
Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. NICCRAT-BRIN Team Sample Collection and WGS
2.2. Ethical Statements on the Use of Samples from Human Participants
2.3. NICCRAT-BRIN Team Bioinformatics Processes
2.4. Lineage and Phylogenetic Analyses
3. Results
3.1. Analyses of the Genomes Deposited by the NICCRAT-BRIN Team
3.2. Profiles of Genomic Sequences Deposited by Indonesia, Neigbouring Countries, and the World
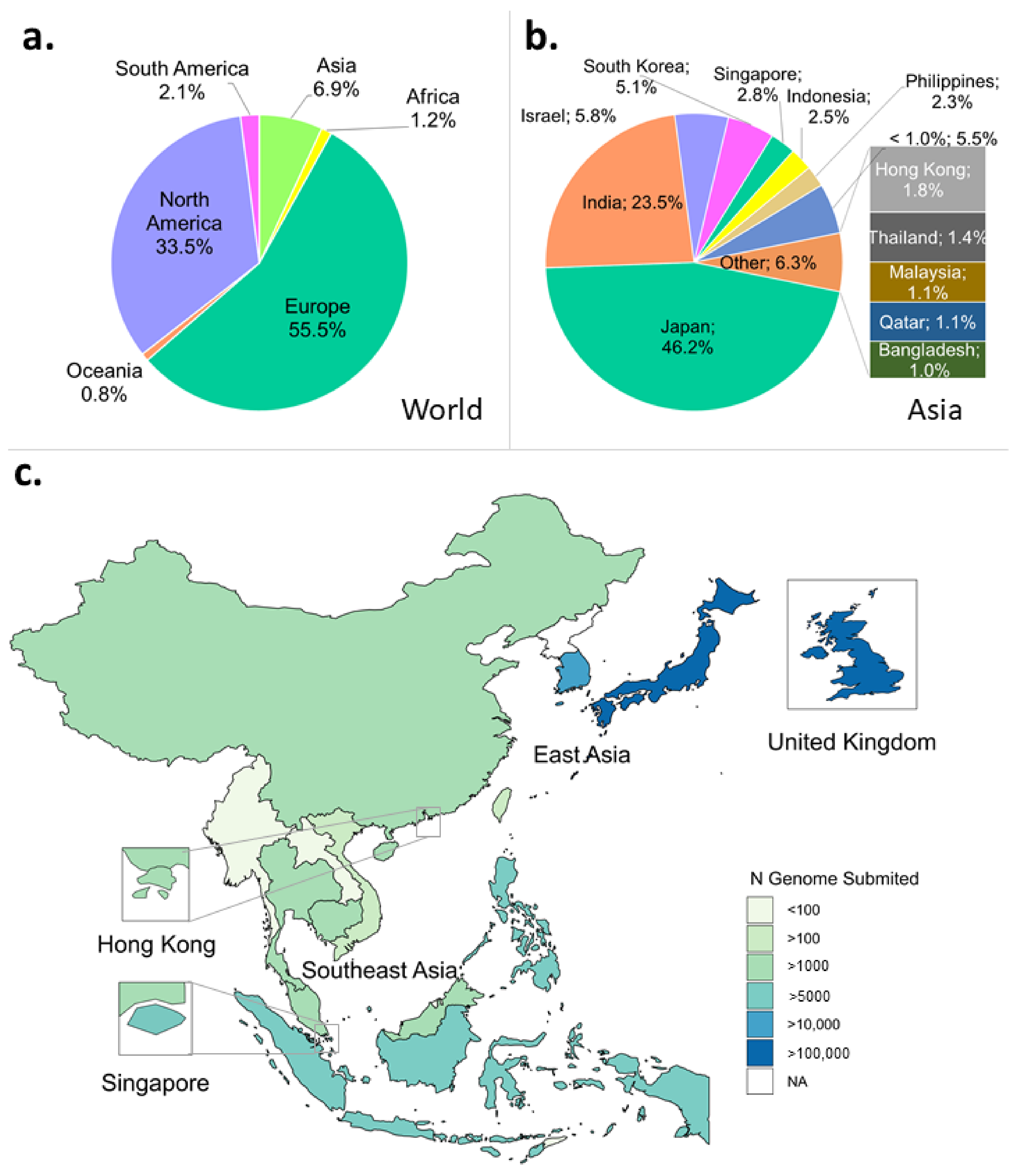
3.2.1. Sequencing Rates
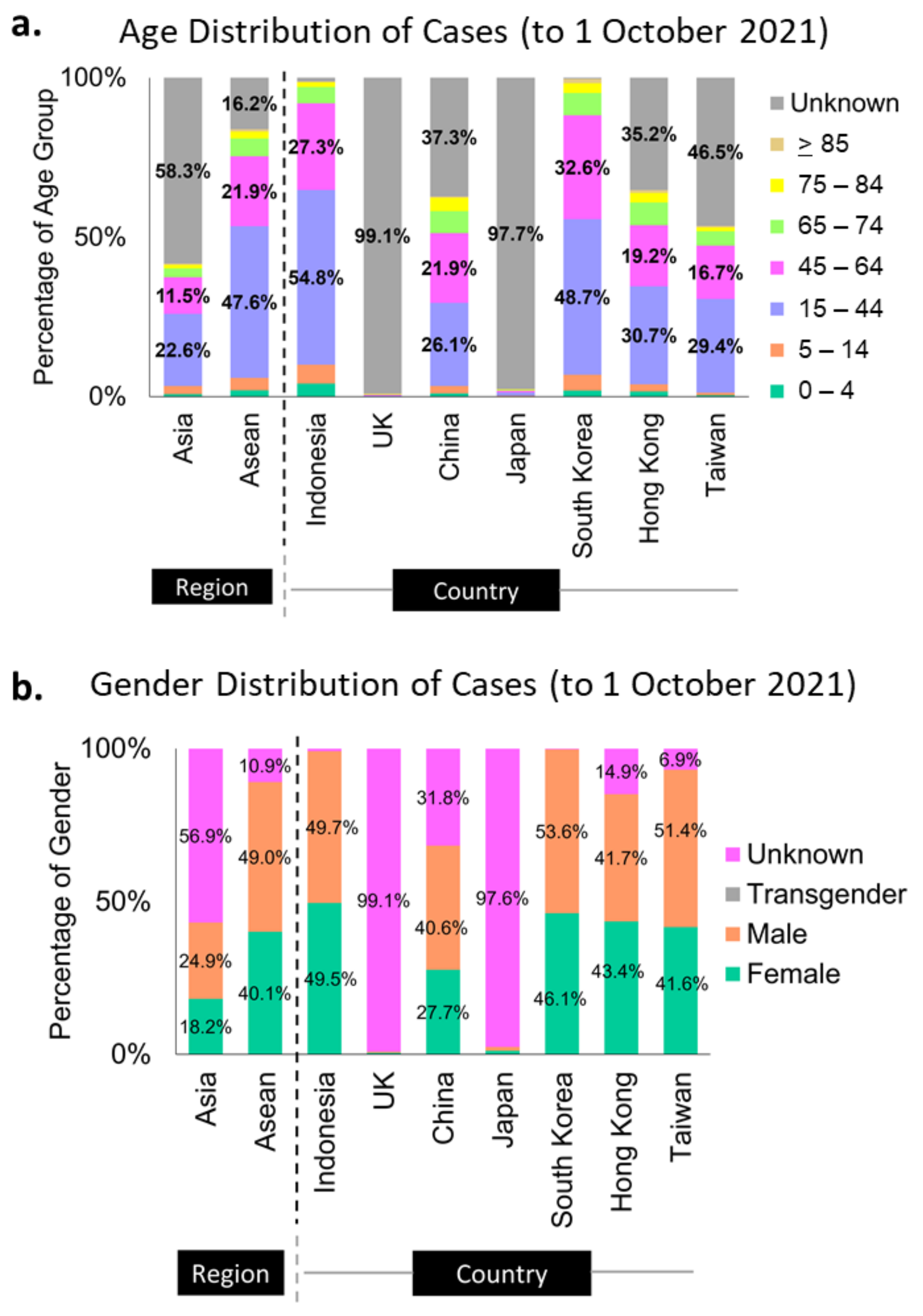
3.2.2. Metadata
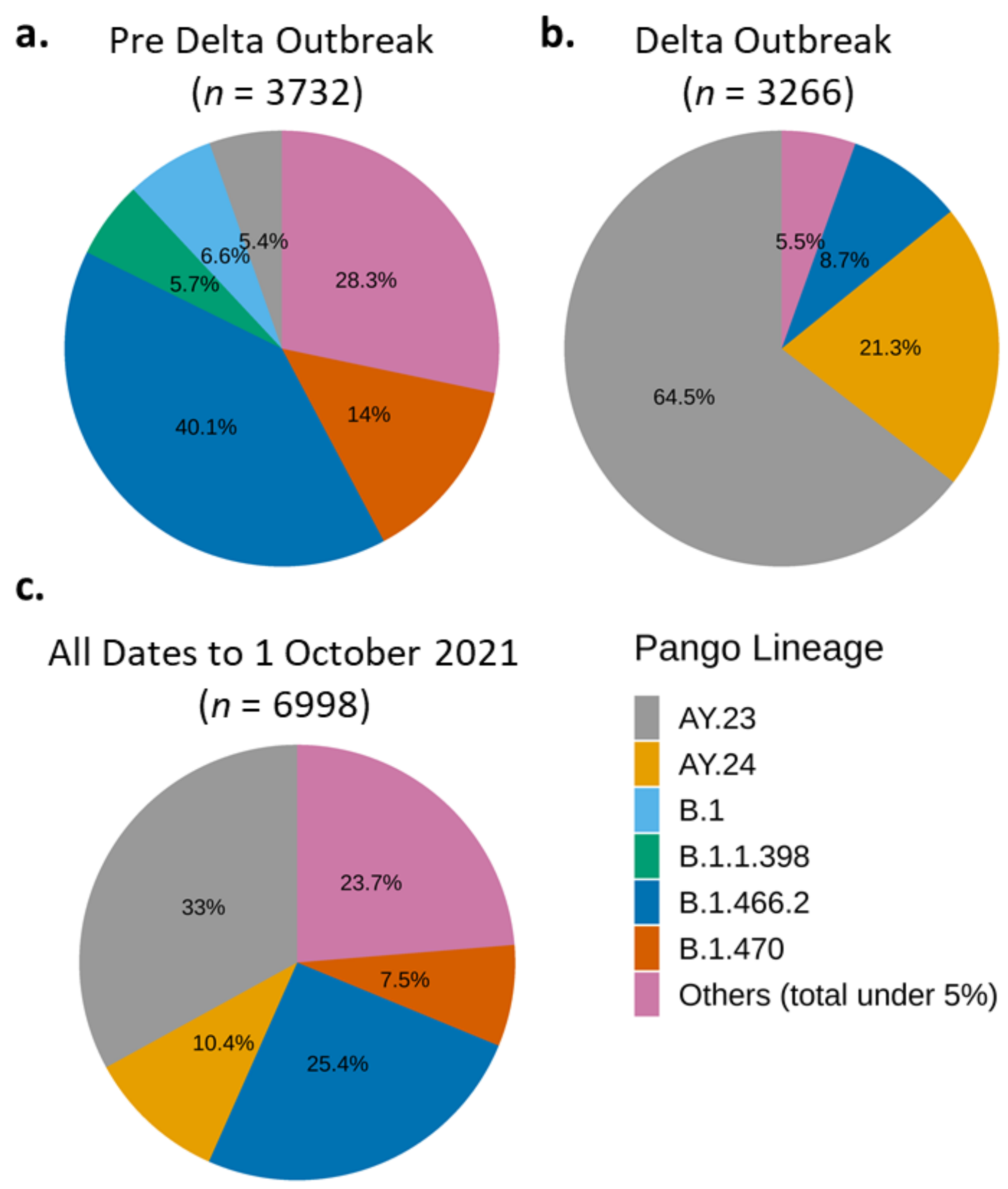
3.2.3. The Distribution of Variants by PANGO Lineages
3.2.4. Mutations and Metadata of the Indonesian Unique Variants
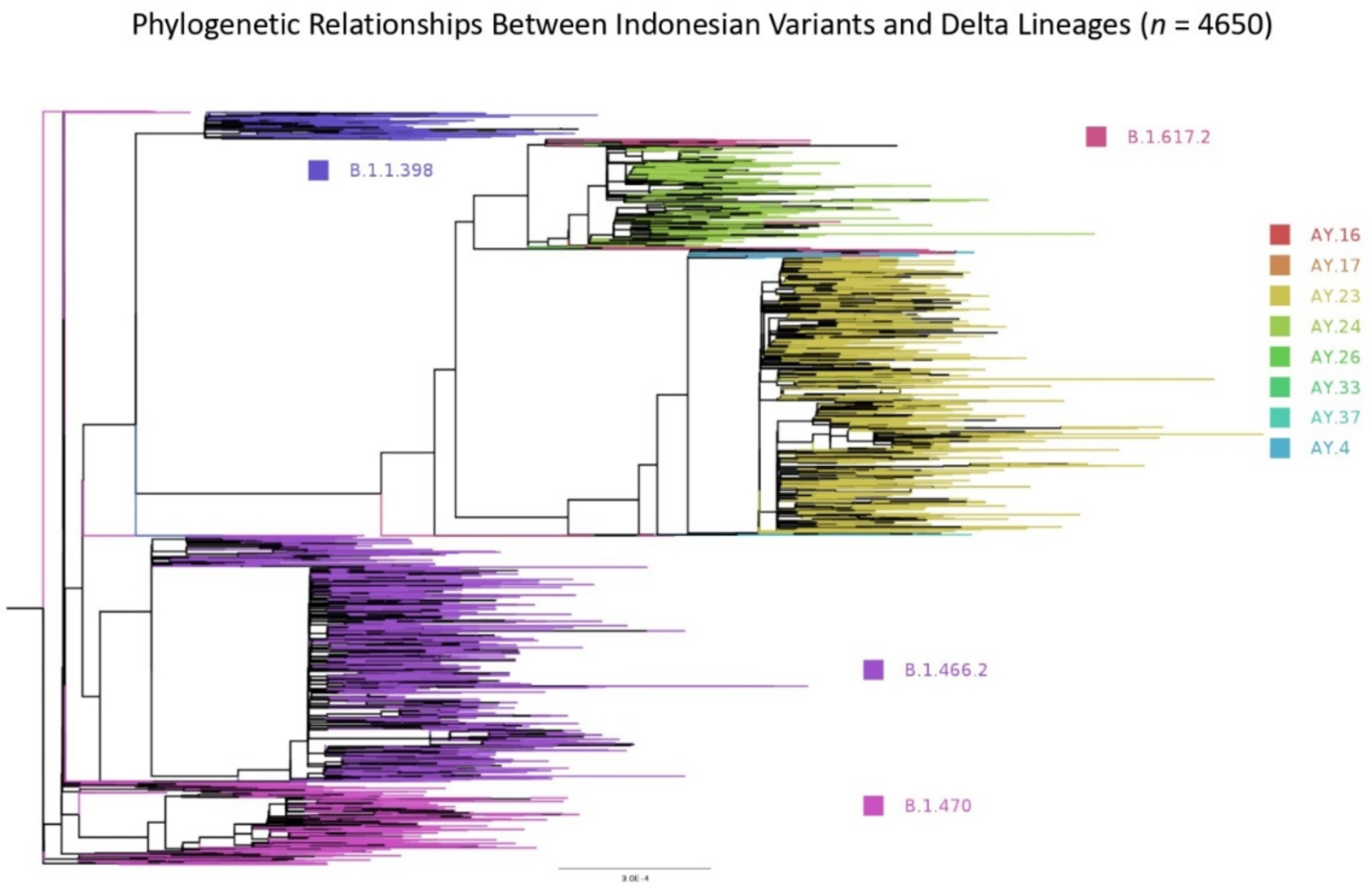
3.2.5. Phylogenetic Relationship Exists between Indonesian Variants and Those Exported to the Neighbouring Countries
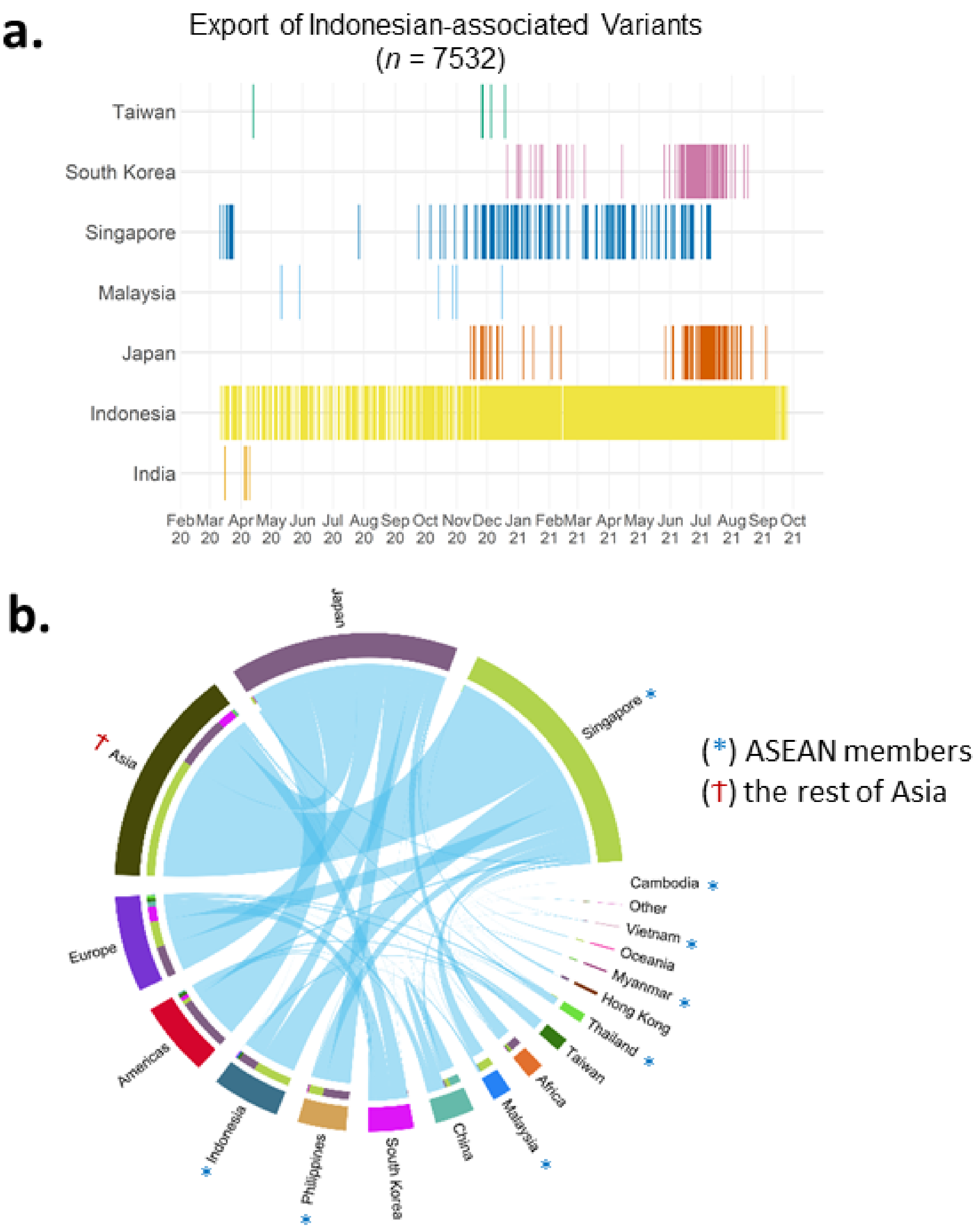
3.2.6. Inter-Nation Transmissions
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BRIN | National Research and Innovation Agency |
| EIJK | Eijkman Research Center for Molecular Biology |
| GSI Lab | Laboratory of Genomik Solidaritas Indonesia |
| IRCVS | Indonesian Research Center for Veterinary Science |
| ITB | Bandung Institute of technology |
| ITD-UNAIR | Research Center for Vaccine Technology and Development, Institute of Tropical Disease, Airlangga University |
| LIPI | Indonesian Institute of Sciences |
| MRINUPH | Mochtar Riady Institute for Nanotechnology-Universitas Pelita Harapan |
| NICCRAT | Nottingham Indonesia Collaboration for Clinical Research and Training |
| NIHRD | National Institute of Health Research and Development |
| RSRM | Raden Mattaher Regional General Hospital |
| SHSIU | Syarif Hidayatullah State Islamic University Jakarta |
| TFRIC | Padjadjaran University |
| TUH | Tanjungpura University Hospital |
| UA | Institute of Tropical Disease-Airlangga University |
| UGM | Gadjah Mada University |
| UHAS | Hasanuddin University |
| UI | University of Indonesia |
| UNS | Sebelas Maret University |
| UNSOED | Jenderal Soedirman University |
| UNSRI | Sriwijaya University |
| UNWAR | Warmadewa University |
| UPNVJ | University of Pembangunan Nasional Veteran Jakarta |
| USK | Syiah Kuala University |
| USU | Sumatera Utara University |
| WJHL | West Java Health Laboratory |
References
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl. Virol. 2021, 2, 428–440. [Google Scholar] [CrossRef]
- Severe Acute Respiratory Syndrome (SARS). Available online: https://www.who.int/health-topics/severe-acute-respiratory-syndrome#tab=tab_1 (accessed on 8 June 2021).
- Middle East respiratory syndrome coronavirus (MERS-CoV). Available online: https://www.who.int/health-topics/middle-east-respiratory-syndrome-coronavirus-mers#tab=tab_1 (accessed on 22 June 2021).
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 22 June 2021).
- World Health Organization (WHO). Naming the Coronavirus Disease (COVID-19) and the Virus That Causes It. 2019. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 22 June 2021).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co-Nucleotide-NCBI. Available online: https://www.ncbi.nlm.nih.gov/nuccore/MN908947 (accessed on 22 June 2021).
- Chinese researchers reveal draft genome of virus implicated in Wuhan pneumonia outbreak|Science|AAAS. Available online: https://www.sciencemag.org/news/2020/01/chinese-researchers-reveal-draft-genome-virus-implicated-wuhan-pneumonia-outbreak (accessed on 22 June 2021).
- Coronavirus disease 2019 (COVID-19) Situation Report–39. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200228-sitrep-39-covid-19.pdf (accessed on 22 June 2021).
- Kilas Balik Kronologi Munculnya Kasus Pertama Covid-19 di Indonesia Halaman all—Kompas.com. Available online: https://megapolitan.kompas.com/read/2021/03/02/05300081/kilas-balik-kronologi-munculnya-kasus-pertama-covid-19-di-indonesia?page=all (accessed on 8 June 2021).
- Indonesia finally reports two coronavirus cases. Scientists worry it has many more|Science|AAAS. Available online: https://www-sciencemag-org.ezproxy.nottingham.ac.uk/news/2020/03/indonesia-finally-reports-two-coronavirus-cases-scientists-worry-it-has-many-more (accessed on 2 July 2021).
- Setiawaty, V.; Kosasih, H.; Mardian, Y.; Ajis, E.; Prasetyowati, E.B.; Siswanto; Karyana, M. The Identification of First COVID-19 Cluster in Indonesia. Am. J. Trop. Med. Hyg. 2020, 103, 2339–2342. [Google Scholar] [CrossRef] [PubMed]
- WHO, Special edition: Proposed working definitions of SARS-CoV-2 Variants of Interest and Variants of Concern, Https://Www.Who.Int/Publications/m/Item/Covid-19-Weekly-Epidemiological-Update. (2021) 4. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20210225_weekly_epi_update_voc-special-edition.pdf (accessed on 8 June 2021).
- Globalization and Disasters: Issues of Public Health, State Capacity and Political Action on Jstor. Available online: https://www.jstor.org/stable/24358435?seq=1#metadata_info_tab_contents (accessed on 8 June 2021).
- GISAID—Submission Tracker Global. Available online: https://www.gisaid.org/test5/submission-tracker-global (accessed on 12 June 2021).
- Bruce, E.A.; Huang, M.-L.; Perchetti, G.A.; Tighe, S.; Laaguiby, P.; Hoffman, J.J.; Gerrard, D.L.; Nalla, A.K.; Wei, Y.; Greninger, A.L.; et al. Direct RT-qPCR detection of SARS-CoV-2 RNA from patient nasopharyngeal swabs without an RNA extraction step. PLOS Biol. 2020, 18, e3000896. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654–1665. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Won, J.; Lee, S.; Park, M.; Kim, T.Y.; Park, M.G.; Choi, B.Y.; Kim, D.; Chang, H.; Kim, V.N.; Lee, V.N.K.A.C.J. Development of a Laboratory-safe and Low-cost Detection Protocol for SARS-CoV-2 of the Coronavirus Disease 2019 (COVID-19). Exp. Neurobiol. 2020, 29, 107–119. [Google Scholar] [CrossRef]
- Guidance on the Inactivation or Removal of Select Agents and Toxins for Future Use Animal and Plant Health Inspection Service (APHIS) Division of Agricultural Select Agents and Toxins. Available online: https://www.selectagents.gov/compliance/guidance/inactivation/index.htm (accessed on 26 May 2021).
- Jung, Y.J.; Park, G.-S.; Moon, J.H.; Ku, K.; Beak, S.-H.; Kim, S.; Park, E.C.; Park, D.; Lee, J.-H.; Byeon, C.W.; et al. Comparative analysis of primer-probe sets for the laboratory confirmation of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Quick, J. nCoV-2019 sequencing protocol v2 (GunIt). Protocols.Io. 2020; p. 1. (accessed on 26 May 2021).
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. Biorxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; FerreiraI, A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; LumbI, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- PANGO lineages. Available online: https://cov-lineages.org/pangolin.html (accessed on 26 May 2021).
- Pater, A.A.; Bosmeny, M.S.; Parasrampuria, M.; Eddington, S.B.; Ovington, K.N.; Barkau, C.L.; White, A.A.; Metz, P.E.; Sylvain, R.J.; Chilamkurthy, R.; et al. High Throughput Nanopore Sequencing of SARS-CoV-2 Viral Genomes from Patient Samples. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Draw Venn Diagram. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn (accessed on 8 November 2021).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.2, a Graphical Viewer of Phylogenetic Trees; University of Edinburg: Edinburgh, UK, 2014. [Google Scholar]
- Population ages 15-64 (% of total population)|Data. Available online: https://data.worldbank.org/indicator/SP.POP.1564.TO.ZS?name_desc=false&view=chart (accessed on 13 June 2021).
- Population ages 0-14 (% of total population)|Data. Available online: https://data.worldbank.org/indicator/SP.POP.0014.TO.ZS?name_desc=false&view=chart (accessed on 13 June 2021).
- The World Bank. Population Ages 65 and Above (% of Total Population). 2017. Available online: https://data.worldbank.org/indicator/SP.POP.65UP.TO.ZS?name_desc=false&view=chart (accessed on 13 June 2021).
- Population, male (% of total population)|Data. Available online: https://data.worldbank.org/indicator/SP.POP.TOTL.MA.ZS?name_desc=false (accessed on 13 June 2021).
- World Health Organization. Tracking SARS-CoV-2 variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants (accessed on 12 June 2021).
- PANGO lineages B.1.466.2. Available online: https://cov-lineages.org/lineages/lineage_B.1.466.2.html (accessed on 14 June 2021).
- PANGO lineages B.1.470. Available online: https://cov-lineages.org/lineages/lineage_B.1.470.html (accessed on 14 June 2021).
- PANGO lineages B.1.1.398. Available online: https://cov-lineages.org/lineages/lineage_B.1.1.398.html (accessed on 14 June 2021).
- Cov-Lineages B.1.468. Available online: https://cov-lineages.org/lineage.html?lineage=B.1.468 (accessed on 7 November 2021).
- Cov-Lineages AY.23. Available online: https://cov-lineages.org/lineage.html?lineage=AY.23 (accessed on 7 November 2021).
- Cov-Lineages AY.24. Available online: https://cov-lineages.org/lineage.html?lineage=AY.24 (accessed on 7 November 2021).
- Kannan, S.R.; Spratt, A.N.; Quinn, T.P.; Heng, X.; Lorson, C.L.; Sönnerborg, A.; Byrareddy, S.N.; Singh, K. Infectivity of SARS-CoV-2: There Is Something More than D614G? J. Neuroimmune Pharmacol. 2020, 15, 574–577. [Google Scholar] [CrossRef]
- Ilmjärv, S.; Abdul, F.; Acosta-Gutiérrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.-H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mudi, S.R. Spike protein D614G and RdRp P323L: The SARS-CoV-2 mutations associated with severity of COVID-19. Genom. Informatics 2020, 18, e44. [Google Scholar] [CrossRef]
- Peckham, H.; de Gruijter, N.M.; Raine, C.; Radziszewska, A.; Ciurtin, C.; Wedderburn, L.R.; Rosser, E.C.; Webb, K.; Deakin, C.T. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Surendra, H.; Elyazar, I.R.; Djaafara, A.B.; Ekawati, L.L.; Saraswati, K.; Adrian, V.; Widyastuti; Oktavia, D.; Salama, N.; Lina, R.N.; et al. Clinical characteristics and mortality associated with COVID-19 in Jakarta, Indonesia: A hospital-based retrospective cohort study. Lancet Reg. Heal.-West. Pac. 2021, 9, 100108. [Google Scholar] [CrossRef]
- NICCRAT-Delivering Impact Partnerships—Asia Business Centre. Available online: https://blogs.nottingham.ac.uk/asiabusinesscentre/2021/05/25/niccrat-delivering-impact-partnerships (accessed on 26 June 2021).
- Nottingham-Indonesia Collaboration in Cancer Research and Training—Asia Business Centre. Available online: https://blogs.nottingham.ac.uk/asiabusinesscentre/2019/10/18/nottingham-indonesia-collaboration-in-cancer-research-and-training (accessed on 6 April 2021).
- Kementerian Kesehatan Republik Indonesia. Available online: https://www.kemkes.go.id/article/view/21010900005/pemerintah-tingkatkan-kapasitas-deteksi-genom-virus-sars-cov-2.html (accessed on 25 May 2021).
- COVID-19 Genomics UK Consortium. Available online: https://www.cogconsortium.uk/ (accessed on 6 April 2021).
- Priesemann, V.; Balling, R.; Brinkmann, M.M.; Ciesek, S.; Czypionka, T.; Eckerle, I.; Giordano, G.; Hanson, C.; Hel, Z.; Hotulainen, P.; et al. An action plan for pan-European defence against new SARS-CoV-2 variants. Lancet 2021, 397, 469–470. [Google Scholar] [CrossRef]
- Alene, M.; Yismaw, L.; Assemie, M.A.; Ketema, D.B.; Mengist, B.; Kassie, B.; Birhan, T.Y. Magnitude of asymptomatic COVID-19 cases throughout the course of infection: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0249090. [Google Scholar] [CrossRef]
- Pollock, A.M.; Lancaster, J. Asymptomatic transmission of covid-19. BMJ 2020, 371. [Google Scholar] [CrossRef]
- Subramanian, R.; He, Q.; Pascual, M. Quantifying asymptomatic infection and transmission of COVID-19 in New York City using observed cases, serology, and testing capacity. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Youth, Delta variant behind UK COVID surge|CIDRAP. Available online: https://www.cidrap.umn.edu/news-perspective/2021/06/youth-delta-variant-behind-uk-covid-surge (accessed on 12 November 2021).
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; Filipe, A.D.S.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187. [Google Scholar] [CrossRef]
- Recent circulation of multiple sublineages of B.1.466.2 in Jakarta, Indonesia · Issue #405 · cov-lineages/pango-designation · GitHub. Available online: https://github.com/cov-lineages/pango-designation/issues/405 (accessed on 29 March 2022).
- Omais, S.; Kharroubi, S.; Zaraket, H. No association between the SARS-CoV-2 variants and mortality rates in the Eastern Mediterranean Region. Gene 2021, 801, 145843. [Google Scholar] [CrossRef]
- Thomas, S. Mapping the Nonstructural Transmembrane Proteins of Severe Acute Respiratory Syndrome Coronavirus 2. J. Comput. Biol. 2021, 28, 909–921. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Mudgal, S.; Yadav, R.; Haokip, H.R.; Pandit, A.; Mary, Y.S. Genomic variation and point mutations analysis of Indian COVID-19 patient samples submitted in GISAID database. J. Indian Chem. Soc. 2021, 98, 100156. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
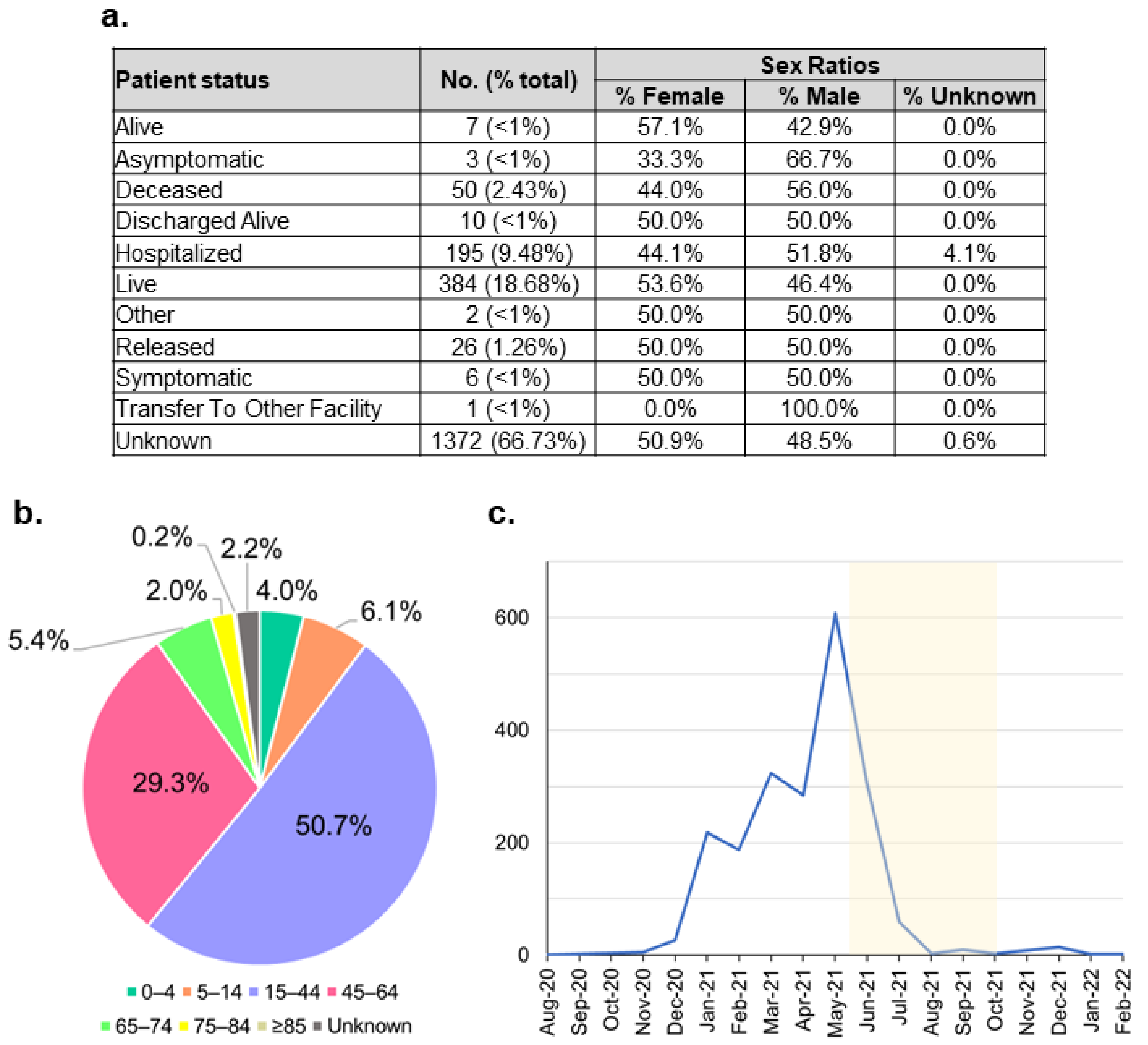
| Country | Patient Status | No. Sequenced Genomes | Recorded Metadata (% Sequenced Genome) | |||
|---|---|---|---|---|---|---|
| Alive | Asymptomatic | Deceased | Hospitalised | |||
| Japan | 114 | 62 | 16 | 9114 | 130,481 | 7.1% |
| South Korea | 25 | - | 9 | - | 14,118 | 0.2% |
| Singapore | - | - | - | 585 | 7979 | 7.3% |
| Philippines | 5090 | 1 | 221 | 20 | 7099 | 75.1% |
| Indonesia | 1689 | 24 | 159 | 764 | 7045 | 37.4% |
| Hong Kong | 601 | - | 26 | 323 | 4997 | 19.0% |
| Thailand | 32 | - | 42 | 10 | 3767 | 2.2% |
| Malaysia | 1876 | 1 | 231 | 128 | 3207 | 69.7% |
| China | 239 | 2 | 1 | 153 | 1347 | 29.3% |
| Cambodia | 1325 | - | - | - | 1165 | 113.7% |
| Vietnam | 312 | - | 6 | 68 | 569 | 67.8% |
| Timor-Leste | - | - | - | - | 357 | 0.0% |
| Taiwan | 25 | - | - | 74 | 245 | 40.4% |
| Myanmar | 2 | - | 1 | 27 | 75 | 40.0% |
| Brunei | - | - | - | - | 38 | 0.0% |
| Laos | - | - | - | - | 23 | 0.0% |
| Total | 11,330 | 90 | 712 | 11,266 | 182,512 | 12.8% |
| VoC | First Detected Case | Total Sequenced Genomes | ||
|---|---|---|---|---|
| Accession ID | Date | Pre-Outbreak (Until 1 June 2021) | Delta Outbreak (2 June–1 October 2021) | |
| B.1.1.7 (Alpha) | hCoV-19/Indonesia/SS-NIHRD-WGS00427/2021 | 5 January 2021 | 23 | 44 |
| B.1.351 (Beta) | hCoV-19/Indonesia/BA-NIHRD-WGS00725/2021 | 25 January 2021 | 4 | 18 |
| B.1.617.2 (Delta) and AY.xx | hCoV-19/Indonesia/JK-NIHRD-WGS00007/2021 | 7 January 2021 | 29 | 3035 |
| Pango Lineage | Other Name | Mutation Type (% Frequency) | Total Genomes | |||
|---|---|---|---|---|---|---|
| NSP12_P323L | NSP4_K35R | NSP6_L37F | Spike_D614G | |||
| AY.xx | Delta 2 | 99.9 | 1.8 | 2.7 | 100.0 | 2385 |
| B.1.1.7 | Alpha | 100.0 | 1.7 | 1.7 | 100.0 | 59 |
| B.1.351 | Beta | 80.0 | 10.0 | 10.0 | 100.0 | 10 |
| B.1.466.2 | - | 100.0 | 2.8 | 5.2 | 100.0 | 1530 |
| B.1.470 | - | 98.9 | 0.4 | 3.0 | 100.0 | 527 |
| B.1.617.2 | Delta (original) | 100.0 | 6.8 | 4.1 | 100.0 | 74 |
| Pango Lineage | Top 10 Mutation Type (% Frequency) | |
|---|---|---|
| B.1.466.2 |
|
|
| B.1.470 |
|
|
| Variant Type (n = 4253) | Age | t-Test p-Value | |||
|---|---|---|---|---|---|
| Minimum | Median | Mean | Maximum | ||
| Indonesian | 0.5 | 38 | 38.9 | 92 | <0.001 |
| Delta (and sub-lineages) | 0.4 | 35 | 36.4 | 91 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cahyani, I.; Putro, E.W.; Ridwanuloh, A.M.; Wibowo, S.; Hariyatun, H.; Syahputra, G.; Akbariani, G.; Utomo, A.R.; Ilyas, M.; Loose, M.; et al. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements. Viruses 2022, 14, 778. https://doi.org/10.3390/v14040778
Cahyani I, Putro EW, Ridwanuloh AM, Wibowo S, Hariyatun H, Syahputra G, Akbariani G, Utomo AR, Ilyas M, Loose M, et al. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements. Viruses. 2022; 14(4):778. https://doi.org/10.3390/v14040778
Chicago/Turabian StyleCahyani, Inswasti, Eko W. Putro, Asep M. Ridwanuloh, Satrio Wibowo, Hariyatun Hariyatun, Gita Syahputra, Gilang Akbariani, Ahmad R. Utomo, Mohammad Ilyas, Matthew Loose, and et al. 2022. "Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements" Viruses 14, no. 4: 778. https://doi.org/10.3390/v14040778