Radiosensitization of Non-Small Cell Lung Cancer Cells by the Plk1 Inhibitor Volasertib Is Dependent on the p53 Status

 , , , , , ,
, , , , , ,  and
and
Abstract
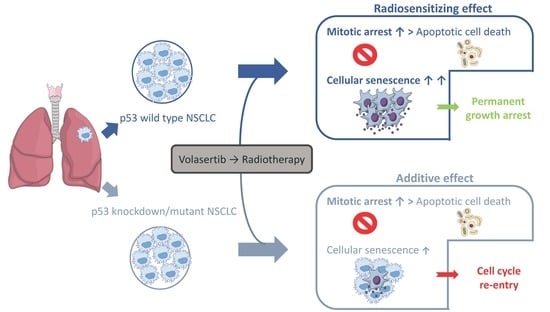
:
1. Introduction
2. Results
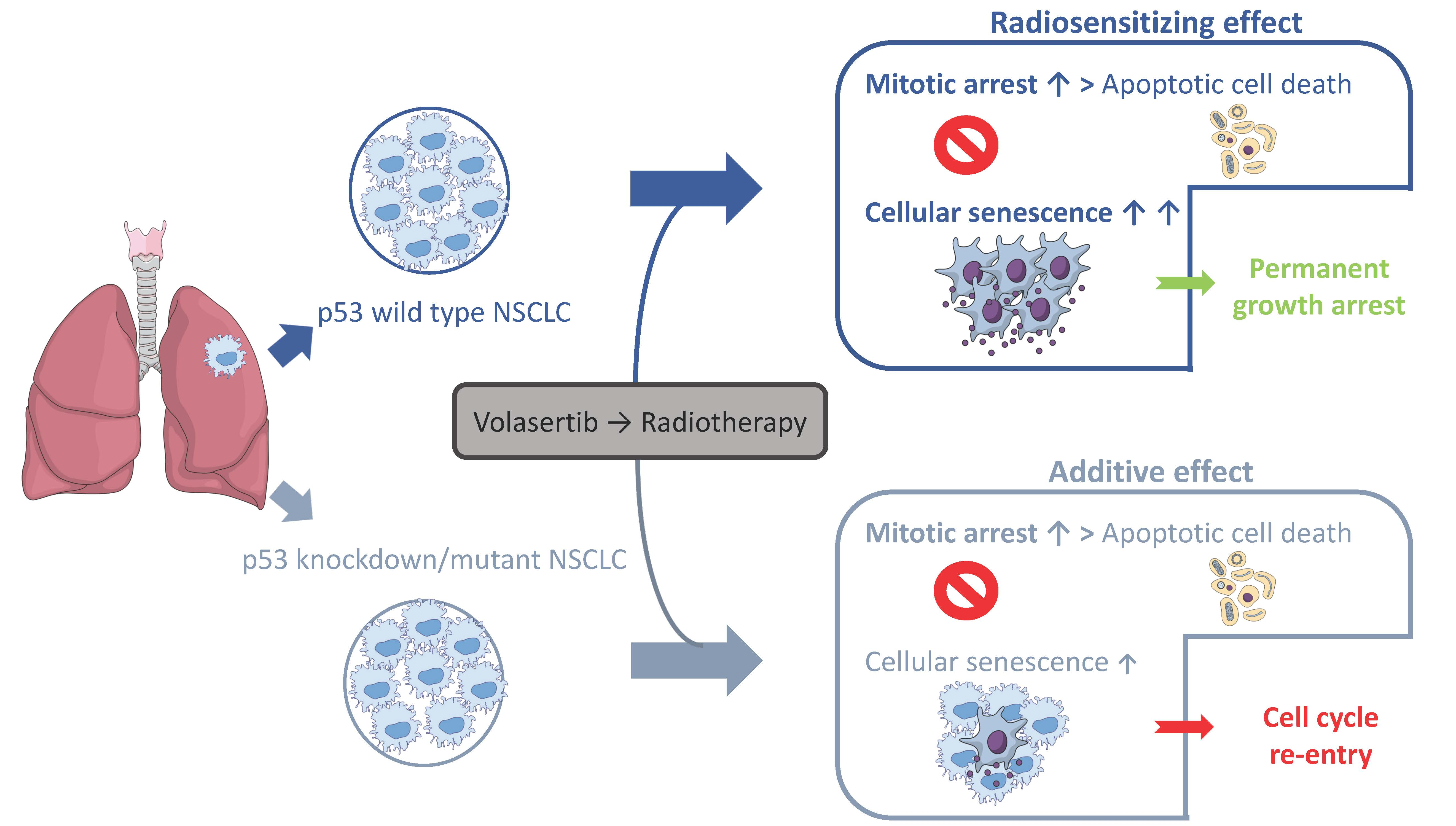
2.1. Volasertib Radiosensitizes p53 Wild Type Cells in a Concentration-Dependent Manner
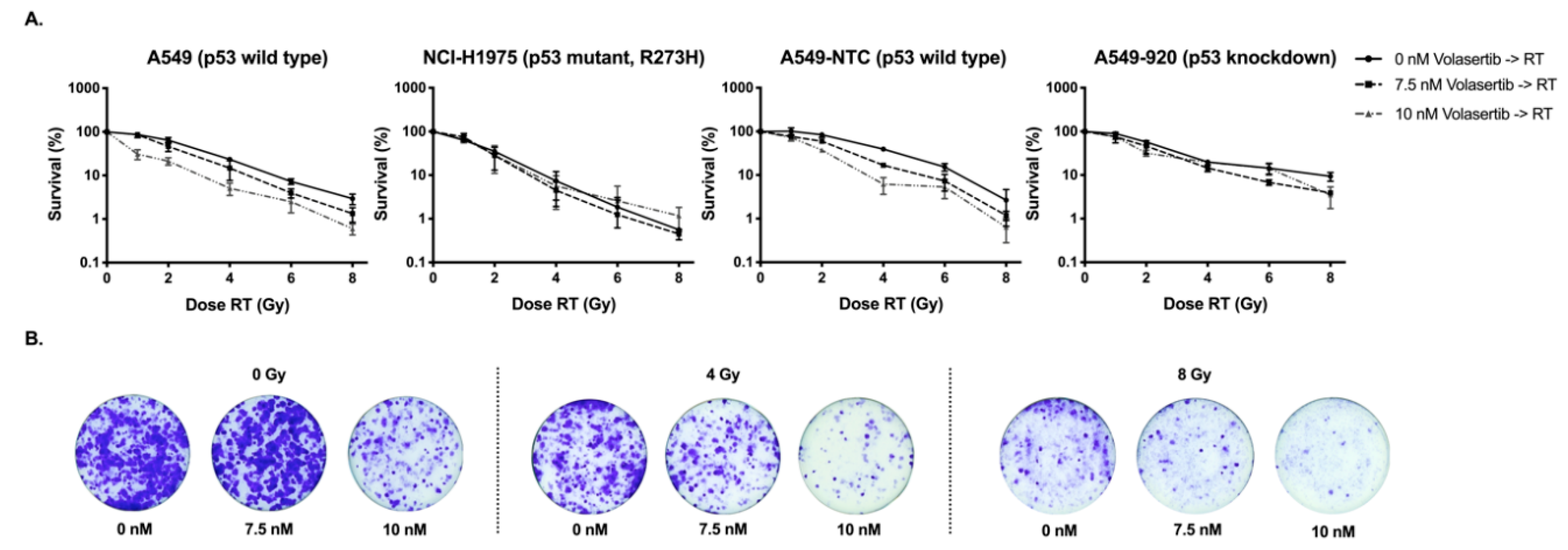
2.2. Treatment with Volasertib Followed by Irradiation Induces Mitotic Arrest
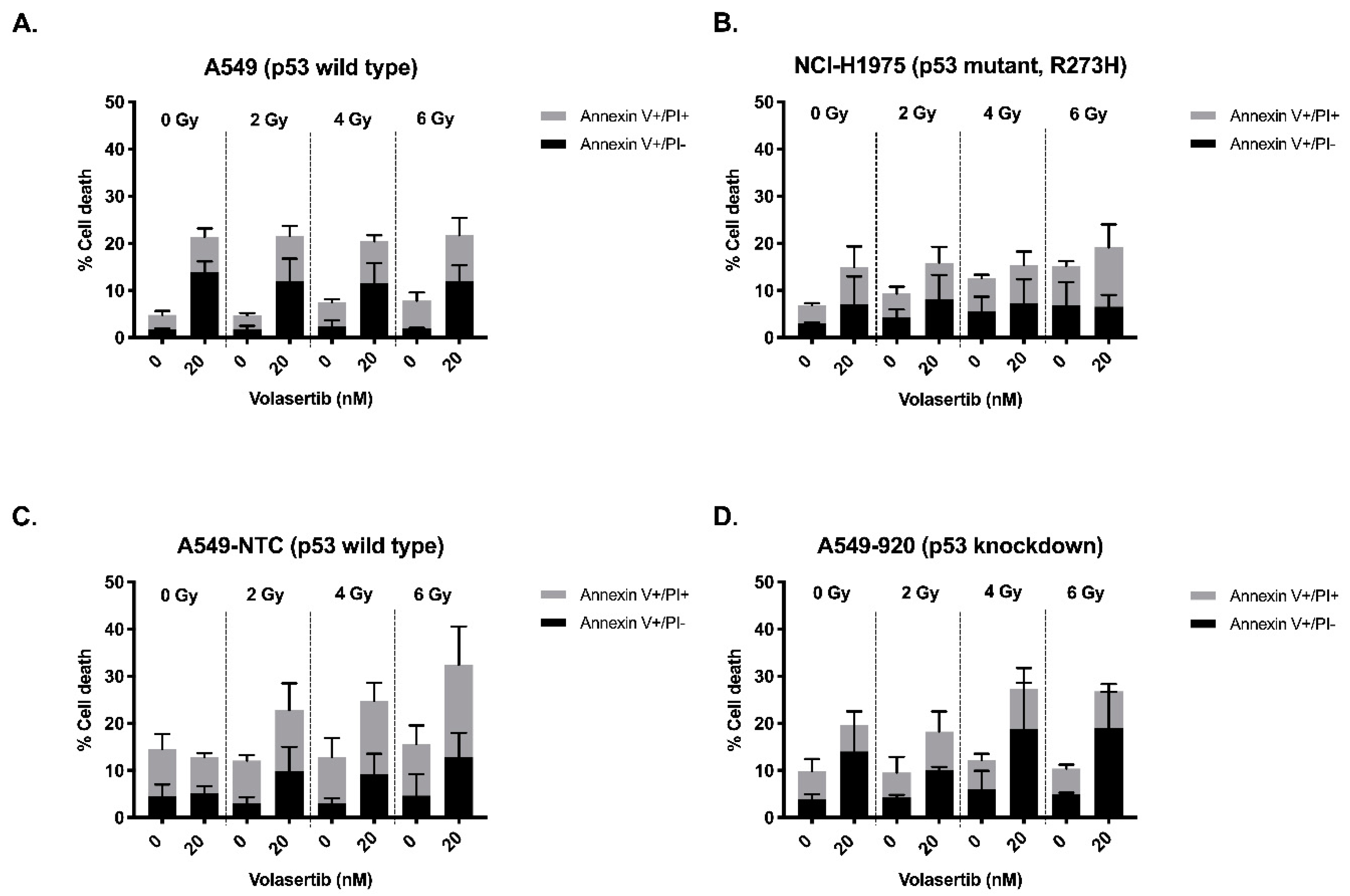
2.3. The Combination of Volasertib with Radiotherapy Does Not Result in Synergistic Induction of Apoptotic Cell Death
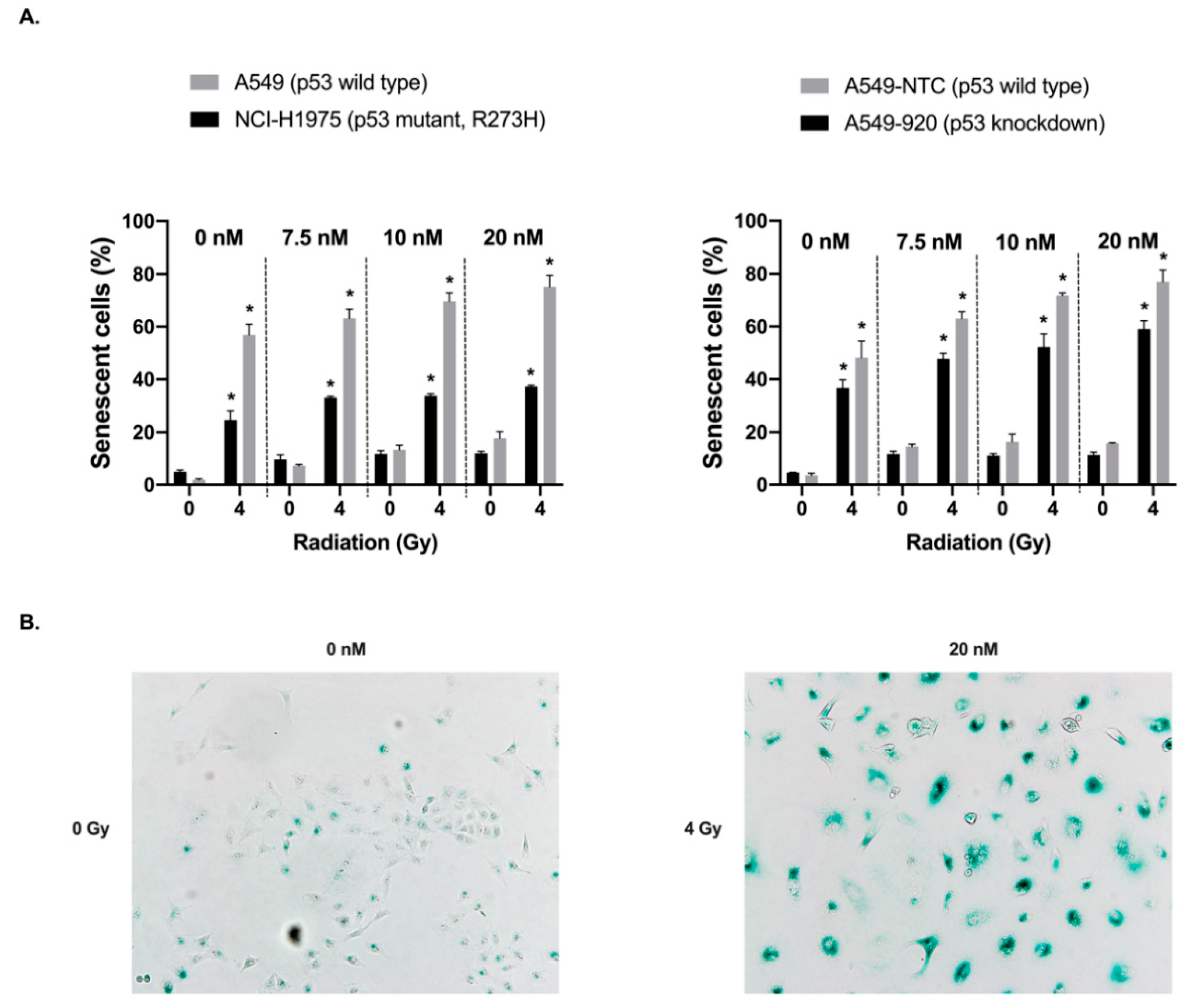
2.4. Treatment with Volasertib Followed by Irradiation Induces Cellular Senescence, Especially in p53 Wild Type Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture Conditions
4.2. Radiation Experiments
4.3. Analysis of Cell Cycle Distribution
4.4. Analysis of Apoptotic Cell Death Induction
4.5. Analysis of Cellular Senescence
4.6. Immunofluorescence Experiments
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jackson, J.R.; Patrick, D.R.; Dar, M.M.; Huang, P.S. Targeted anti-mitotic therapies: Can we improve on tubulin agents? Nat. Rev. Cancer 2007, 7, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Lardon, F.; Deschoolmeester, V.; De Pauw, I.; Vermorken, J.B.; Specenier, P.; Pauwels, P.; Peeters, M.; Wouters, A. Spotlight on Volasertib: Preclinical and Clinical Evaluation of a Promising Plk1 Inhibitor. Med. Res. Rev. 2016, 36, 749–786. [Google Scholar] [CrossRef]
- Hyun, S.Y.; Hwang, H.I.; Jang, Y.J. Polo-like kinase-1 in DNA damage response. BMB Rep. 2014, 47, 249–255. [Google Scholar] [CrossRef]
- Mandal, R.; Strebhardt, K. Plk1: Unexpected roles in DNA replication. Cell Res. 2013, 23, 1251–1253. [Google Scholar] [CrossRef]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rubsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef]
- Wang, Z.X.; Xue, D.; Liu, Z.L.; Lu, B.B.; Bian, H.B.; Pan, X.; Yin, Y.M. Overexpression of polo-like kinase 1 and its clinical significance in human non-small cell lung cancer. Int. J. Biochem. Cell Biol. 2012, 44, 200–210. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Deben, C.; Op de Beeck, K.; Deschoolmeester, V.; Hermans, C.; De Pauw, I.; Jacobs, J.; Van Schil, P.; Vermorken, J.B.; Pauwels, P.; et al. Towards Prognostic Profiling of Non-Small Cell Lung Cancer: New Perspectives on the Relevance of Polo-Like Kinase 1 Expression, the TP53 Mutation Status and Hypoxia. J. Cancer 2017, 8, 1441–1452. [Google Scholar] [CrossRef]
- Ueda, A.; Oikawa, K.; Fujita, K.; Ishikawa, A.; Sato, E.; Ishikawa, T.; Kuroda, M.; Kanekura, K. Therapeutic potential of PLK1 inhibition in triple-negative breast cancer. Lab. Invest. 2019, 99, 1275–1286. [Google Scholar] [CrossRef]
- Stratmann, J.A.; Sebastian, M. Polo-like kinase 1 inhibition in NSCLC: Mechanism of action and emerging predictive biomarkers. Lung Cancer (Auckl.) 2019, 10, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, L.; Yao, Y.; Lu, G.; Xu, L.; Zhou, J. Polo-like kinase 1 inhibitor BI 6727 induces DNA damage and exerts strong antitumor activity in small cell lung cancer. Cancer Lett. 2018, 436, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Lubbert, M.; Fiedler, W.; Fouillard, L.; Haaland, A.; Brandwein, J.M.; Lepretre, S.; Reman, O.; Turlure, P.; Ottmann, O.G.; et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood 2014, 124, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Yamauchi, T.; Kiyoi, H.; Sakura, T.; Hata, T.; Ando, K.; Watabe, A.; Harada, A.; Taube, T.; Miyazaki, Y.; et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with acute myeloid leukemia. Cancer Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; Lardon, F.; Rolfo, C.; Pauwels, P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit. Rev. Oncol. Hematol. 2016, 99, 63–73. [Google Scholar] [CrossRef]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv1–iv21. [Google Scholar] [CrossRef]
- Medema, R.H.; Lin, C.C.; Yang, J.C. Polo-like kinase 1 inhibitors and their potential role in anticancer therapy, with a focus on NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6459–6466. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Deben, C.; De Pauw, I.; Lambrechts, H.; Hermans, C.; Deschoolmeester, V.; Jacobs, J.; Specenier, P.; Pauwels, P.; Vermorken, J.B.; et al. In vitro study of the Polo-like kinase 1 inhibitor volasertib in non-small-cell lung cancer reveals a role for the tumor suppressor p53. Mol. Oncol. 2019, 13, 1196–1213. [Google Scholar] [CrossRef] [PubMed]
- Lund-Andersen, C.; Patzke, S.; Nahse-Kumpf, V.; Syljuasen, R.G. PLK1-inhibition can cause radiosensitization or radioresistance dependent on the treatment schedule. Radiother. Oncol. 2014, 110, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Sanhaji, M.; Kreis, N.N.; Zimmer, B.; Berg, T.; Louwen, F.; Yuan, J. P53 is not directly relevant to the response of Polo-like kinase 1 inhibitors. Cell Cycle 2012, 11, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Greshock, J.; Laquerre, S.; Gilmartin, A.G.; Jing, J.; Richter, M.; Zhang, X.; Bleam, M.; Halsey, W.; Hughes, A.; et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol. Cancer 2010, 9, 2079–2089. [Google Scholar] [CrossRef]
- Yao, D.; Gu, P.; Wang, Y.; Luo, W.; Chi, H.; Ge, J.; Qian, Y. Inhibiting Polo-like Kinase 1 (PLK1) enhances radiosensitization via modulating DNA repair proteins in non-small cell lung cancer. Biochem. Cell Biol. 2017, 96, 317–325. [Google Scholar] [CrossRef]
- Vinod, S.K. International patterns of radiotherapy practice for non-small cell lung cancer. Semin. Radiat. Oncol. 2015, 25, 143–150. [Google Scholar] [CrossRef]
- Chen, J.L.; Chen, J.P.; Huang, Y.S.; Tsai, Y.C.; Tsai, M.H.; Jaw, F.S.; Cheng, J.C.; Kuo, S.H.; Shieh, M.J. Radiosensitization in esophageal squamous cell carcinoma: Effect of polo-like kinase 1 inhibition. Strahlenther. Onkol. 2016, 192, 260–268. [Google Scholar] [CrossRef]
- Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Donson, A.M.; Knipstein, J.; Dubuc, A.; Taylor, M.D.; Handler, M.H.; Foreman, N.K.; et al. Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer 2012, 12, 80. [Google Scholar] [CrossRef]
- Bogado, R.F.; Pezuk, J.A.; de Oliveira, H.F.; Tone, L.G.; Brassesco, M.S. BI 6727 and GSK461364 suppress growth and radiosensitize osteosarcoma cells, but show limited cytotoxic effects when combined with conventional treatments. Anti-Cancer Drugs 2015, 26, 56–63. [Google Scholar] [CrossRef]
- Brassesco, M.S.; Pezuk, J.A.; Morales, A.G.; de Oliveira, J.C.; Roberto, G.M.; da Silva, G.N.; Francisco de Oliveira, H.; Scrideli, C.A.; Tone, L.G. In vitro targeting of Polo-like kinase 1 in bladder carcinoma: Comparative effects of four potent inhibitors. Cancer Biol. 2013, 14, 648–657. [Google Scholar] [CrossRef]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Oliveira, H.F.; Scrideli, C.A.; Tone, L.G. Inhibition of polo-like kinase 1 induces cell cycle arrest and sensitizes glioblastoma cells to ionizing radiation. Cancer Biother. Radiopharm. 2013, 28, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandle, A.T.; Kramp, T.; Kil, W.J.; Halthore, A.; Gehlhaus, K.; Shankavaram, U.; Tofilon, P.J.; Caplen, N.J.; Camphausen, K. Inhibition of polo-like kinase 1 in glioblastoma multiforme induces mitotic catastrophe and enhances radiosensitisation. Eur. J. Cancer 2013, 49, 3020–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodel, F.; Keppner, S.; Capalbo, G.; Bashary, R.; Kaufmann, M.; Rodel, C.; Strebhardt, K.; Spankuch, B. Polo-like kinase 1 as predictive marker and therapeutic target for radiotherapy in rectal cancer. Am. J. Pathol. 2010, 177, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Amani, V.; Prince, E.W.; Alimova, I.; Balakrishnan, I.; Birks, D.; Donson, A.M.; Harris, P.; Levy, J.M.; Handler, M.; Foreman, N.K.; et al. Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016, 16, 647. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.M.; Li, M.; Gao, D.; Liu, L.H.; Lang, Y.; Yang, S.M.; Ou, H.; Huang, B.; Zhou, P.K.; Shang, Z.F. The vanillin derivative 6-bromine-5-hydroxy-4-methoxybenzaldehyde induces aberrant mitotic progression and enhances radio-sensitivity accompanying suppression the expression of PLK1 in esophageal squamous cell carcinoma. Toxicol. Appl. Pharm. 2018, 348, 76–84. [Google Scholar] [CrossRef]
- Brassesco, M.S.; Pezuk, J.A.; Salomao, K.B.; Roberto, G.M.; Scrideli, C.A.; Tone, L.G. PLK1 Inhibition Radiosensitizes Breast Cancer Cells, but Shows Low Efficacy as Monotherapy or in Combination with other Cytotoxic Drugs. Anticancer Agents. Med. Chem. 2018, 18, 1252–1257. [Google Scholar] [CrossRef]
- Inoue, M.; Yoshimura, M.; Kobayashi, M.; Morinibu, A.; Itasaka, S.; Hiraoka, M.; Harada, H. PLK1 blockade enhances therapeutic effects of radiation by inducing cell cycle arrest at the mitotic phase. Sci. Rep. 2015, 5, 15666. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Park, S.Y.; Nguyen, N.; Ezhilarasan, R.; Martinez-Ledesma, E.; Wu, S.; Henry, V.; Piao, Y.; Tiao, N.; Brunell, D.; et al. The polo-like kinase 1 inhibitor volasertib synergistically increases radiation efficacy in glioma stem cells. Oncotarget 2018, 9, 10497–10509. [Google Scholar] [CrossRef] [Green Version]
- Kadletz, L.; Bigenzahn, J.; Thurnher, D.; Stanisz, I.; Erovic, B.M.; Schneider, S.; Schmid, R.; Seemann, R.; Birner, P.; Heiduschka, G. Evaluation of Polo-like kinase 1 as a potential therapeutic target in Merkel cell carcinoma. Head Neck 2016, 38, E1918–E1925. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Liu, C.J.; Huang, Y.C.; Wu, S.H.; Fang, H.W.; Chen, Y.J. BI2536 induces mitotic catastrophe and radiosensitization in human oral cancer cells. Oncotarget 2018, 9, 21231–21243. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Kummer, B.; Deparade, A.; Eicheler, W.; Pfitzmann, D.; Yaromina, A.; Kunz-Schughart, L.A.; Baumann, M. Simultaneous PLK1 inhibition improves local tumour control after fractionated irradiation. Radiother. Oncol. 2013, 108, 422–428. [Google Scholar] [CrossRef] [PubMed]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharm. 2019, 162, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Mori, I.; Nakayama, Y.; Miyakoda, M.; Kodama, S.; Watanabe, M. Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat. Res. 2001, 155, 248–253. [Google Scholar] [CrossRef]
- Driscoll, D.L.; Chakravarty, A.; Bowman, D.; Shinde, V.; Lasky, K.; Shi, J.; Vos, T.; Stringer, B.; Amidon, B.; D’Amore, N.; et al. Plk1 inhibition causes post-mitotic DNA damage and senescence in a range of human tumor cell lines. PLoS ONE 2014, 9, e111060. [Google Scholar] [CrossRef]
- Chou, Y.S.; Yen, C.C.; Chen, W.M.; Lin, Y.C.; Wen, Y.S.; Ke, W.T.; Wang, J.Y.; Liu, C.Y.; Yang, M.H.; Chen, T.H.; et al. Cytotoxic mechanism of PLK1 inhibitor GSK461364 against osteosarcoma: Mitotic arrest, apoptosis, cellular senescence, and synergistic effect with paclitaxel. Int. J. Oncol. 2016, 48, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, J.H.; Kim, J.R. Downregulation of Polo-like kinase 1 induces cellular senescence in human primary cells through a p53-dependent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1145–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharm. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Deben, C.; Wouters, A.; Op de Beeck, K.; van Den Bossche, J.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Wouters, A.; Pauwels, B.; Lambrechts, H.A.; Pattyn, G.G.; Ides, J.; Baay, M.; Meijnders, P.; Lardon, F.; Vermorken, J.B. Counting clonogenic assays from normoxic and anoxic irradiation experiments manually or by using densitometric software. Phys. Med. Biol. 2010, 55, N167–N178. [Google Scholar] [CrossRef]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A549 | ID50 | DEF |
|---|---|---|
| 0 nM volasertib → RT | 2.64 ± 0.20 | / |
| 7.5 nM volasertib → RT | 2.02 ± 1.05 | 1.32 ± 0.12 |
| 10 nM volasertib → RT | 0.66 ± 0.07 | 4.07 ± 0.59 |
| A549-NTC | ID50 | DEF |
| 0 nM volasertib → RT | 3.53 ± 0.28 | / |
| 7.5 nM volasertib → RT | 2.26 ± 0.19 | 1.56 ± 0.07 |
| 10 nM volasertib → RT | 1.59 ± 0.25 | 2.24 ± 0.21 |
| A549-920 | ID50 | DEF |
| 0 nM volasertib → RT | 2.45 ± 0.19 | / |
| 7.5 nM volasertib → RT | 1.80 ± 0.61 | 1.44 ± 0.39 |
| 10 nM volasertib → RT | 1.63 ± 0.18 | 1.50 ± 0.07 |
| NCI-H1975 | ID50 | DEF |
| 0 nM volasertib → RT | 1.43 ± 0.04 | / |
| 7.5 nM volasertib → RT | 1.52 ± 0.37 | 0.97 ± 0.26 |
| 10 nM volasertib → RT | 1.48 ± 0.43 | 1.02 ± 0.33 |
| Cell Percentages in Each Phase of the Cell Cycle | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | Condition | Sub-G1 | G0/G1 | S | G2/M | NCI-H1975 | Sub-G1 | G0/G1 | S | G2/M |
| 0 nM volasertib → 0 Gy | 0.09 ± 0.09 | 67.88 ± 1.71 | 14.78 ± 1.40 | 17.48 ± 0.48 | 1.42 ± 1.10 | 58.73 ± 4.46 | 14.30 ± 1.99 | 25.70 ± 5.98 | ||
| 7.5 nM volasertib → 0 Gy | 0.82 ± 1.21 | 64.90 ± 2.71 | 13.23 ± 1.75 | 20.73 ± 0.74 | 1.14 ± 0.62 | 57.83 ± 4.26 | 13.45 ± 2.11 | 27.48 ± 5.58 | ||
| 12.5 nM volasertib → 0 Gy | 2.65 ± 3.64 | 61.20 ± 3.35 | 12.73 ± 1.41 | 23.18 ± 1.50 | 2.02 ± 1.37 | 53.85 ± 3.16 | 12.64 ± 4.80 | 31.48 ± 5.62 | ||
| 20 nM volasertib → 0 Gy | 3.96 ± 1.02 | 49.13 ± 3.37 | 10.95 ± 1.54 | 35.10 ± 5.94 | 2.06 ± 1.25 | 50.18 ± 5.06 | 12.93 ± 2.15 | 34.63 ± 2.07 | ||
| 0 nM volasertib → 2 Gy | 1.12 ± 1.45 | 73.40 ± 0.22 | 8.95 ± 1.45 | 16.73 ± 2.34 | 1.61 ± 1.15 | 58.55 ± 8.01 | 12.87 ± 2.15 | 27.00 ± 8.37 | ||
| 7.5 nM volasertib → 2 Gy | 0.58 ± 0.37 | 64.93 ± 4.25 | 8.76 ± 1.61 | 25.88 ± 5.12 | 1.98 ± 1.50 | 54.70 ± 6.07 | 10.43 ± 1.66 | 33.03 ± 5.86 | ||
| 12.5 nM volasertib → 2 Gy | 2.10 ± 2.32 | 59.63 ± 8.45 | 8.03 ± 1.58 | 29.88 ± 7.97 | 2.12 ± 1.88 | 48.80 ± 2.69 | 10.32 ± 1.54 | 38.53 ± 5.33 | ||
| 20 nM volasertib → 2 Gy | 2.53 ± 0.88 | 44.00 ± 6.08 | 7.65 ± 1.33 | 44.97 ± 6.80 | 3.23 ± 1.61 | 35.33 ± 3.07 | 11.62 ± 3.15 | 50.50 ± 8.51 | ||
| 0 nM volasertib → 4 Gy | 0.39 ± 0.47 | 67.33 ± 3.39 | 5.33 ± 0.74 | 27.05 ± 4.10 | 2.37 ± 2.44 | 49.95 ± 7.47 | 8.24 ± 2.14 | 39.33 ± 5.70 | ||
| 7.5 nM volasertib → 4 Gy | 1.45 ± 0.95 | 53.78 ± 8.69 | 5.31 ± 0.73 | 39.58 ± 8.43 | 2.11 ± 0.89 | 39.23 ± 5.69 | 7.05 ± 2.83 | 51.15 ± 5.68 | ||
| 12.5 nM volasertib → 4 Gy | 1.22 ± 1.44 | 49.55 ± 9.11 | 5.06 ± 1.02 | 43.98 ± 8.01 | 2.27 ± 1.86 | 34.55 ± 5.12 | 7.67 ± 2.80 | 55.68 ± 5.98 | ||
| 20 nM volasertib → 4 Gy | 3.15 ± 0.26 | 36.77 ± 7.93 | 6.84 ± 2.43 | 53.03 ± 8.89 | 3.44 ± 1.15 | 25.00 ± 7.86 | 9.43 ± 2.95 | 62.48 ± 8.40 | ||
| 0 nM volasertib → 6 Gy | 0.29 ± 0.21 | 55.88 ± 5.54 | 3.94 ± 0.42 | 39.80 ± 5.53 | 1.83 ± 0.86 | 34.30 ± 6.00 | 5.76 ± 1.40 | 58.30 ± 5.26 | ||
| 7.5 nM volasertib → 6 Gy | 0.68 ± 0.69 | 45.13 ± 8.70 | 3.32 ± 0.23 | 50.83 ± 8.25 | 2.04 ± 0.96 | 25.15 ± 4.72 | 6.38 ± 2.56 | 66.20 ± 5.66 | ||
| 12.5 nM volasertib → 6 Gy | 1.08 ± 1.05 | 41.90 ± 7.49 | 4.44 ± 0.79 | 52.55 ± 6.91 | 1.52 ± 1.26 | 17.57 ± 2.02 | 5.34 ± 1.79 | 75.50 ± 5.11 | ||
| 20 nM volasertib → 6 Gy | 3.56 ± 1.02 | 33.40 ± 7.44 | 5.82 ± 1.51 | 57.93 ± 6.83 | 2.01 ± 0.88 | 18.95 ± 10.97 | 6.86 ± 2.87 | 72.10 ± 12.08 | ||
| A549-NTC | Condition | Sub-G1 | G0/G1 | S | G2/M | A549-920 | Sub-G1 | G0/G1 | S | G2/M |
| 0 nM volasertib → 0 Gy | 0.27 ± 0.31 | 66.30 ± 2.26 | 16.30 ± 3.24 | 16.90 ± 0.62 | 0.45 ± 0.16 | 66.67 ± 0.83 | 15.83 ± 2.59 | 16.77 ± 2.55 | ||
| 7.5 nM volasertib → 0 Gy | 0.85 ± 0.38 | 61.03 ± 3.95 | 14.50 ± 3.80 | 23.20 ± 1.73 | 3.06 ± 1.17 | 57.20 ± 3.66 | 17.00 ± 3.46 | 22.57 ± 4.47 | ||
| 12.5 nM volasertib → 0 Gy | 1.37 ± 1.02 | 57.07 ± 3.74 | 13.57 ± 1.53 | 28.87 ± 2.71 | 2.43 ± 0.37 | 56.13 ± 2.12 | 17.07 ± 4.37 | 24.87 ± 6.37 | ||
| 20 nM volasertib → 0 Gy | 3.25 ± 1.60 | 30.97 ± 5.85 | 8.48 ± 0.73 | 57.00 ± 5.02 | 4.15 ± 2.40 | 35.93 ± 4.47 | 14.77 ± 3.01 | 45.37 ± 5.17 | ||
| 0 nM volasertib → 2 Gy | 0.24 ± 0.20 | 74.77 ± 5.05 | 9.69 ± 4.05 | 15.70 ± 1.13 | 0.47 ± 0.33 | 68.10 ± 0.79 | 13.37 ± 3.35 | 18.27 ± 3.04 | ||
| 7.5 nM volasertib → 2 Gy | 0.87 ± 0.46 | 62.97 ± 3.58 | 8.83 ± 2.66 | 27.70 ± 3.13 | 1.81 ± 0.66 | 53.23 ± 2.90 | 12.97 ± 4.06 | 32.30 ± 3.97 | ||
| 12.5 nM volasertib → 2 Gy | 1.37 ± 0.79 | 57.90 ± 3.40 | 8.16 ± 3.18 | 33.40 ± 3.11 | 2.09 ± 1.00 | 48.90 ± 2.75 | 11.00 ± 3.80 | 37.90 ± 7.04 | ||
| 20 nM volasertib → 2 Gy | 2.32 ± 2.28 | 26.90 ± 5.39 | 6.21 ± 1.37 | 65.93 ± 4.31 | 2.31 ± 0.81 | 26.30 ± 4.71 | 12.96 ± 4.27 | 58.27 ± 5.77 | ||
| 0 nM volasertib → 4 Gy | 0.72 ± 0.53 | 69.77 ± 2.14 | 4.44 ± 1.35 | 25.33 ± 1.97 | 0.54 ± 0.22 | 63.33 ± 2.53 | 8.76 ± 1.37 | 27.97 ± 3.71 | ||
| 7.5 nM volasertib → 4 Gy | 0.85 ± 0.55 | 54.83 ± 3.51 | 4.67 ± 0.57 | 39.87 ± 3.65 | 1.31 ± 0.57 | 43.70 ± 1.51 | 8.84 ± 2.03 | 46.10 ± 1.28 | ||
| 12.5 nM volasertib → 4 Gy | 0.77 ± 0.44 | 45.53 ± 8.47 | 4.46 ± 1.33 | 49.60 ± 9.61 | 1.87 ± 0.18 | 37.67 ± 2.49 | 10.36 ± 3.07 | 50.30 ± 2.59 | ||
| 20 nM volasertib → 4 Gy | 1.71 ± 1.21 | 26.80 ± 1.92 | 4.24 ± 0.40 | 67.87 ± 3.09 | 2.05 ± 0.84 | 21.17 ± 3.88 | 12.88 ± 4.43 | 63.97 ± 4.80 | ||
| 0 nM volasertib → 6 Gy | 0.40 ± 0.36 | 56.40 ± 3.16 | 3.29 ± 0.56 | 40.30 ± 3.73 | 0.62 ± 0.04 | 52.07 ± 3.74 | 6.59 ± 0.83 | 41.17 ± 4.63 | ||
| 7.5 nM volasertib → 6 Gy | 0.58 ± 0.67 | 43.17 ± 3.43 | 2.96 ± 0.27 | 53.37 ± 3.56 | 1.01 ± 0.54 | 33.80 ± 0.96 | 7.63 ± 1.53 | 57.83 ± 1.11 | ||
| 12.5 nM volasertib → 6 Gy | 0.82 ± 0.68 | 40.77 ± 3.43 | 3.30 ± 0.42 | 55.33 ± 3.75 | 1.21 ± 0.47 | 31.10 ± 4.34 | 7.72 ± 2.88 | 60.03 ± 6.72 | ||
| 20 nM volasertib → 6 Gy | 1.98 ± 0.70 | 22.57 ± 3.69 | 4.67 ± 0.73 | 71.20 ± 3.13 | 1.91 ± 1.34 | 16.80 ± 6.34 | 9.36 ± 4.49 | 71.60 ± 10.06 | ||
| Percentages of Living and Dead Cells | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | Condition | Annexin V−/PI− | Annexin V+/PI− | Annexin V+/PI+ | Annexin V−/PI+ | NCI−H1975 | Annexin V−/PI− | Annexin V+/PI− | Annexin V+/PI+ | Annexin V−/PI+ |
| 0 nM volasertib → 0 Gy | 94.01 ± 1.33 | 1.71 ± 0.30 | 3.16 ± 0.81 | 0.65 ± 0.21 | 92.53 ± 0.80 | 3.07 ± 0.17 | 3.87 ± 0.41 | 0.57 ± 0.38 | ||
| 20 nM volasertib → 0 Gy | 70.30 ± 7.71 | 13.97 ± 2.22 | 7.44 ± 1.76 | 1.61 ± 0.62 | 83.37 ± 12.04 | 7.12 ± 5.87 | 7.90 ± 4.33 | 1.60 ± 1.90 | ||
| 0 nM volasertib → 2 Gy | 93.10 ± 2.37 | 1.86 ± 0.66 | 2.88 ± 0.50 | 0.62 ± 0.11 | 89.87 ± 1.50 | 4.27 ± 1.69 | 5.07 ± 1.46 | 0.79 ± 0.44 | ||
| 20 nM volasertib → 2 Gy | 76.00 ± 7.45 | 11.97 ± 4.79 | 9.61 ± 2.11 | 2.46 ± 1.16 | 81.77 ± 10.48 | 8.13 ± 5.18 | 7.76 ± 3.36 | 2.35 ± 2.05 | ||
| 0 nM volasertib → 4 Gy | 93.06 ± 1.94 | 2.46 ± 1.27 | 5.13 ± 0.59 | 1.30 ± 0.10 | 86.33 ± 2.54 | 5.50 ± 3.20 | 7.13 ± 0.74 | 1.01 ± 0.36 | ||
| 20 nM volasertib → 4 Gy | 77.87 ± 5.44 | 11.6 ± 4.26 | 8.82 ± 1.31 | 1.76 ± 0.82 | 80.83 ± 9.55 | 7.32 ± 5.02 | 8.08 ± 2.89 | 3.78 ± 2.76 | ||
| 0 nM volasertib → 6 Gy | 90.96 ± 9.18 | 1.96 ± 0.16 | 5.90 ± 1.73 | 1.64 ± 0.27 | 83.57± 4.74 | 6.94 ± 4.84 | 8.29 ± 1.00 | 1.21 ± 0.89 | ||
| 20 nM volasertib → 6 Gy | 67.20 ± 9.18 | 11.94 ± 3.40 | 9.80 ± 3.65 | 1.49 ± 0.79 | 77.57 ± 09.33 | 6.56 ± 2.49 | 12.60 ± 4.88 | 2.68 ± 1.22 | ||
| A549-NTC | Condition | Annexin V−/PI− | Annexin V+/PI− | Annexin V+/PI+ | Annexin V−/PI+ | A549-920 | Annexin V−/PI− | Annexin V+/PI− | Annexin V+/PI+ | Annexin V−/PI+ |
| 0 nM volasertib → 0 Gy | 84.68 ± 5.37 | 4.59 ± 2.45 | 9.94 ± 3.24 | 0.80 ± 0.19 | 89.20 ± 2.70 | 3.97 ± 0.99 | 5.90 ± 2.59 | 0.93 ± 0.46 | ||
| 20 nM volasertib → 0 Gy | 85.38 ± 1.95 | 5.24 ± 1.43 | 7.62 ± 0.79 | 1.79 ± 0.62 | 79.33 ± 8.57 | 14.03 ± 8.47 | 5.56 ± 3.01 | 1.08 ± 0.50 | ||
| 0 nM volasertib → 2 Gy | 86.96 ± 2.26 | 3.05 ± 1.29 | 9.04 ± 1.20 | 0.96 ± 0.15 | 89.73 ± 3.34 | 4.28 ± 0.53 | 5.28 ± 3.35 | 0.69 ± 0.18 | ||
| 20 nM volasertib → 2 Gy | 74.08 ± 10.20 | 9.94 ± 5.09 | 12.88 ± 5.69 | 3.12 ± 0.93 | 81.40 ± 0.95 | 10.16 ± 0.62 | 8.09 ± 4.27 | 0.98 ± 0.15 | ||
| 0 nM volasertib → 4 Gy | 84.72 ± 6.11 | 3.04 ± 1.09 | 9.83 ± 4.04 | 2.38 ± 1.28 | 87.15 ± 5.18 | 6.03 ± 3.89 | 6.16 ± 1.37 | 0.64 ± 0.19 | ||
| 20 nM volasertib → 4 Gy | 69.75 ± 6.38 | 9.15 ± 4.42 | 15.65 ± 3.78 | 5.46 ± 1.50 | 77.00 ± 2.78 | 18.85 ± 9.77 | 8.44 ± 4.43 | 1.22 ± 0.14 | ||
| 0 nM volasertib → 6 Gy | 82.50 ± 6.90 | 4.65 ± 4.52 | 10.96 ± 3.94 | 1.89 ± 0.74 | 88.73 ± 3.09 | 4.92 ± 0.37 | 5.49 ± 0.83 | 0.85 ± 0.36 | ||
| 20 nM volasertib → 6 Gy | 63.34 ± 10.45 | 12.88 ± 5.09 | 19.54 ± 8.13 | 4.23 ± 2.23 | 72.18 ± 8.98 | 19.00 ± 7.67 | 7.83 ± 1.53 | 0.99 ± 0.15 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van den Bossche, J.; Domen, A.; Peeters, M.; Deben, C.; De Pauw, I.; Jacobs, J.; De Bruycker, S.; Specenier, P.; Pauwels, P.; Vermorken, J.B.; et al. Radiosensitization of Non-Small Cell Lung Cancer Cells by the Plk1 Inhibitor Volasertib Is Dependent on the p53 Status. Cancers 2019, 11, 1893. https://doi.org/10.3390/cancers11121893
Van den Bossche J, Domen A, Peeters M, Deben C, De Pauw I, Jacobs J, De Bruycker S, Specenier P, Pauwels P, Vermorken JB, et al. Radiosensitization of Non-Small Cell Lung Cancer Cells by the Plk1 Inhibitor Volasertib Is Dependent on the p53 Status. Cancers. 2019; 11(12):1893. https://doi.org/10.3390/cancers11121893
Chicago/Turabian StyleVan den Bossche, Jolien, Andreas Domen, Marc Peeters, Christophe Deben, Ines De Pauw, Julie Jacobs, Sven De Bruycker, Pol Specenier, Patrick Pauwels, Jan Baptist Vermorken, and et al. 2019. "Radiosensitization of Non-Small Cell Lung Cancer Cells by the Plk1 Inhibitor Volasertib Is Dependent on the p53 Status" Cancers 11, no. 12: 1893. https://doi.org/10.3390/cancers11121893