
A Novel Aeromonas popoffii Phage AerP_220 Proposed to Be a Member of a New Tolavirus Genus in the Autographiviridae Family

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Host Strain Isolation, Culture Conditions and Susceptibility Testing
2.2. Phage Isolation and Propagation
2.3. Phage Plaques and Phage Particles Morphology
2.4. Biological Properties of the AerP_220 Phage and Host Range Study
2.5. AerP_220 DNA Purification and Complete Genome Sequencing
2.6. Genome Analysis
2.7. Phylogenetic Analysis of Phage Proteins
3. Results and Discussion
3.1. Bacterial Host Isolation and Susceptibility Testing
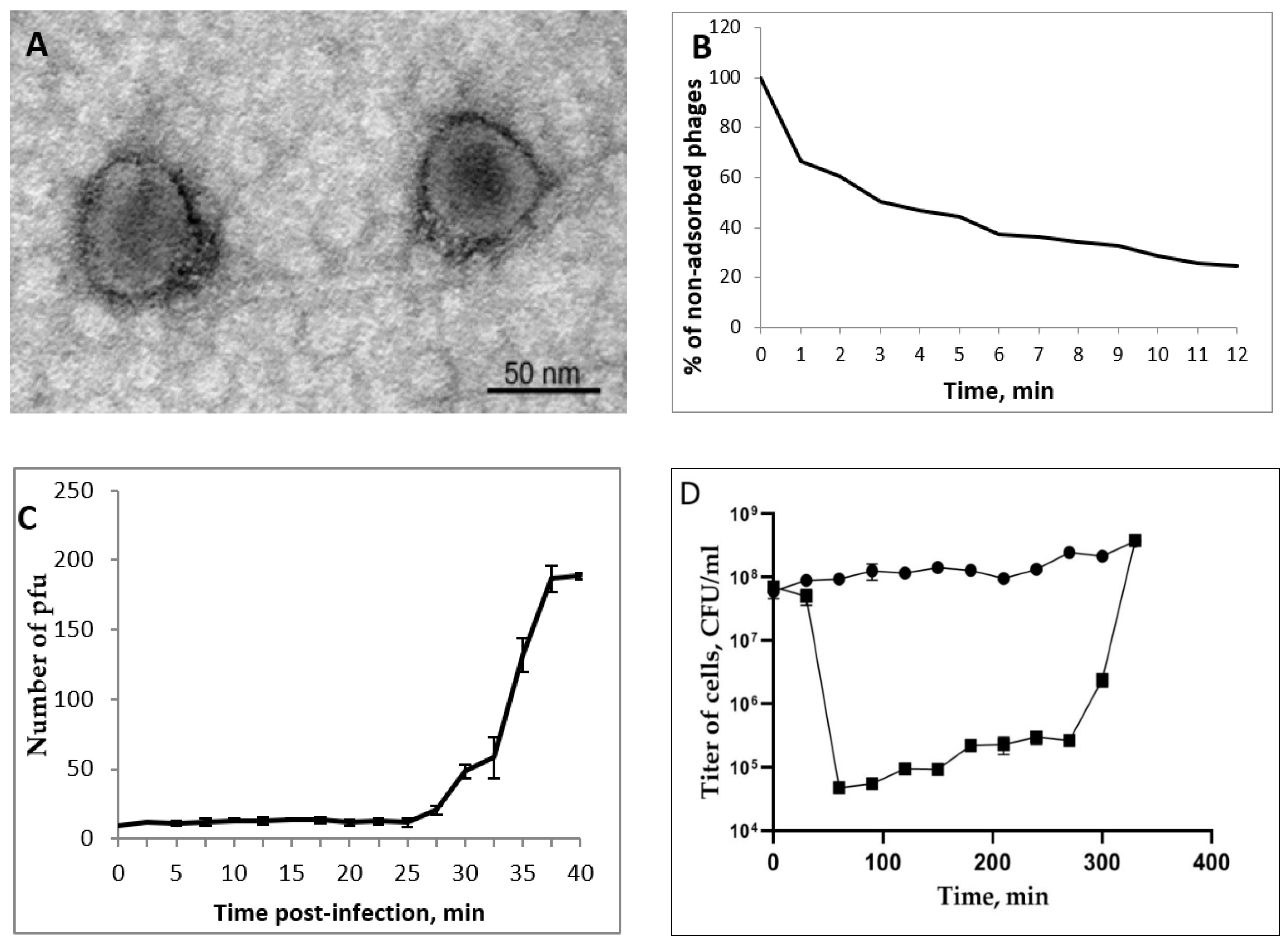
3.2. AerP_220 Plaques and Phage Morphology
3.3. AerP_220 Biological Properties and Host Range
3.4. Genome Characteristics
3.5. Comparative Analysis of the AerP_220 Genome with Other Genomes
3.6. Phylogenetic Analysis of AerP_220 Proteins
3.7. Protein-Based Similarity Networks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernández-Bravo, A.; Figueras, M.J. An update on the genus Aeromonas: Taxonomy, epidemiology, and pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoel, S.; Vadstein, O.; Jakobsen, A.N. The significance of mesophilic Aeromonas spp. in minimally processed ready-to-eat seafood. Microorganisms 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.L.; Shaw, J.G. Aeromonas spp. clinical microbiology and disease. J. Infect. 2011, 62, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, S.F.; Anjaria, D.; Mohr, A.; Livingston, D.H. Necrotizing fasciitis and sepsis caused by Aeromonas hydrophila after crush injury of the lower extremity. Surg. Infect. 2008, 9, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.R.; Clark, M.E.; Bowen, D.K.; Uratake, D.; Ayubi, F.; Katras, T.; Kellicut, D.C. Necrotizing soft tissue infection following a peripheral bypass. Ann. Vasc. Surg. 2015, 29, 843.e17–843.e22. [Google Scholar] [CrossRef]
- Verriere, B.; Sabatier, B.; Carbonnelle, E.; Mainardi, J.L.; Prognon, P.; Whitaker, I.; Lantieri, L.; Hivelin, M. Medicinal leech therapy and Aeromonas spp. infection. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.T.; Fernández-Bravo, A.; Sanchis, M.; Mayayo, E.; Figueras, M.J.; Charette, S.J. Investigation of the virulence and genomics of Aeromonas salmonicida strains isolated from human patients. Infect. Genet. Evol. 2019, 68, 1–9. [Google Scholar] [CrossRef]
- Sinclair, H.A.; Heney, C.; Sidjabat, H.E.; George, N.M.; Bergh, H.; Anuj, S.N.; Nimmo, G.R.; Paterson, D.L. Genotypic and phenotypic identification of Aeromonas species and CphA-mediated carbapenem resistance in Queensland, Australia. Diagn. Microbiol. Infect. Dis. 2016, 85, 98–101. [Google Scholar] [CrossRef]
- Rosso, F.; Cedano, J.A.; Parra-Lara, L.G.; Sanz, A.M.; Toala, A.; Velez, J.F.; Hormaza, M.P.; Moncada, P.A.; Correa, A. Emerging carbapenem-resistant Aeromonas spp. infections in Cali, Colombia. Braz. J. Infect. Dis. 2019, 23, 336–342. [Google Scholar] [CrossRef]
- Fauzi, N.N.F.N.M.; Hamdan, R.H.; Mohamed, M.; Ismail, A.; Mat Zin, A.A.; Mohamad, N.F.A. Prevalence, antibiotic susceptibility, and presence of drug resistance genes in Aeromonas spp. isolated from freshwater fish in Kelantan and Terengganu states, Malaysia. Vet. World 2021, 14, 2064–2072. [Google Scholar] [CrossRef]
- Patel, K.M.; Svestka, M.; Sinkin, J.; Ruff, P., 4th. Ciprofloxacin-resistant Aeromonas hydrophila infection following leech therapy: A case report and review of the literature. J. Plast. Reconstr. Aesthetic Surg. 2013, 66, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Huys, G.; Kämpfer, P.; Altwegg, M.; Kersters, I.; Lamb, A.; Coopman, R.; Lüthy-Hottenstein, J.; Vancanneyt, M.; Janssen, P.; Kersters, K. Aeromonas popoffii sp. nov., a mesophilic bacterium isolated from drinking water production plants and reservoirs. Int. J. Syst. Bacteriol. 1997, 47, 1165–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, L.; Figueras, M.J.; Chacón, M.R.; Vila, J.; Marco, F.; Martinez-Murcia, A.J.; Guarro, J. Potential virulence and antimicrobial susceptibility of Aeromonas popoffii recovered from freshwater and seawater. FEMS Immunol. Med. Microbiol. 2002, 32, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.T.; Bollet, C.; Tercian, S.; Drancourt, M.; Raoult, D. Aeromonas popoffii urinary tract infection. J. Clin. Microbiol. 2004, 42, 5427–5428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Son, J.S.; Choresca, C.H.; Shin, S.P.; Han, J.E.; Jun, J.W.; Kang, D.H.; Oh, C.; Heo, S.J.; Park, S.C. Complete genome sequence of bacteriophage phiAS7, a T7-like virus that infects Aeromonas salmonicida subsp. salmonicida. J. Virol. 2012, 86, 2894–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.T.; Paquet, V.E.; Bernatchez, A.; Tremblay, D.M.; Moineau, S.; Charette, S.J. Characterization and diversity of phages infecting Aeromonas salmonicida subsp. salmonicida. Sci. Rep. 2017, 7, 7054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, N.; Paquet, V.E.; Chehreghani, M.; Moineau, S.; Charette, S.J. Phage cocktail development against Aeromonas salmonicida subsp. salmonicida strains is compromised by a prophage. Viruses 2021, 13, 2241. [Google Scholar] [CrossRef]
- Park, S.Y.; Han, J.E.; Kwon, H.; Park, S.C.; Kim, J.H. Recent insights into Aeromonas salmonicida and its bacteriophages in aquaculture: A comprehensive review. J. Microbiol. Biotechnol. 2020, 30, 1443–1457. [Google Scholar] [CrossRef]
- Bujak, K.; Decewicz, P.; Kitowicz, M.; Radlinska, M. Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family. Viruses 2022, 14, 1016. [Google Scholar] [CrossRef]
- Anand, T.; Bera, B.C.; Virmani, N.; Vaid, R.K.; Vashisth, M.; Tripathi, B.N. Isolation and characterization of a novel, T7-like phage against Aeromonas veronii. Virus Genes 2018, 54, 160–164. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D. Bacteriophage λ and its vectors. In Molecular Cloning, 3rd ed.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001; Volume 1, pp. 2.25–2.106. [Google Scholar]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; pp. 69–76. [Google Scholar] [CrossRef]
- Kropinski, A.M. Measurement of the rate of attachment of bacteriophage to cells. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; Volume 1, pp. 151–155. [Google Scholar] [CrossRef]
- Pajunen, M.; Kiljunen, S.; Skurnik, M. Bacteriophage phiYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 2000, 182, 5114–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, Y.J.; Lee, Y.R.; Jung, H.H.; Lee, J.; Ko, G.; Cho, Y.H. Antibacterial Efficacy of phages against Pseudomonas aeruginosa infections in mice and Drosophila melanogaster. Antimicrob. Agents Chemother. 2009, 53, 2469–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, E. Phage host range and efficiency of plating. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; Volume 1, pp. 141–149. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of staphylococcal phage K: A new lineage of Myoviridae infecting gram-positive bacteria with a low GC content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Moriyama, E.N. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.W. Phage classification and characterization. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; Volume 1, pp. 127–140. [Google Scholar] [CrossRef]
- Merino, S.; Camprubí, S.; Tomás, J.M. Characterization of an O-antigen bacteriophage from Aeromonas hydrophila. Can. J. Microbiol. 1992, 38, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Paquet, V.E.; Vincent, A.T.; Moineau, S.; Charette, S.J. Beyond the A-layer: Adsorption of lipopolysaccharides and characterization of bacteriophage-insensitive mutants of Aeromonas salmonicida subsp. salmonicida. Mol. Microbiol. 2019, 112, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Leduc, G.R.; Paquet, V.E.; Vincent, A.T.; Charette, S.J. Characterization of bacteriophage T7-Ah reveals its lytic activity against a subset of both mesophilic and psychrophilic Aeromonas salmonicida strains. Arch. Virol. 2021, 166, 521–533. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, S.; Wang, D.; Zhao, J.; Xu, L.; Liu, H.; Lu, T.; Mou, Z. Genomic characterization of a novel virulent phage infecting the Aeromonas hydrophila isolated from rainbow trout (Oncorhynchus mykiss). Virus Res. 2019, 273, 197764. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Islam, M.S.; Raz, A.; Liu, Y.; Elbassiony, K.R.A.; Dong, X.; Zhou, P.; Zhou, Y.; Li, J. Complete genome sequence of Aeromonas phage ZPAH7 with halo zones, isolated in China. Microbiol. Resour. Announc. 2019, 8, e01678-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin, W.J.; Lin, N.T.; Tseng, Y.H.; Weng, S.F. Genomic characterization of the novel Aeromonas hydrophila phage Ahp1 suggests the derivation of a new subgroup from phiKMV-Like family. PLoS ONE 2016, 11, e0162060. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; Yu, M.S.; Tseng, T.T.; Lin, L.C. Molecular characterization of Ahp2, a lytic bacteriophage of Aeromonas hydrophila. Viruses 2021, 13, 477. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Homologous Genes | Proposed Tolavirinae Subfamily, n = 7 | Colwellvirinae, n = 14 | Moulineuxvirinae, n = 25 | |
|---|---|---|---|---|
| Proposed Tolavirus Genus, n = 2 | Gajwadongvirus Genus, n = 3 | |||
| Between members of the genera | 41 | 41 | ||
| Between members of the proposed subfamily | 26 | 18 | 17 | |
| Between members of the subfamilies | 13 | |||
| Proposed Tolavirinae Subfamily, n = 7 | Colwellvirinae Subfamily, n = 14 | Moulineuxvirinae Subfamily, n = 25 | |
|---|---|---|---|
| proposed Tolavirinae | 0.26 < SG < 0.85 | 0.16 < SG < 0.21 | 0.14 < SG < 0.2 |
| Colwellvirinae | 0.16 < SG < 0.21 | 0.28 < SG < 0.97 | 0.14 < SG < 0.22 |
| Moulineuxvirinae | 0.14 < SG < 0.2 | 0.14 < SG < 0.22 | 0.3 < SG < 0.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morozova, V.; Kozlova, Y.; Jdeed, G.; Tikunov, A.; Ushakova, T.; Bardasheva, A.; Zhirakovskaia, E.; Poletaeva, Y.; Ryabchikova, E.; Tikunova, N.V. A Novel Aeromonas popoffii Phage AerP_220 Proposed to Be a Member of a New Tolavirus Genus in the Autographiviridae Family. Viruses 2022, 14, 2733. https://doi.org/10.3390/v14122733
Morozova V, Kozlova Y, Jdeed G, Tikunov A, Ushakova T, Bardasheva A, Zhirakovskaia E, Poletaeva Y, Ryabchikova E, Tikunova NV. A Novel Aeromonas popoffii Phage AerP_220 Proposed to Be a Member of a New Tolavirus Genus in the Autographiviridae Family. Viruses. 2022; 14(12):2733. https://doi.org/10.3390/v14122733
Chicago/Turabian StyleMorozova, Vera, Yuliya Kozlova, Ghadeer Jdeed, Artem Tikunov, Tatyana Ushakova, Alevtina Bardasheva, Elena Zhirakovskaia, Yuliya Poletaeva, Elena Ryabchikova, and Nina V. Tikunova. 2022. "A Novel Aeromonas popoffii Phage AerP_220 Proposed to Be a Member of a New Tolavirus Genus in the Autographiviridae Family" Viruses 14, no. 12: 2733. https://doi.org/10.3390/v14122733