
Multiple Novel Human Norovirus Recombinants Identified in Wastewater in Pretoria, South Africa by Next-Generation Sequencing

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Recovery
2.2. Nucleic Acid Extraction and Virus Detection
2.3. Molecular Typing of Noroviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Primer/Probe | Sequence (5′–3′) * | Polarity | Location | Amplification Conditions | |
| Norovirus GI | QNIF4 a | CGCTGGATGCGNTTCCAT | + | 5291–5308 † | 50 °C, 20 min 95 °C, 5 min ×45 cycles 95 °C, 15 s 55 °C, 30 s 65 °C, 30 s | |
| NV1LCR b | CCTTAGACGCCATCATCATTTAC | − | 5354–5376 † | |||
| NVGG1 b | FAM-TGGACAGGAGAYCGCRATCT-TAMRA | + | 5321–5340 † | |||
| Norovirus GII | QNIF2 c | ATGTTCAGRTGGATGAGRTTCTCWGA | + | 5012–5037 * | 50 °C, 20 min 95 °C, 5 min ×45 cycles 95 °C, 15 s 60 °C, 30 s 65 °C, 30 s | |
| COG2R d | TCGACGCCATCTTCATTCACA | − | 5080–5100 * | |||
| QNIFS c | FAM -AGCACGTGGGAGGGCGATCG-TAMRA | + | 5042–5061 * | |||
| Mengovirus | Mengo110F e | GCGGGTCCTGCCGAAAGT | + | 110–127 ❄ | 50 °C, 20 min 95 °C, 5 min ×45 cycles 95 °C, 15 s 60 °C, 30 s 65 °C, 30 s | |
| Mengo209R e | GAAGTAACATATAGACAGACGCACAC | − | 245–270 ❄ | |||
| Mengo147 e | MGB-ATCACATTACTGGCCGAAGC-TAMRA | + | 208–227 ❄ | |||
| Target | Primer | Sequence (5′–3′) * | Polarity | Location | Amplification conditions | |
| 1st round | 2nd round | |||||
| Norovirus GI BC region | JV12Y f | ATACCACTATGATGCAGAYTA | + | 4552–4572 † | ×40 cycles 95 °C, 15 s 50 °C, 30 s 72 °C, 1 min 72 °C, 5 min | ×40 cycles 95 °C, 15 s 50 °C, 30 s 72 °C, 45 s 72 °C, 5 min |
| MON432Tx g | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGGACICGYGGICCYAAYCA | + | 5093–5112 † | |||
| G1SKRTx h | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCAACCCARCCATTRTACA | − | 5653–5671 † | |||
| Norovirus GII BC region | JV12Y f | ATACCACTATGATGCAGAYTA | + | 4279–4299 * | ×40 cycles 95 °C, 15 s 55 °C, 30 s 72 °C, 1 min 72 °C, 5 min | ×40 cycles 95 °C, 15 s 55 °C, 30 s 72 °C, 45 s 72 °C, 5 min |
| MON431Tx g | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGGACIAGRGGICCYAAYCA | + | 4821–4840 * | |||
| G2SKRTx h | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCRCCNGCATRHCCRTTRTACAT | − | 5367–5389 * | |||
| Norovirus GI BC region for viruses with P7 P-type | JV12Y f | ATACCACTATGATGCAGAYTA | + | 4552–4572 † | ×40 cycles 95 °C, 15 s 50 °C, 30 s 72 °C, 1 min 72 °C, 5 min | ×40 cycles 95 °C, 15 s 50 °C, 30 s 72 °C, 1 min 72 °C, 5 min |
| GIP7F i | CTGATGTGGAATTTGACCCAATAAAG | + | 4890–4915 † | |||
| G1SKR h | CCAACCCARCCATTRTACA | − | 5653–5671 † | |||
2.4. DNA Library Preparation and Illumina Sequencing
2.5. NGS Data Analysis
2.6. Sanger Sequencing
2.7. Norovirus Phylogeny and Recombination Analysis
2.8. Statistical Analyses
3. Results
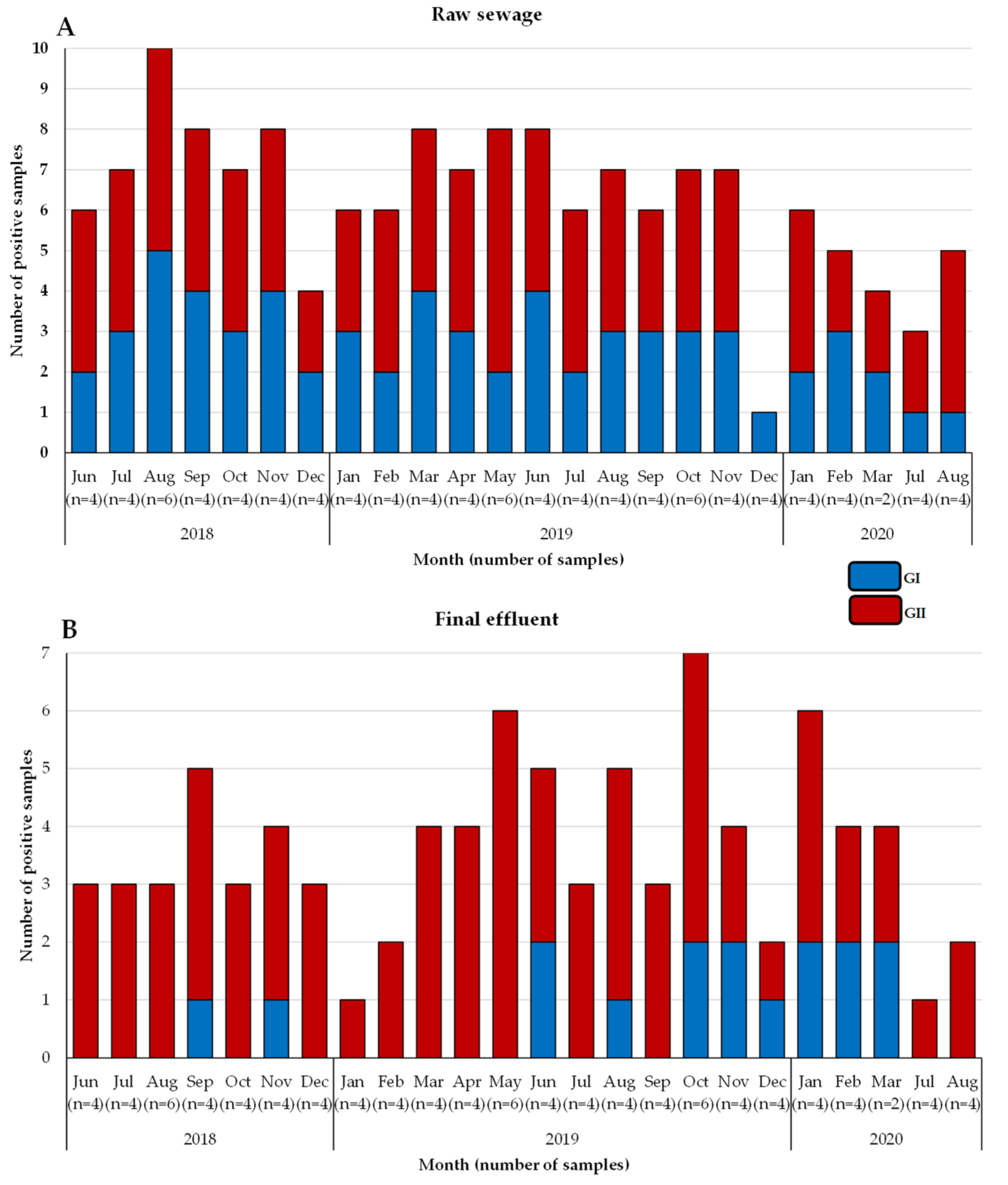
3.1. Detection of Noroviruses
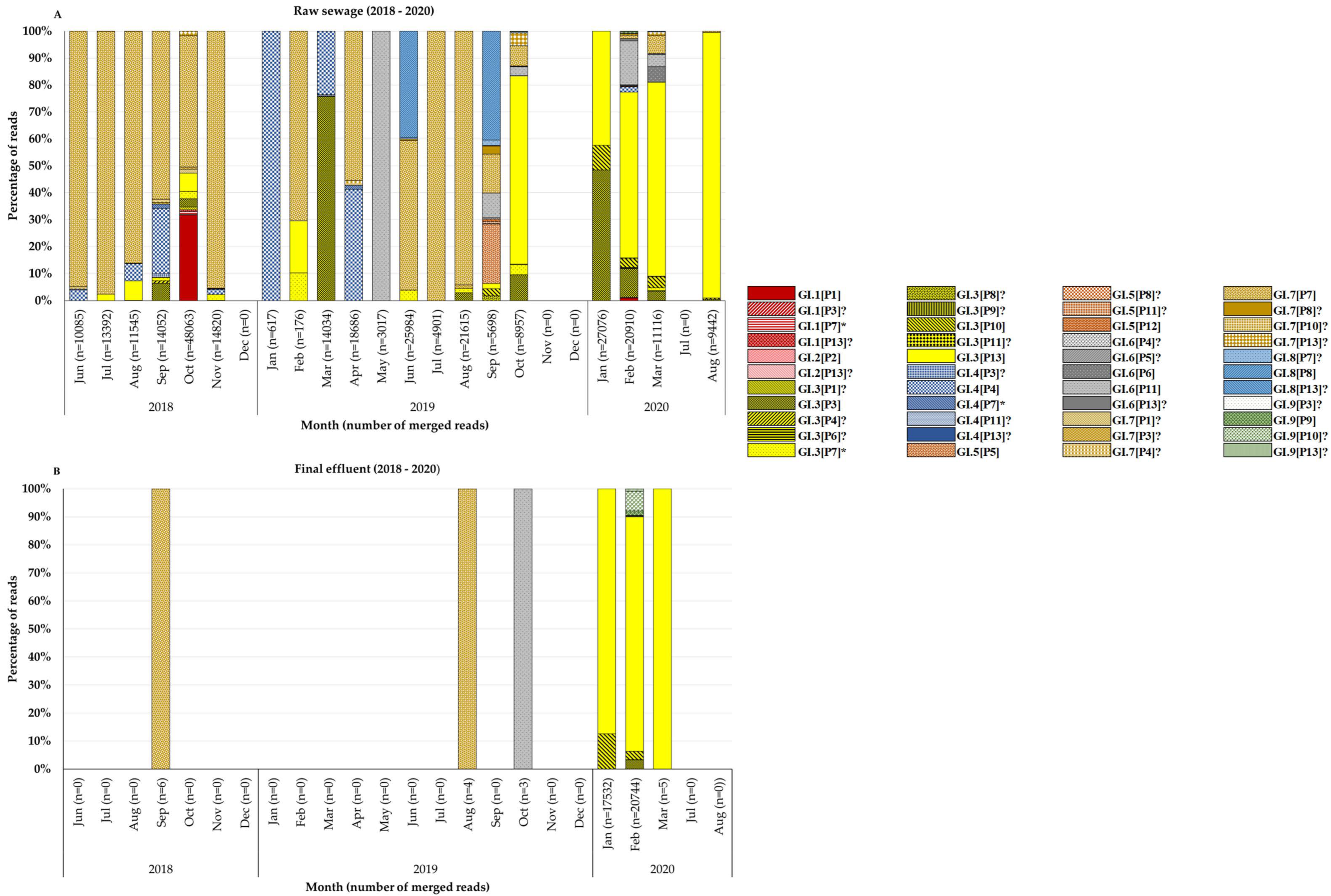
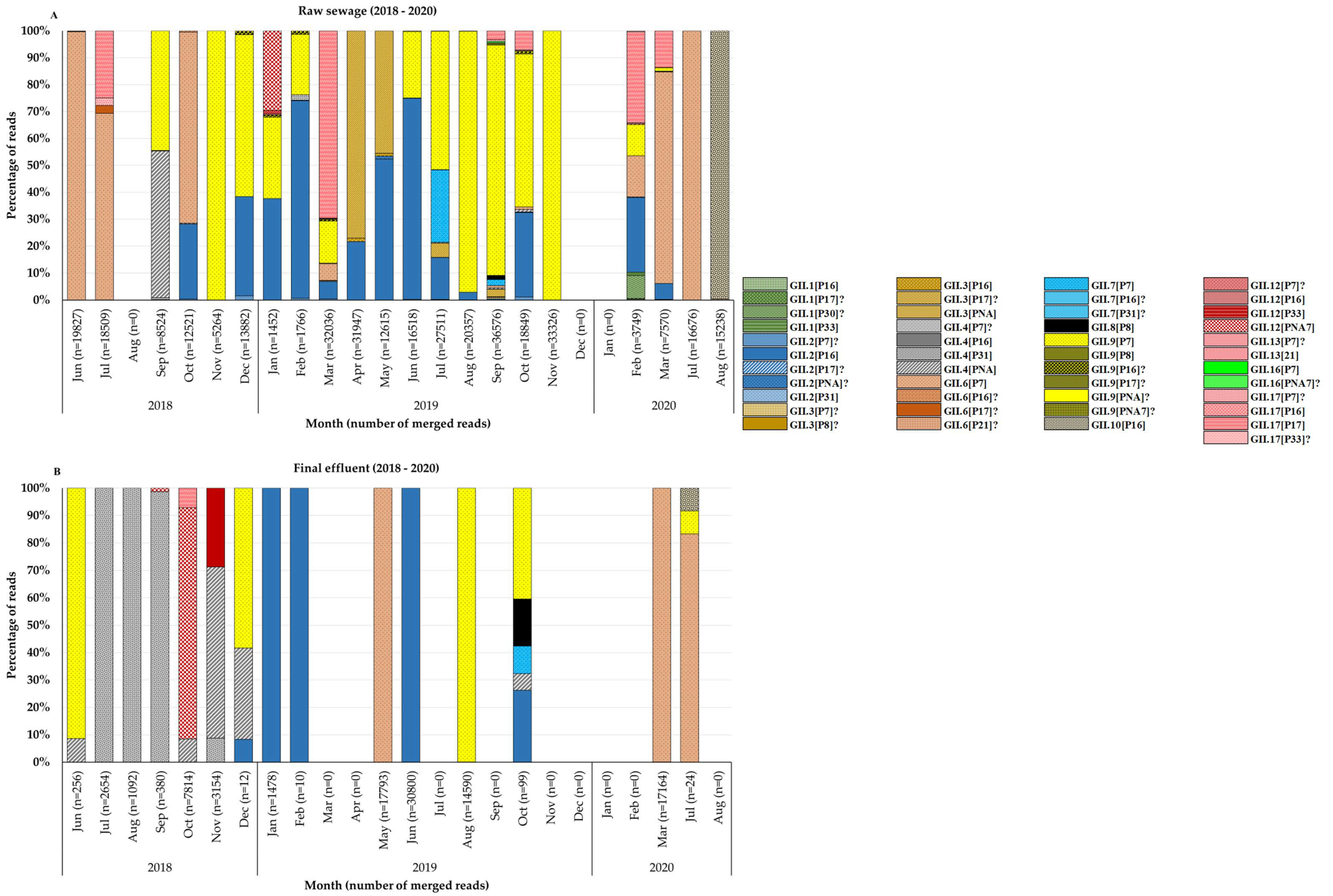
3.2. Molecular Typing of Noroviruses
3.3. Recombination Analysis
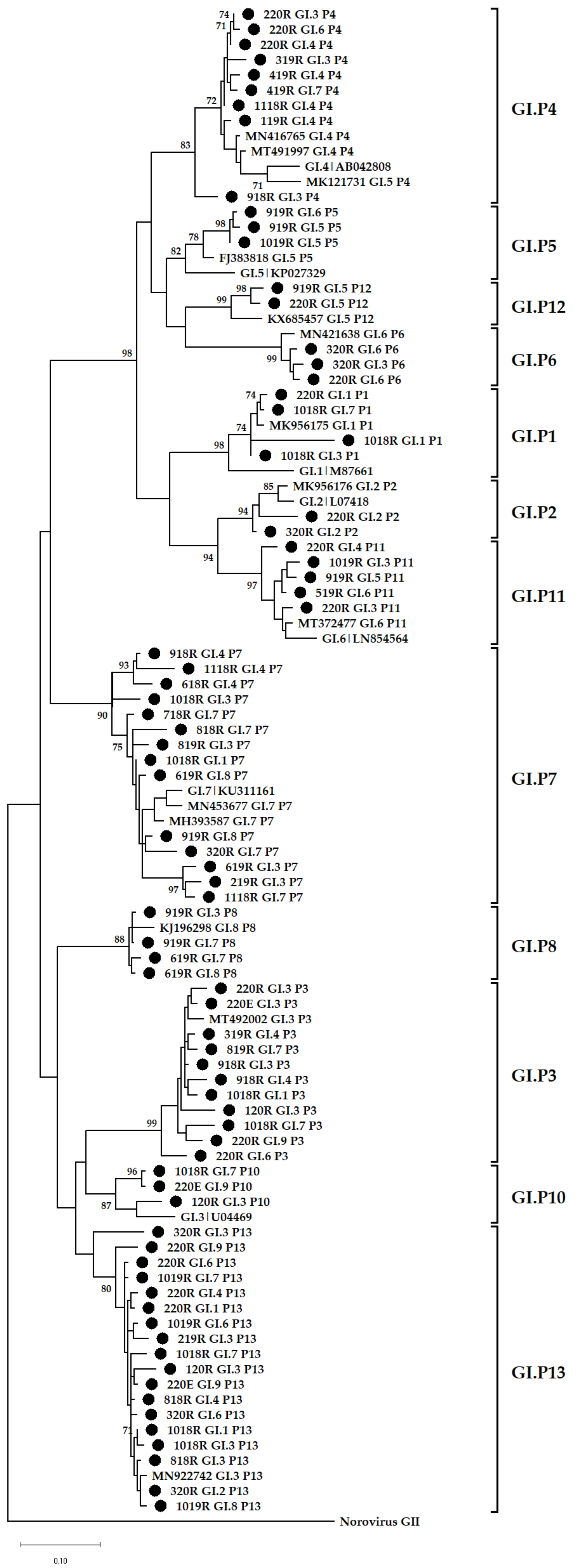
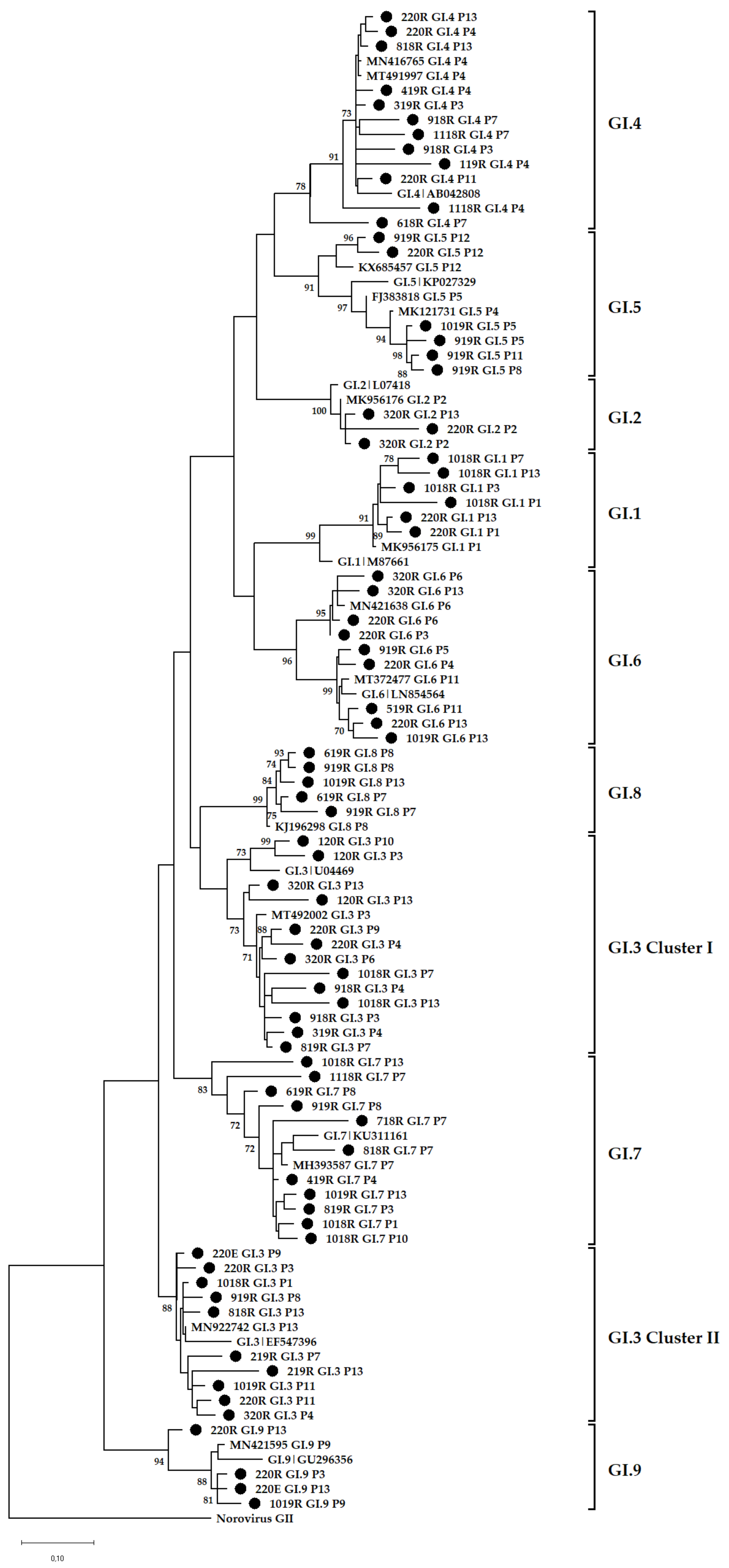
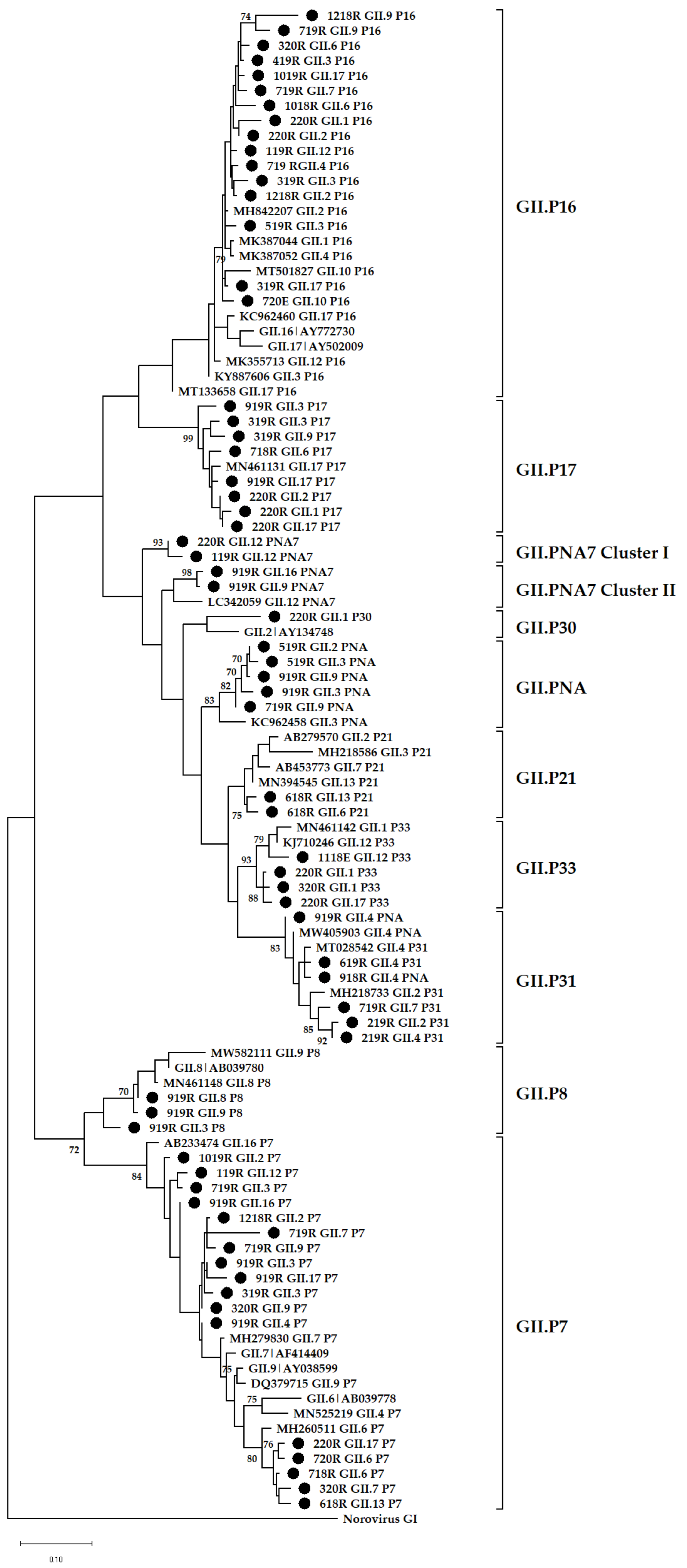
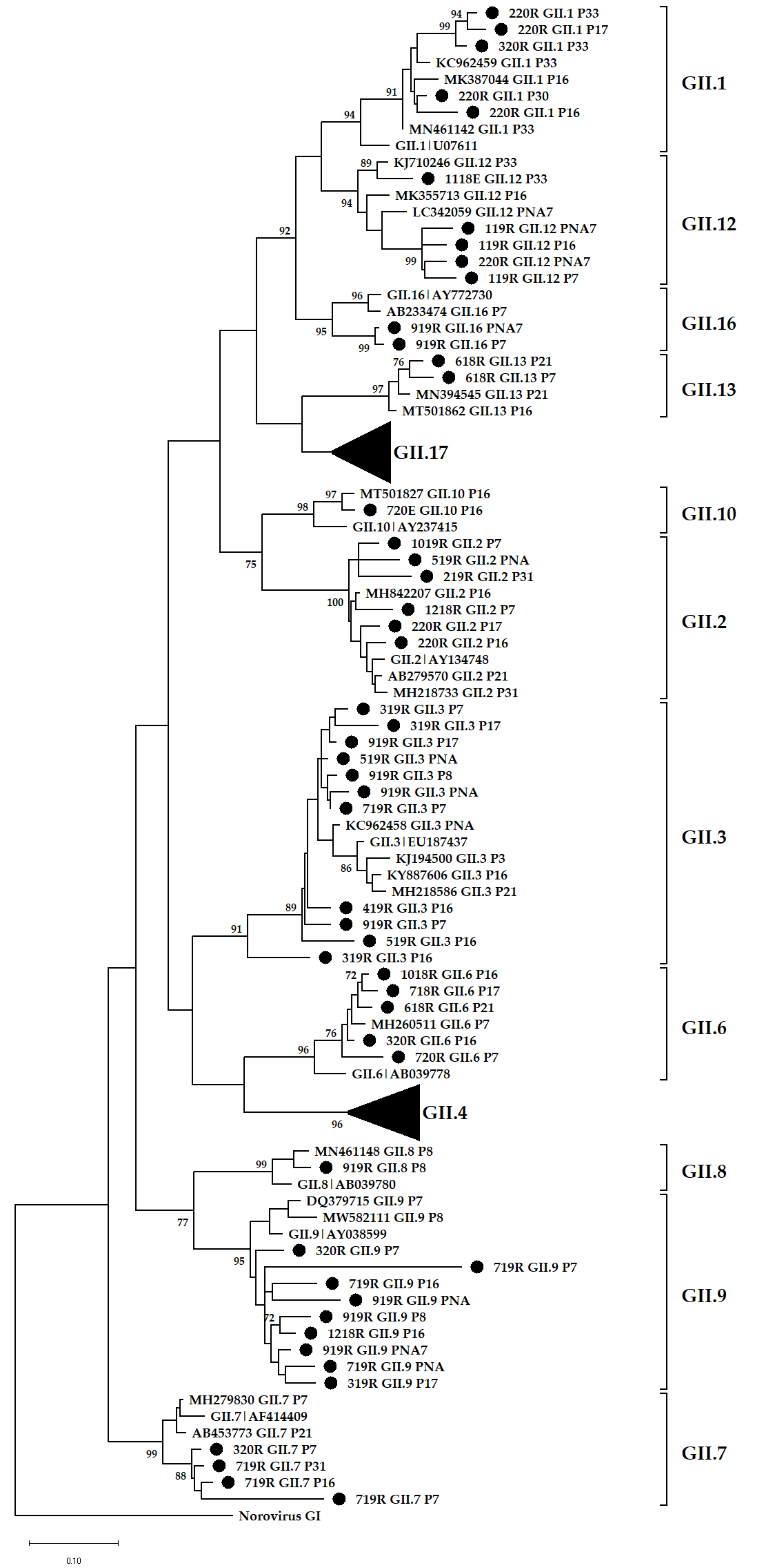
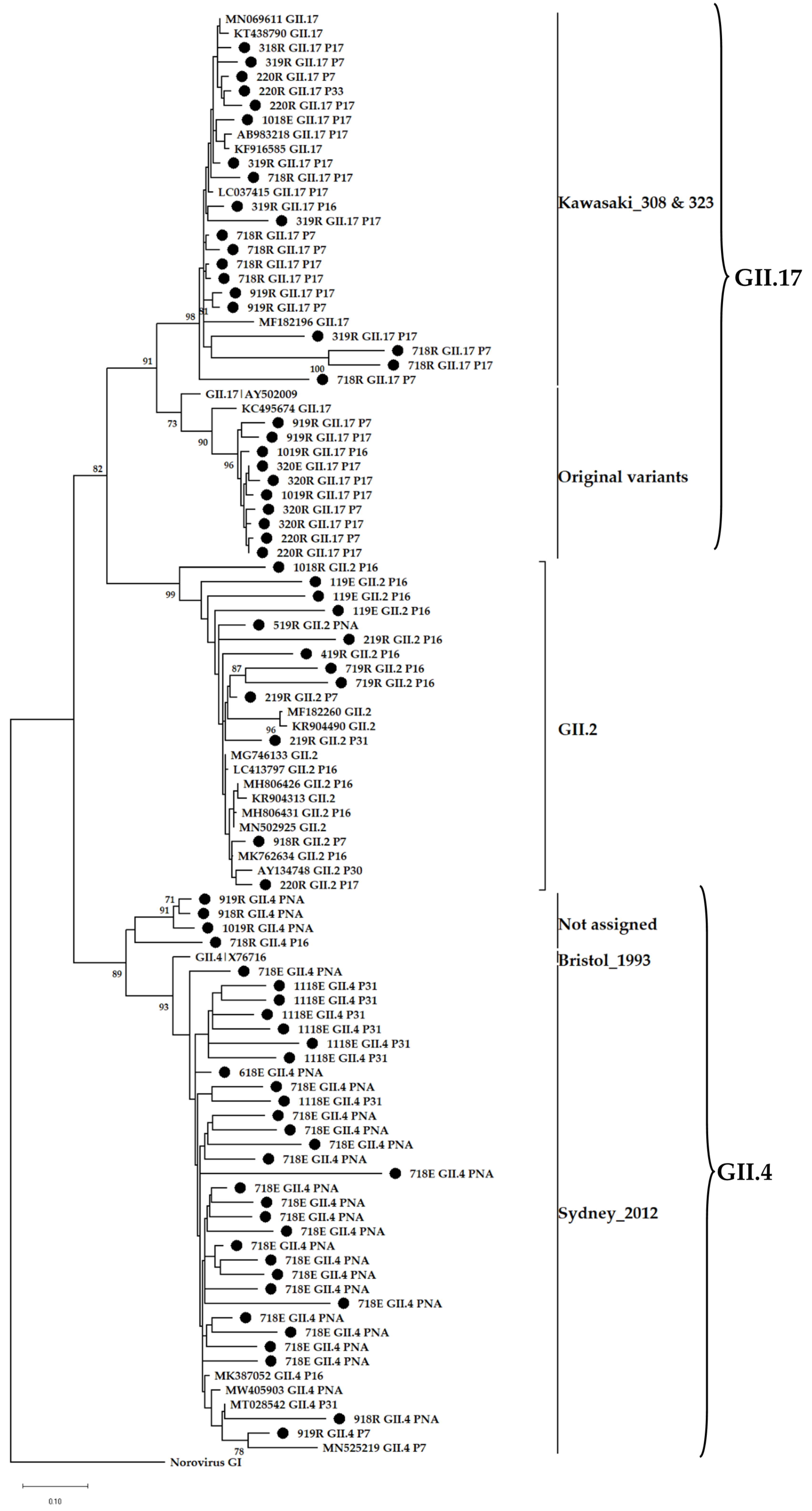
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopman, B.A.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The vast and varied global burden of norovirus: Prospects for prevention and control. PLoS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef]
- Burke, R.M.; Hall, A.J. Global Burden of Norovirus. In Norovirus; Melhem, N., Ed.; Springer: Cham, Switzerland, 2019; pp. 1–29. [Google Scholar]
- Huys, A.; Grau, K.R.; Karst, S.M. Development of oral rotavirus and norovirus vaccines. In Mucosal Vaccines, 2nd ed.; Hiroshi, K., David, W.P., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 699–712. [Google Scholar]
- Riera-Montes, M.; O’ryan, M.; Verstraeten, T. Norovirus and rotavirus disease severity in children: Systematic review and meta-analysis. Pediatr. Infect. Dis. J. 2018, 37, 501–505. [Google Scholar] [CrossRef]
- Lindsay, L.; Wolter, J.; De Coster, I.; Van Damme, P.; Verstraeten, T. A decade of norovirus disease risk among older adults in upper-middle- and high-income countries: A systematic review. BMC Infect. Dis. 2015, 15, 425. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Shah, D.; Breuer, J. Viral gastrointestinal infections and norovirus genotypes in a paediatric UK hospital, 2014–2015. J. Clin. Virol. 2016, 84, 1–6. [Google Scholar] [CrossRef]
- Kampmeier, S.; Pettke, A.; Kossow, A.; Willems, S.; Mellmann, A. Norovirus infections in a tertiary care centre−individual cases do not necessarily lead to an outbreak. J. Clin. Virol. 2016, 84, 39–41. [Google Scholar] [CrossRef]
- Lopman, B.; Gastanaduy, P.; Park, G.W.; Hall, A.J.; Parashar, U.D.; Vinjé, J. Environmental transmission of norovirus gastroenteritis. Curr. Opin. Virol. 2012, 2, 96–102. [Google Scholar] [CrossRef]
- Marsh, Z.; Shah, M.P.; Wikswo, M.E.; Barclay, L.; Kisselburgh, H.; Kambhampati, A.; Cannon, J.L.; Parashar, U.D.; Vinjé, J.; Hall, A.J. Epidemiology of foodborne norovirus outbreaks–United States, 2009–2015. Food Safety 2018, 6, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Malik, Y.S.; Kobayashi, N. Therapeutics and immunoprophylaxis against noroviruses and rotaviruses: The past, present, and future. Curr. Drug Metab. 2018, 19, 170–191. [Google Scholar] [CrossRef]
- Riddle, M.S.; Walker, R.I. Status of vaccine research and development for norovirus. Vaccine 2016, 34, 2895–2899. [Google Scholar] [CrossRef] [Green Version]
- Salim, O.; Lambden, P.R.; Clarke, I.N. Norovirus. In The Springer Index of Viruses; Tidona, C., Darai, G., Eds.; Springer: New York, NY, USA, 2011; pp. 245–250. [Google Scholar]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.A. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Silva, L.D.; Junior, E.C.S.; Bandeira, R.S.; Rodrigues, E.A.M.; Lucena, M.S.S.; Costa ST, P.; Gabbay, Y.B. Molecular epidemiology and temporal evolution of norovirus associated with acute gastroenteritis in Amazonas state, Brazil. BMC Infect. Dis. 2018, 18, 147. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.; Sosnovtsev, S.V.; Green, K.Y. Static and evolving norovirus genotypes: Implications for epidemiology and immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Koo, E.S.; Kim, M.S.; Choi, J.D.; Shin, Y.; Jeong, Y.S. Re-emergence of a GII.4 norovirus Sydney 2012 variant equipped with GII.P16 RdRp and its predominance over novel variants of GII.17 in South Korea in 2016. Food Environ. Virol. 2017, 9, 168–178. [Google Scholar] [CrossRef]
- Barreira, D.M.P.G.; Fumian, T.M.; Tonini, M.A.L.; Volpini, L.P.B.; Santos, R.P.; Ribeiro, A.L.C.; Leite, J.P.G.; Souza, M.T.B.D.M.E.; Brasil, P.; da Cunha, D.C.; et al. Detection and molecular characterization of the novel recombinant norovirus GII.P16-GII.4 Sydney in southeastern Brazil in 2016. PLoS ONE 2017, 12, e0189504. [Google Scholar] [CrossRef] [Green Version]
- Mabasa, V.V.; Meno, K.D.; Taylor, M.B.; Mans, J. Environmental surveillance for noroviruses in selected South African wastewaters 2015–2016: Emergence of the novel GII.17. Food Environ. Virol. 2018, 10, 16–28. [Google Scholar] [CrossRef]
- Fumian, T.M.; Fioretti, J.M.; Lun, J.H.; Dos Santos, I.A.; White, P.A.; Miagostovich, M.P. Detection of norovirus epidemic genotypes in raw sewage using next generation sequencing. Environ. Int. 2019, 123, 282–291. [Google Scholar] [CrossRef]
- La Rosa, G.; Della Libera, S.; Iaconelli, M.; Proroga, Y.; De Medici, D.; Martella, V.; Suffredini, E. Detection of norovirus GII.17 Kawasaki 2014 in shellfish, marine water and underwater sewage discharges in Italy. Food Environ. Virol. 2017, 9, 326–333. [Google Scholar] [CrossRef]
- Murray, T.Y.; Mans, J.; Taylor, M.B. Human calicivirus diversity in wastewater in South Africa. J. Appl. Microbiol. 2013, 114, 1843–1853. [Google Scholar] [CrossRef] [Green Version]
- Cantalupo, P.G.; Pipas, J.M. Detecting viral sequences in NGS data. Curr. Opin. Virol. 2019, 39, 41–48. [Google Scholar] [CrossRef]
- Makhura, D. Profile: City of Tshwane. Provincial Gazette. Available online: https://www.cogta.gov.za/ddm/wp-content/uploads/2020/08/2nd-Take_Final_DistrictProfile_TSHWANE2306-1-002 (accessed on 7 January 2020).
- Bofill-Mas, S.; Hundesa, A.; Calgua, B.; Rusiñol, M.; de Motes, C.M.; Girones, R. Cost-effective method for microbial source tracking using specific human and animal viruses. J. Vis. Exp. 2011, 3, e2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachida, S.; Taylor, M.B. Potentially infectious novel hepatitis A virus strains detected in selected treated wastewater discharge sources, South Africa. Viruses 2020, 12, 1468. [Google Scholar] [CrossRef] [PubMed]
- ISO 15216-1:2017; Microbiology of the Dood Chain—Horizontal Method for Determination of Hepatitis A Virus and Norovirus Using Real-Time RT-PCR—Part 1: Method for Quantification. ISO: Bengaluru, Bengaluru, 2017.
- Stals, A.; Mathijs, E.; Baert, L.; Botteldoorn, N.; Denayer, S.; Mauroy, A.; Scipioni, A.; Daube, G.; Dierick, K.; Herman, L. Molecular detection and genotyping of noroviruses. Food Environ. Virol. 2012, 4, 153–167. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.K.; Le Saux, J.C.; Parnaudeau, S.; Pommepuy, M.; Elimelech, M.; Le Guyader, F.S. Evaluation of removal of noroviruses during wastewater treatment, using real-time reverse transcription-PCR: Different behaviors of genogroups I and II. Appl. Environ. Microbiol. 2007, 73, 7891–7897. [Google Scholar] [CrossRef] [Green Version]
- Svraka, S.; Duizer, E.; Vennema, H.; de Bruin, E.; van der Veer, B.; Dorresteijn, B.; Koopmans, M. Etiological role of viruses in outbreaks of acute gastroenteritis in The Netherlands from 1994 through 2005. J. Clin. Microbiol. 2007, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Loisy, F.; Atmar, R.L.; Guillon, P.; Le Cann, P.; Pommepuy, M.; Le Guyader, F.S. Real-time RT-PCR for norovirus screening in shellfish. J. Virol. Methods 2005, 123, 1–7. [Google Scholar] [CrossRef]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Pintó, R.M.; Costafreda, M.I.; Bosch, A. Risk assessment in shellfish-borne outbreaks of hepatitis A. Appl. Environ. Microbiol. 2009, 75, 7350–7355. [Google Scholar] [CrossRef] [Green Version]
- Vennema, H.; De Bruin, E.; Koopmans, M. Rational optimization of generic primers used for Norwalk-like virus detection by reverse transcriptase polymerase chain reaction. J. Clin. Virol. 2002, 25, 233–235. [Google Scholar] [CrossRef]
- Beuret, C. A simple method for isolation of enteric viruses (noroviruses and enteroviruses) in water. J. Virol. Methods 2003, 107, 1–8. [Google Scholar] [CrossRef]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J. Virol. Methods 2002, 100, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. Map: A fast, Accurate, Splice-Aware Aligner (No. LBNL-7065E); Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.; Penaranda, S.; Oberste, M.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Tubatsi, G.; Kebaabetswe, L.P. Detection of enteric viruses from wastewater and river water in Botswana. Food Environ. Virol. 2022, 14, 157–169. [Google Scholar] [CrossRef]
- Kiulia, N.; Mans, J.; Mwenda, J.; Taylor, M. Norovirus GII.17 predominates in selected surface water sources in Kenya. Food Environ. Virol. 2014, 6, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittigul, L.; Rupprom, K.; Che-Arsae, M.; Pombubpa, K.; Thongprachum, A.; Hayakawa, S.; Ushijima, H. Occurrence of noroviruses in recycled water and sewage sludge: Emergence of recombinant norovirus strains. J. Appl. Microbiol. 2019, 126, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Neyvaldt, J.; Enache, L.; Sikora, P.; Mattsson, A.; Johansson, A.; Lindh, M.; Bergstedt, O.; Norder, H. Variations among viruses in influent water and effluent water at a wastewater plant over one year as assessed by quantitative PCR and metagenomics. Appl. Environ. Microbiol. 2020, 86, e02020–e02073. [Google Scholar] [CrossRef] [PubMed]
- Sano, D.; Amarasiri, M.; Hata, A.; Watanabe, T.; Katayama, H. Risk management of viral infectious diseases in wastewater reclamation and reuse. Environ. Int. 2016, 91, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromeans, T.; Park, G.W.; Costantini, V.; Lee, D.; Wang, Q.; Farkas, T.; Lee, A.; Vinjé, J. Comprehensive comparison of cultivable norovirus surrogates in response to different inactivation and disinfection treatments. Appl. Environ. Microbiol. 2014, 80, 5743–5751. [Google Scholar] [CrossRef] [Green Version]
- Kendra, J.A.; Tohma, K.; Parra, G.I. Global and regional circulation trends of norovirus genotypes and recombinants, 1995–2019: A comprehensive review of sequences from public databases. Rev. Med. Virol. 2022, 32, e2354. [Google Scholar] [CrossRef]
- Cantelli, C.P.; da Silva, M.F.M.; Fumian, T.M.; da Cunha, D.C.; de Andrade, J.d.S.R.; Malta, F.C.; Fialho, A.M.; de Moraes, M.T.B.; Brasil, P.; Miagostovich, M.P.; et al. High genetic diversity of noroviruses in children from a community-based study in Rio de Janeiro, Brazil, 2014–2018. Arch. Virol. 2019, 164, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.-C.; Hsu, J.-K.; Hu, S.-C.; Wu, C.-Y.; Wang, Y.-C.; Lin, J.-H. Molecular epidemiology of GI.3 norovirus outbreaks from acute gastroenteritis surveillance system in Taiwan, 2015–2019. BioMed Res. Int. 2020, 2020, 4707538. [Google Scholar] [CrossRef]
- Esteves, A.; Nordgren, J.; Tavares, C.; Fortes, F.; Dimbu, R.; Saraiva, N.; Istrate, C. Genetic diversity of norovirus in children under 5 years of age with acute gastroenteritis from Angola. Epidemiol. Infect. 2018, 146, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Lysén, M.; Thorhagen, M.; Brytting, M.; Hjertqvist, M.; Andersson, Y.; Hedlund, K.-O. Genetic diversity among food-borne and waterborne norovirus strains causing outbreaks in Sweden. J. Clin. Microbiol. 2009, 47, 2411–2418. [Google Scholar] [CrossRef]
- Nenonen, N.P.; Hannoun, C.; Larsson, C.U.; Bergström, T. Marked genomic diversity of norovirus genogroup I strains in a waterborne outbreak. Appl. Environ. Microbiol. 2012, 78, 1846–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruggink, L.; Catton, M.; Marshall, J. A norovirus intervariant GII.4 recombinant in Victoria, Australia, June 2016: The next epidemic variant? Eurosurveillance 2016, 21, 30353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, J.H.; Hewitt, J.; Sitabkhan, A.; Eden, J.-S.; Tuipulotu, D.E.; Netzler, N.E.; Morrell, L.; Merif, J.; Jones, R.; Huang, B. Emerging recombinant noroviruses identified by clinical and wastewater screening. Emerg. Microbes Infect. 2018, 7, 50. [Google Scholar] [CrossRef]
- Cannon, J.L.; Bonifacio, J.; Bucardo, F.; Buesa, J.; Bruggink, L.; Chan, M.C.-W.; Fumian, T.M.; Giri, S.; Gonzalez, M.D.; Hewitt, J. Global trends in norovirus genotype distribution among children with acute gastroenteritis. Emerg. Infect. Dis. 2021, 27, 1438. [Google Scholar] [CrossRef] [PubMed]
- Mabasa, V.V. Characterisation and histo-blood group antigen binding profiles of South African norovirus genotype II strains. Master’s Thesis, University of Pretoria, Pretoria, South Africa, August 2017. [Google Scholar]
- Parra, G.I. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019, 5, vez048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.C.; Lee, N.; Hung, T.-N.; Kwok, K.; Cheung, K.; Tin, E.K.; Lai, R.W.; Nelson EA, S.; Leung, T.F.; Chan, P.K. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 2015, 6, 10061. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sun, L.; Fang, L.; Yang, F.; Mo, Y.; Lao, J.; Zheng, H.; Tan, X.; Lin, H.; Rutherford, S. Gastroenteritis outbreaks caused by norovirus GII.17, Guangdong Province, China, 2014–2015. Emerg. Infect. Dis. 2015, 21, 1240. [Google Scholar] [CrossRef]
- Matsushima, Y.; Ishikawa, M.; Shimizu, T.; Komane, A.; Kasuo, S.; Shinohara, M.; Nagasawa, K.; Kimura, H.; Ryo, A.; Okabe, N. Genetic analyses of GII. 17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Eurosurveillance 2015, 20, 21173. [Google Scholar] [CrossRef] [Green Version]
- Parra, G.I.; Green, K.Y. Genome of emerging norovirus GII.17, United States, 2014. Emerg. Infect. Dis. 2015, 21, 1477. [Google Scholar] [CrossRef]
- Medici, M.; Tummolo, F.; Calderaro, A.; Chironna, M.; Giammanco, G.; De Grazia, S.; Arcangeletti, M.; De Conto, F.; Chezzi, C.; Martella, V. Identification of the novel Kawasaki 2014 GII.17 human norovirus strain in Italy, 2015. Eurosurveillance 2015, 20, 30010. [Google Scholar] [CrossRef]
- Dinu, S.; Nagy, M.; Negru, D.G.; Popovici, E.D.; Zota, L.; Oprișan, G. Molecular identification of emergent GII.P17-GII.17 norovirus genotype, Romania, 2015. Eurosurveillance 2016, 21, 30141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabbaraju, K.; Wong, A.A.; Tipples, G.A.; Pang, X.-L. Emergence of a novel recombinant norovirus GII.P16-GII.12 strain causing gastroenteritis, Alberta, Canada. Emerg. Infect. Dis. 2019, 25, 1556. [Google Scholar] [CrossRef] [PubMed]
- Fumian, T.M.; Ferreira, F.C.; de Andrade, J.d.S.R.; Canal, N.; Silva Gomes, G.; Teixeira, L.B.; Miagostovich, M.P. Norovirus foodborne outbreak associated with the consumption of ice pop, Southern Brazil, 2020. Food Environ. Virol. 2021, 13, 553–559. [Google Scholar] [CrossRef]
- Mans, J.; Murray, T.Y.; Nadan, S.; Netshikweta, R.; Page, N.A.; Taylor, M.B. Norovirus diversity in children with gastroenteritis in South Africa from 2009 to 2013: GII. 4 variants and recombinant strains predominate. Epidemiol. Infect. 2016, 144, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, N.A.; Groome, M.J.; Nadan, S.; Netshikweta, R.; Keddy, K.H.; Poonsamy, B.; Cohen, C. Norovirus epidemiology in South African children <5 years hospitalised for diarrhoeal illness between 2009 and 2013. Epidemiol. Infect. 2017, 145, 1942–1952. [Google Scholar] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabasa, V.V.; van Zyl, W.B.; Ismail, A.; Allam, M.; Taylor, M.B.; Mans, J. Multiple Novel Human Norovirus Recombinants Identified in Wastewater in Pretoria, South Africa by Next-Generation Sequencing. Viruses 2022, 14, 2732. https://doi.org/10.3390/v14122732
Mabasa VV, van Zyl WB, Ismail A, Allam M, Taylor MB, Mans J. Multiple Novel Human Norovirus Recombinants Identified in Wastewater in Pretoria, South Africa by Next-Generation Sequencing. Viruses. 2022; 14(12):2732. https://doi.org/10.3390/v14122732
Chicago/Turabian StyleMabasa, Victor Vusi, Walda Brenda van Zyl, Arshad Ismail, Mushal Allam, Maureen Beatrice Taylor, and Janet Mans. 2022. "Multiple Novel Human Norovirus Recombinants Identified in Wastewater in Pretoria, South Africa by Next-Generation Sequencing" Viruses 14, no. 12: 2732. https://doi.org/10.3390/v14122732