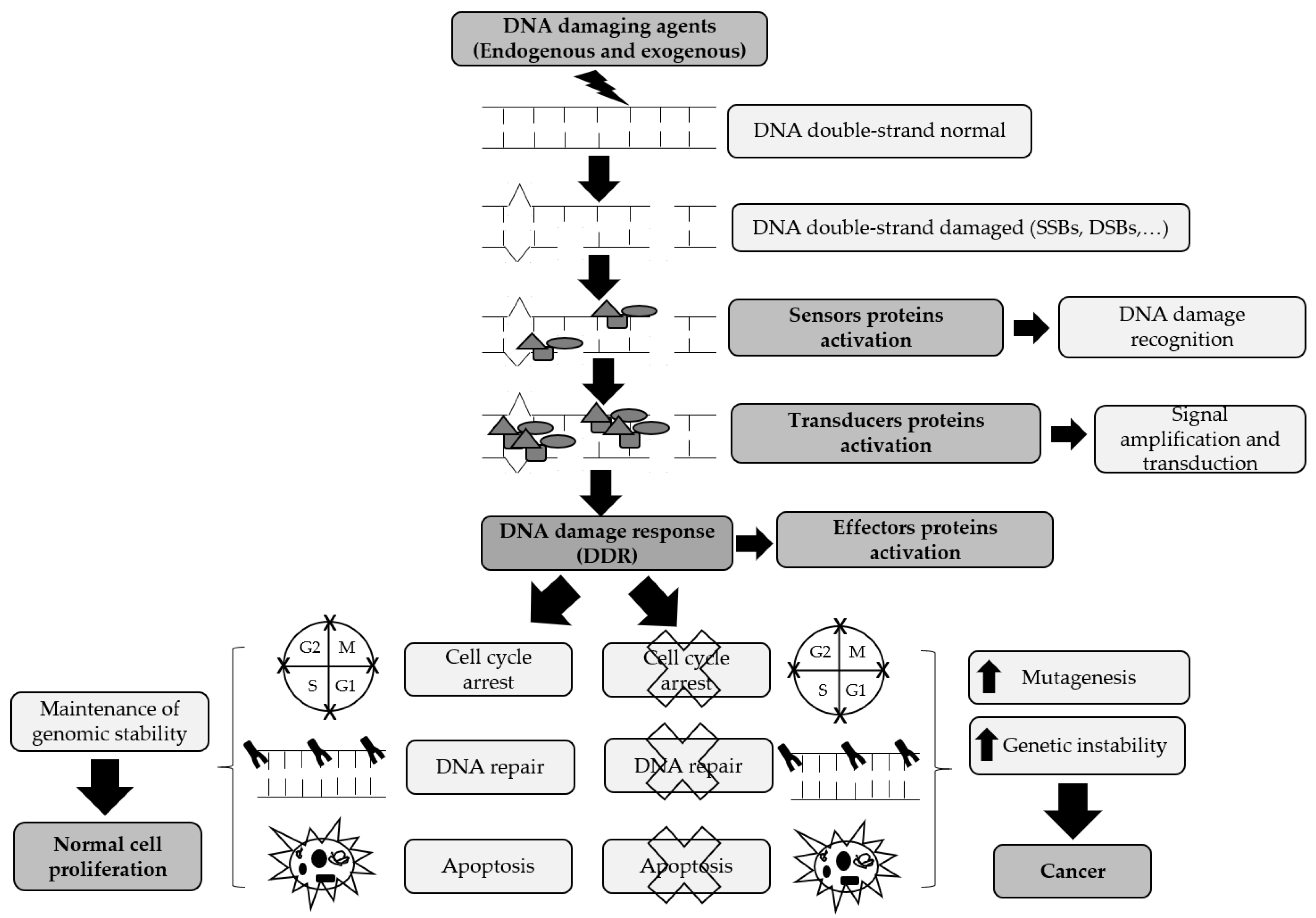
2. DNA Damage Signaling Pathways
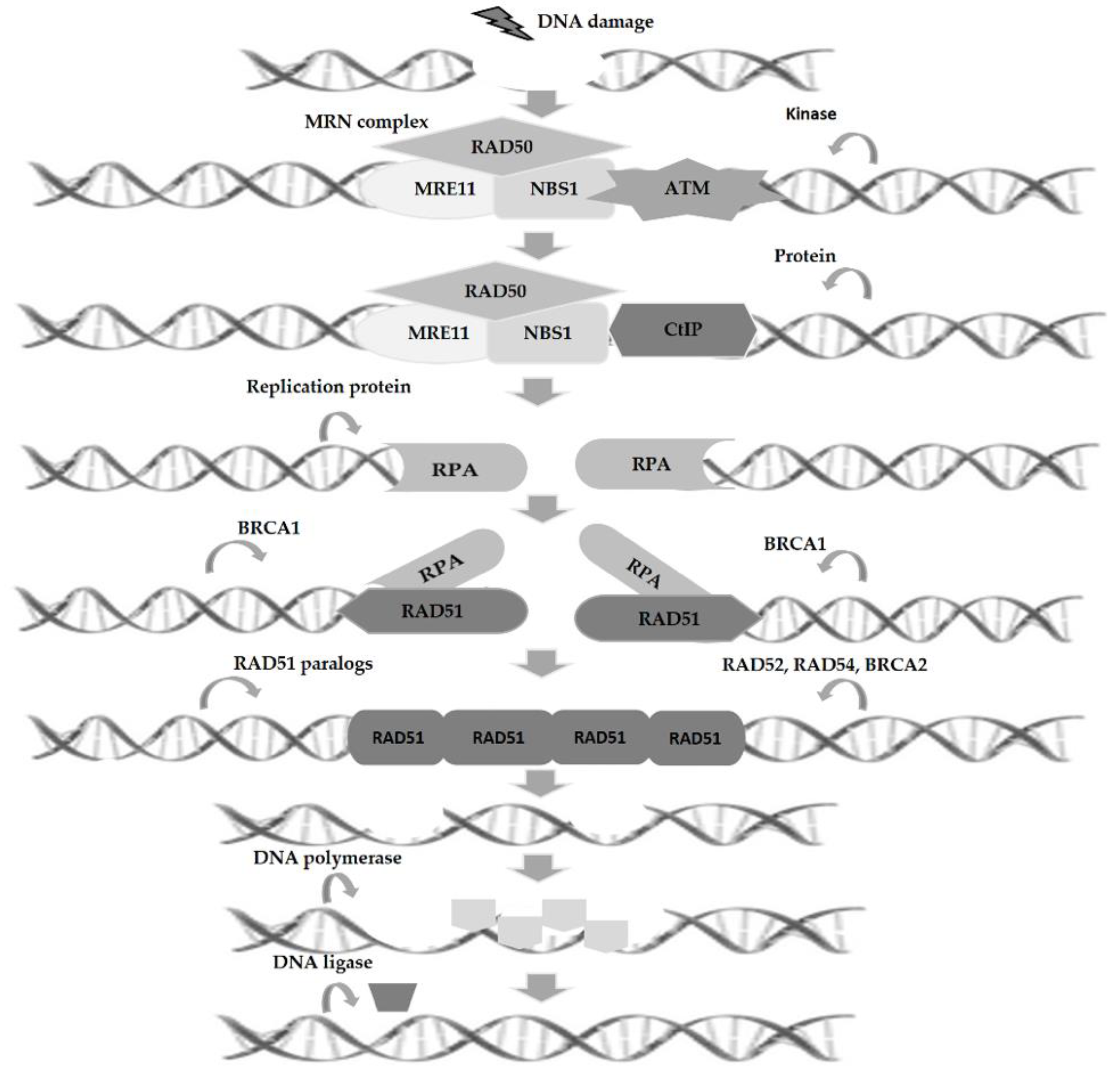
3. Homologous Recombination and DNA Repair
4. RAD52 Functions and Its Implications in Carcinogenesis
4.1. RAD52 Proprieties and Functions
4.2. RAD52 Expression and Regulation
4.3. RAD52 in Carcinogenesis
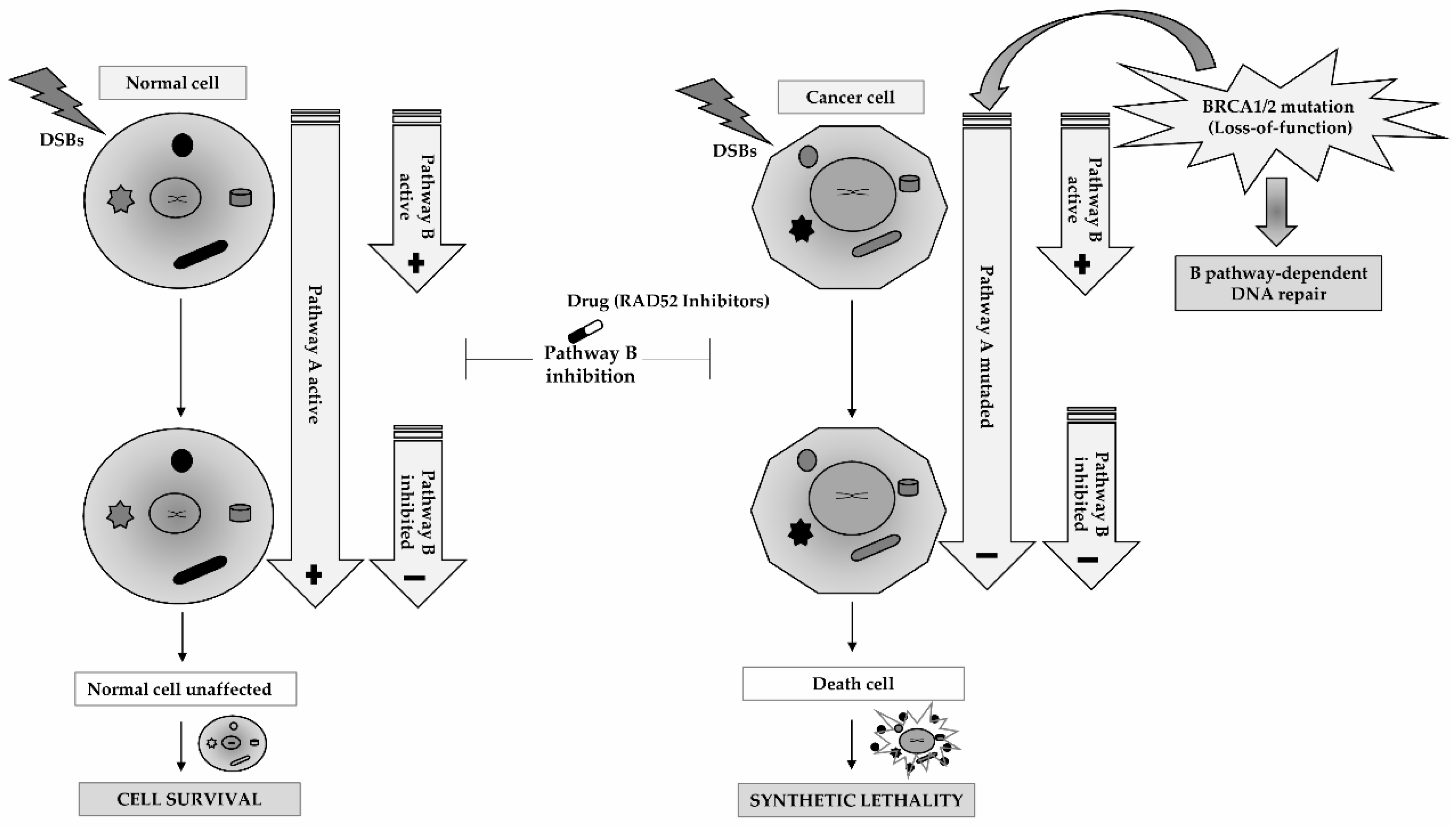
5. RAD52 as a Molecular Target for Cancer Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.; You, M. Corrupting the DNA damage response: A critical role for Rad52 in tumor cell survival. Aging 2017, 9, 1647–1659. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA Damage, Aging, and Cancer. New Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Boil. 2011, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Kinsella, T.J. Understanding DNA Damage Response and DNA Repair Pathways: Applications to More Targeted Cancer Therapeutics. Semin. Oncol. 2009, 36, S42–S51. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Boil. 2009, 21, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Jeggo, P.A. DNA breakage and repair. Adv. Genet. 1998, 38, 185–218. [Google Scholar]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Boil. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: What is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar] [CrossRef] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.A.; McGrew, D.A.; Knight, K.L. Identification of Residues Important for DNA Binding in the Full-length Human Rad52 Protein. J. Mol. Boil. 2005, 345, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.C.; Egelman, E.H. The human Rad52 protein exists as a heptameric ring. Curr. Boil. 2000, 10, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Asleson, E.N.; Okagaki, R.J.; Livingston, D.M. A core activity associated with the N terminus of the yeast RAD52 protein is revealed by RAD51 overexpression suppression of C-terminal rad52 truncation alleles. Genetics 1999, 153, 681–692. [Google Scholar] [PubMed]
- Krejci, L.; Song, B.; Bussen, W.; Mortensen, U.H.; Rothstein, R.; Sung, P. Interaction with Rad51 Is Indispensable for Recombination Mediator Function of Rad52*. J. Boil. Chem. 2002, 277, 40132–40141. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Denison, K.; Lobb, R.; Gatewood, J.M.; Chen, D.J. The human and mouse homologs of the yeast RAD52 gene: cDNA cloning, sequence analysis, assignment to human chromosome 12p12.2–p13, and mRNA expression in mouse tissues. Genomics 1995, 25, 199–206. [Google Scholar] [CrossRef]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef]
- Lok, B.H.; Powell, S.N. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012, 18, 6400–6406. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Shi, J.; Chatterjee, N.; Rotunno, M.; Wang, Y.; Pesatori, A.C.; Consonni, D. Inherited variation at chromosome 12p13.33, including RAD52, influences the risk of squamous cell lung carcinoma. Cancer Discov. 2012, 2, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genom. 2002, 3, 8. [Google Scholar]
- Sugiyama, T.; Kowalczykowski, S.C. Rad52 Protein Associates with Replication Protein A (RPA)-Single-stranded DNA to Accelerate Rad51-mediated Displacement of RPA and Presynaptic Complex Formation. J. Boil. Chem. 2002, 277, 31663–31672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiol. Mol. Boil. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, M.J.; West, S.C. DNA Repair Synthesis Facilitates RAD52-Mediated Second-End Capture during DSB Repair. Mol. Cell 2008, 29, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Bressan, D.A.; Shinohara, M.; Haber, J.E.; Shinohara, A. In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. EMBO J. 2004, 23, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sugiyama, T.; Kowalczykowski, S.C. DNA Annealing Mediated by Rad52 and Rad59 Proteins. J. Boil. Chem. 2006, 281, 15441–15449. [Google Scholar] [CrossRef] [Green Version]
- Parsons, C.A.; Baumann, P.; Van Dyck, E.; West, S.C. Precise binding of single-stranded DNA termini by human RAD52 protein. EMBO J. 2000, 19, 4175–4181. [Google Scholar] [CrossRef] [Green Version]
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.C.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Kumaravel, T.S.; Jalali, F.; Marrano, P.; Squire, J.A.; Bristow, R.G. Defective DNA Strand Break Repair after DNA Damage in Prostate Cancer Cells: Implications for Genetic Instability and Prostate Cancer Progression. Cancer Res. 2004, 64, 8526–8533. [Google Scholar] [CrossRef]
- Ghosh, S.; Krishna, M. Role of Rad52 in fractionated irradiation induced signaling in A549 lung adenocarcinoma cells. Mutat. Res. Mol. Mech. Mutagen. 2012, 729, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Aylott, M.C.; Fisher, K.J.; Lynch, A.M.; Gooderham, N.J. DNA damage responses after exposure to DNA-based products. J. Gene Med. 2006, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.-J.; Lee, E.Y.-H.; Maizels, N. Localization and dynamic relocalization of mammalian Rad52 during the cell cycle and in response to DNA damage. Curr. Boil. 1999, 9, 975–978. [Google Scholar] [CrossRef] [Green Version]
- Barlow, J.H.; Rothstein, R. Timing is everything: Cell cycle control of Rad52. Cell Div. 2010, 5, 7. [Google Scholar] [CrossRef]
- Barlow, J.H.; Lisby, M.; Rothstein, R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell 2008, 30, 73–85. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63. [Google Scholar] [CrossRef]
- Kitao, H.; Yuan, Z.-M. Regulation of Ionizing Radiation-induced Rad52 Nuclear Foci Formation by c-Abl-mediated Phosphorylation. J. Boil. Chem. 2002, 277, 48944–48948. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.; Okuno, Y.; Yoo, J.; Ha, T.; Spies, M. Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 2011, 30, 3368–3382. [Google Scholar] [CrossRef] [Green Version]
- Altmannova, V.; Eckert-Boulet, N.; Arneric, M.; Kolesar, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejci, L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010, 38, 4708–4721. [Google Scholar] [CrossRef] [Green Version]
- Ohuchi, T.; Seki, M.; Branzei, D.; Maeda, D.; Ui, A.; Ogiwara, H.; Tada, S.; Enomoto, T. Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair 2008, 7, 879–889. [Google Scholar] [CrossRef]
- Choi, B.H.; Chen, Y.; Dai, W. Chromatin PTEN is involved in DNA damage response partly through regulating Rad52 sumoylation. Function of a membrane-embedded domain evolutionarily multiplied in the GPI lipid anchor pathway proteins PIG-B, PIG-M, PIG-U, PIG-W, PIG-V, and PIG-Z. Cell Cycle 2013, 12, 3442–3447. [Google Scholar] [CrossRef]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Boil. 2018, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Hironaka, K.; Factor, V.M.; Calvisi, D.F.; Conner, E.A.; Thorgeirsson, S.S. Dysregulation of DNA repair pathways in a transforming growth factor alpha/c-myc transgenic mouse model of accelerated hepatocarcinogenesis. Lab. Investig. 2003, 83, 643. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-Deficient Mice: A Paradigm of Ataxia Telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Treuner, K.; Helton, R.; Barlow, C. Loss of Rad52 partially rescues tumorigenesis and T-cell maturation in Atm-deficient mice. Oncogene 2004, 23, 4655–4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef]
- Lieberman, R.; Xiong, D.; James, M.; Han, Y.; Amos, C.I.; Wang, L. Functional characterization of RAD52 as a lung cancer susceptibility gene in the 12p13.33 locus. Mol. Carcinog. 2016, 55, 953–963. [Google Scholar] [CrossRef]
- Gossage, L.; Madhusudan, S. Cancer pharmacogenomics: Role of DNA repair genetic polymorphisms in individualizing cancer therapy. Mol. Diagn. Ther. 2007, 11. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, W.; Li, Y.; Wu, J.; Shi, G. Pharmacogenetics of DNA repair gene polymorphisms in non-small-cell lung carcinoma patients on platinum-based chemotherapy. Genet. Mol. Res. 2014, 13, 228–236. [Google Scholar] [CrossRef]
- Shi, T.-Y.; Yang, G.; Tu, X.-Y.; Yang, J.-M.; Qian, J.; Wu, X.-H.; Zhou, X.-Y.; Cheng, X.; Wei, Q. RAD52 Variants Predict Platinum Resistance and Prognosis of Cervical Cancer. PLoS ONE 2012, 7, e50461. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Boil. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. New Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic Type | Description |
|---|---|
| Name | Radiation sensitive 52 |
| Symbol | RAD52 |
| Molecular weight | 46169 Da |
| Size | 418 amino acids |
| Structure | Heptameric rings. Two domains (NTD and CTD) |
| Map locus | Chromosome 12p12.2-13 |
| Coding protein | DNA repair protein RAD52 homolog |
| Function | DBSs repair by promoting the annealing of complementary single-stranded DNA and by RAD51 recombinase stimulation |
| Primary localization | Nucleus |
| Protein interactions | ABL1, RPA2, RAD51 |
| Described polymorphisms | rs1060499, rs11571493, rs11571496, rs11571497, rs139916251 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira, A.; Fernandes, M.; Catarino, R.; Medeiros, R. RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy. Cancers 2019, 11, 1622. https://doi.org/10.3390/cancers11111622
Nogueira A, Fernandes M, Catarino R, Medeiros R. RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy. Cancers. 2019; 11(11):1622. https://doi.org/10.3390/cancers11111622
Chicago/Turabian StyleNogueira, Augusto, Mara Fernandes, Raquel Catarino, and Rui Medeiros. 2019. "RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy" Cancers 11, no. 11: 1622. https://doi.org/10.3390/cancers11111622