
The Evolutionary Landscape of SARS-CoV-2 Variant B.1.1.519 and Its Clinical Impact in Mexico City

 ,
,  , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Methods
2.1. Sample Preparation
2.1.1. Participants
2.1.2. Sample Collection
2.1.3. SARS-CoV-2 RNA Extraction
2.1.4. RT-qPCR
2.2. Sequencing
2.2.1. Oxford Nanopore-Sequencing
2.2.2. Oxford-Nanopore Raw Data Processing and Sequencing Data Quality Assessment
2.2.3. Illumina Sequencing
2.2.4. Illumina Raw Data Processing and Sequencing Data Quality Assessment
2.3. Genomic Data Collection
2.4. Effective Reproduction Number Estimation for Variants B.1.1.222 and B.1.1.519
2.5. Haplotype Analysis for Variant B.1.1.519
2.6. Phylogenetic Analysis
2.7. Clinical Data Collection
2.8. Statistical Analysis
3. Results
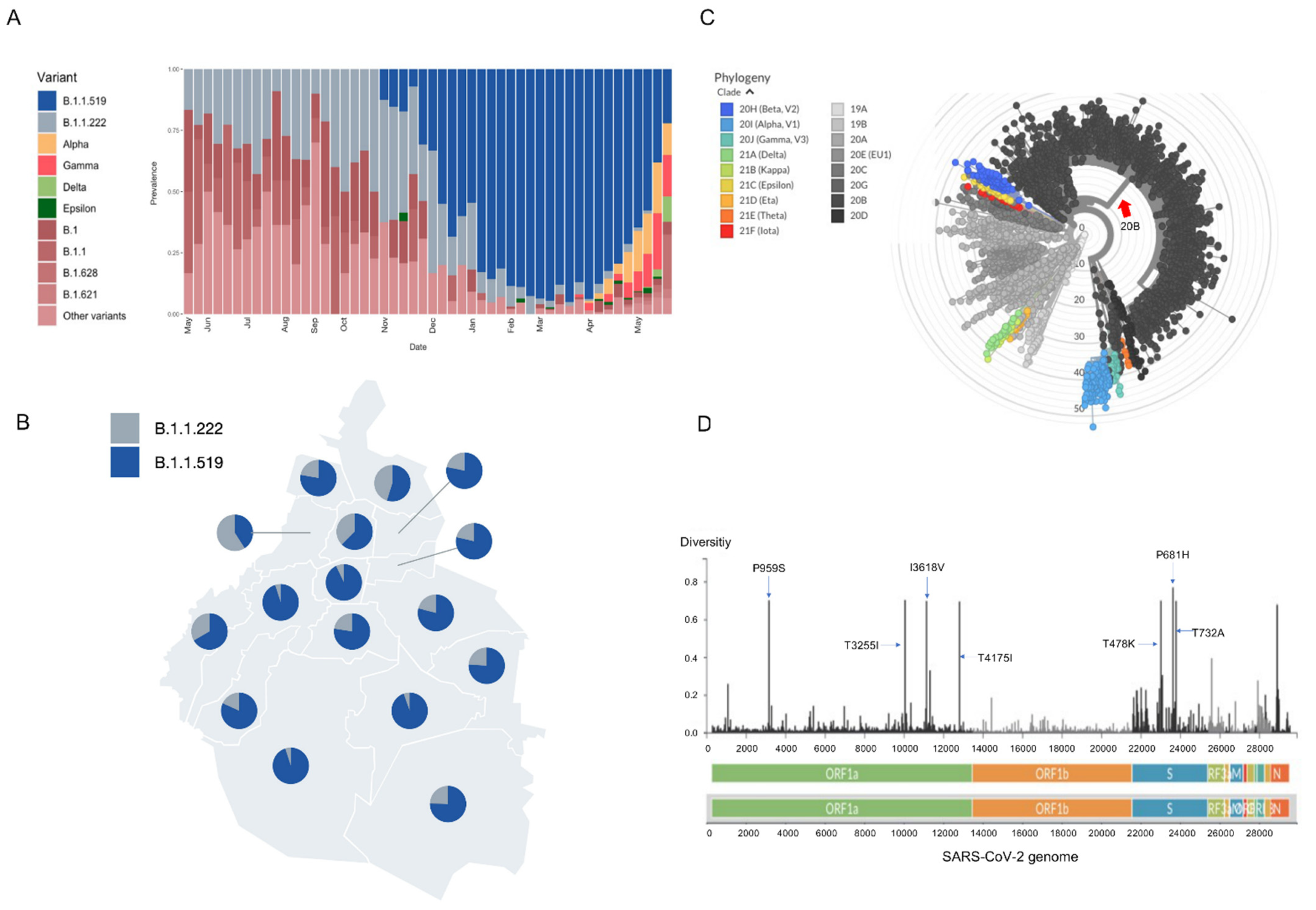
3.1. Identification of Variant B.1.1.519 in Mexico City
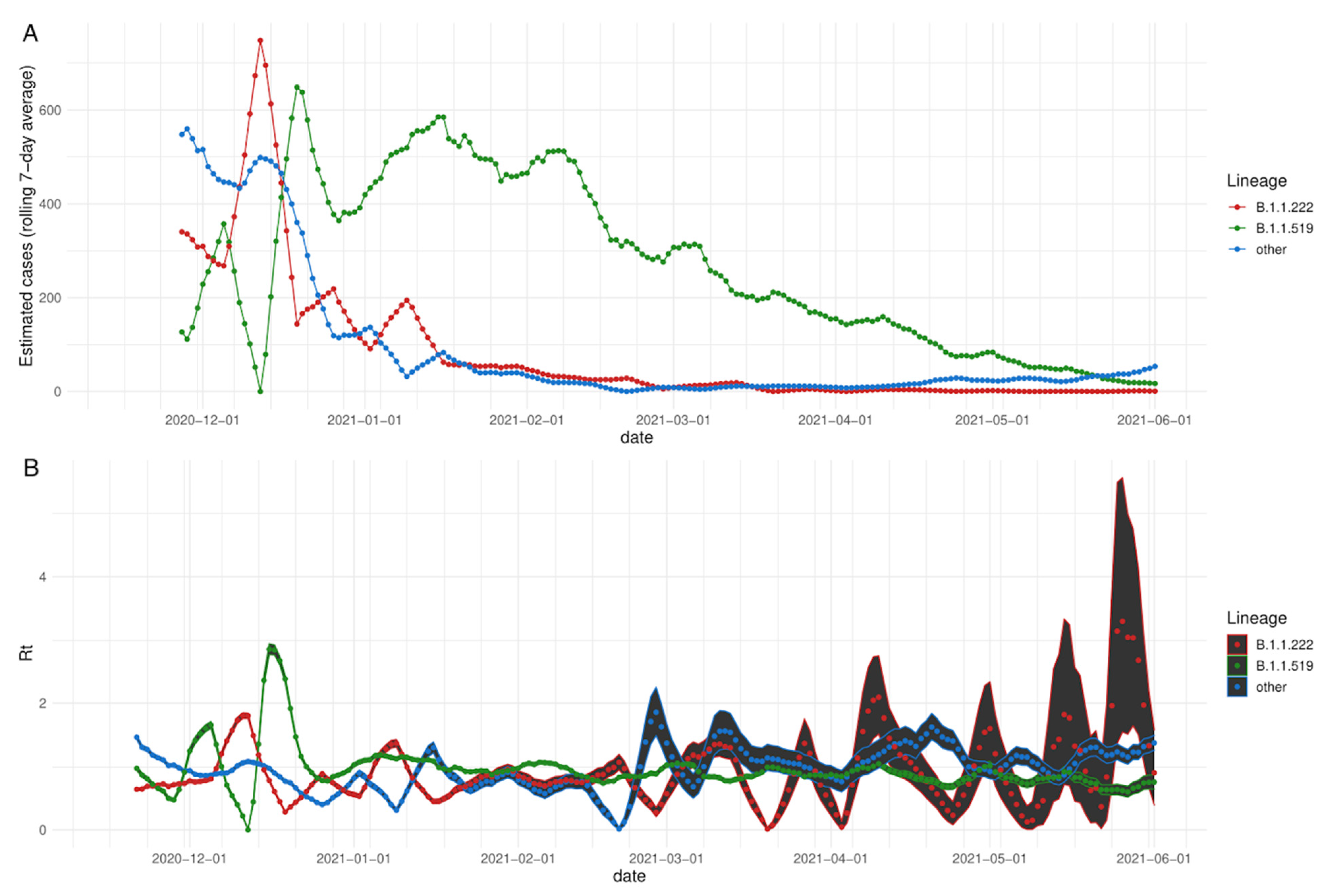
3.2. Rt: Effective Reproduction Number
3.3. Genomic Findings
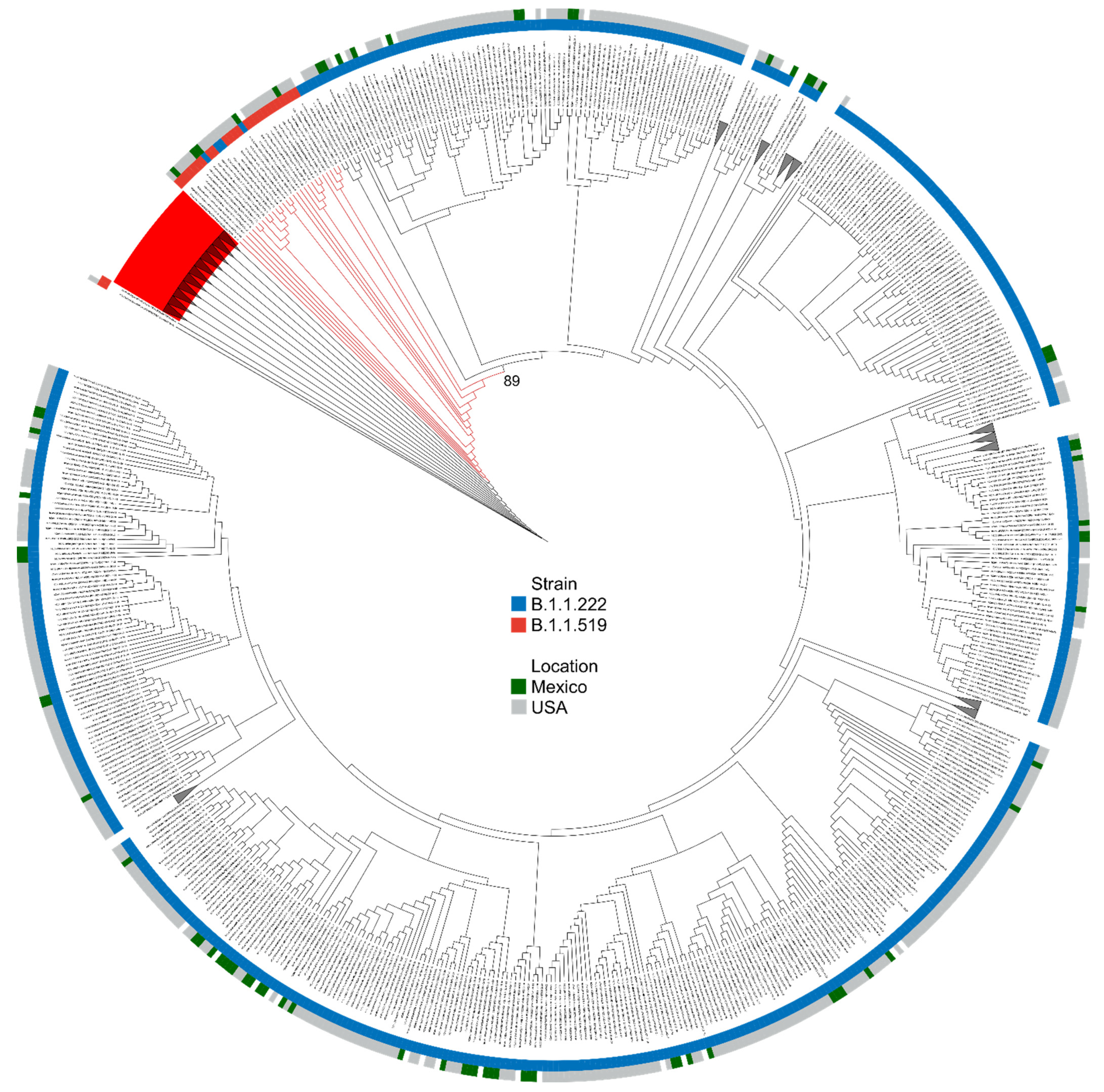
3.3.1. Phylogenetic Analysis
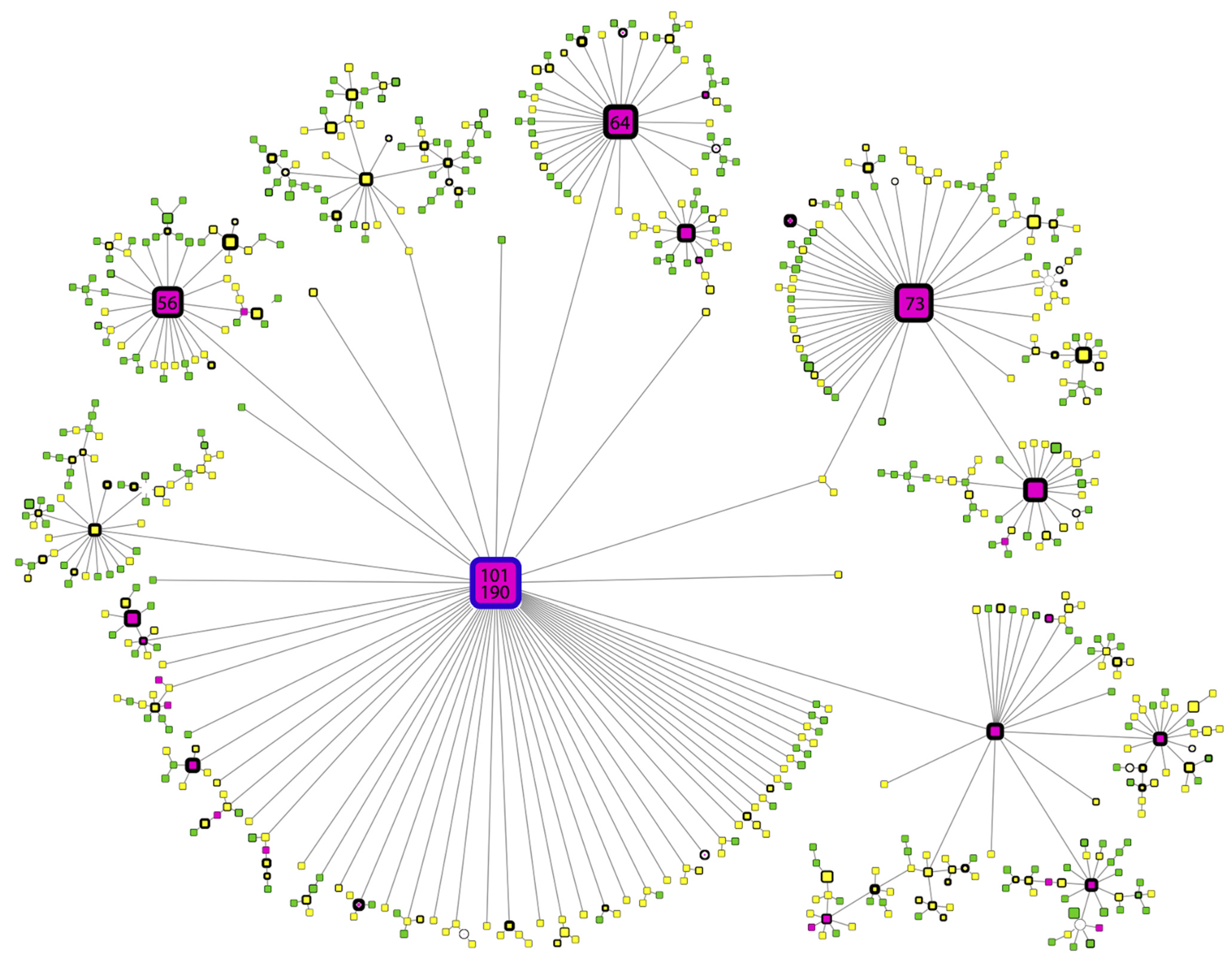
3.3.2. Haplotype Analysis
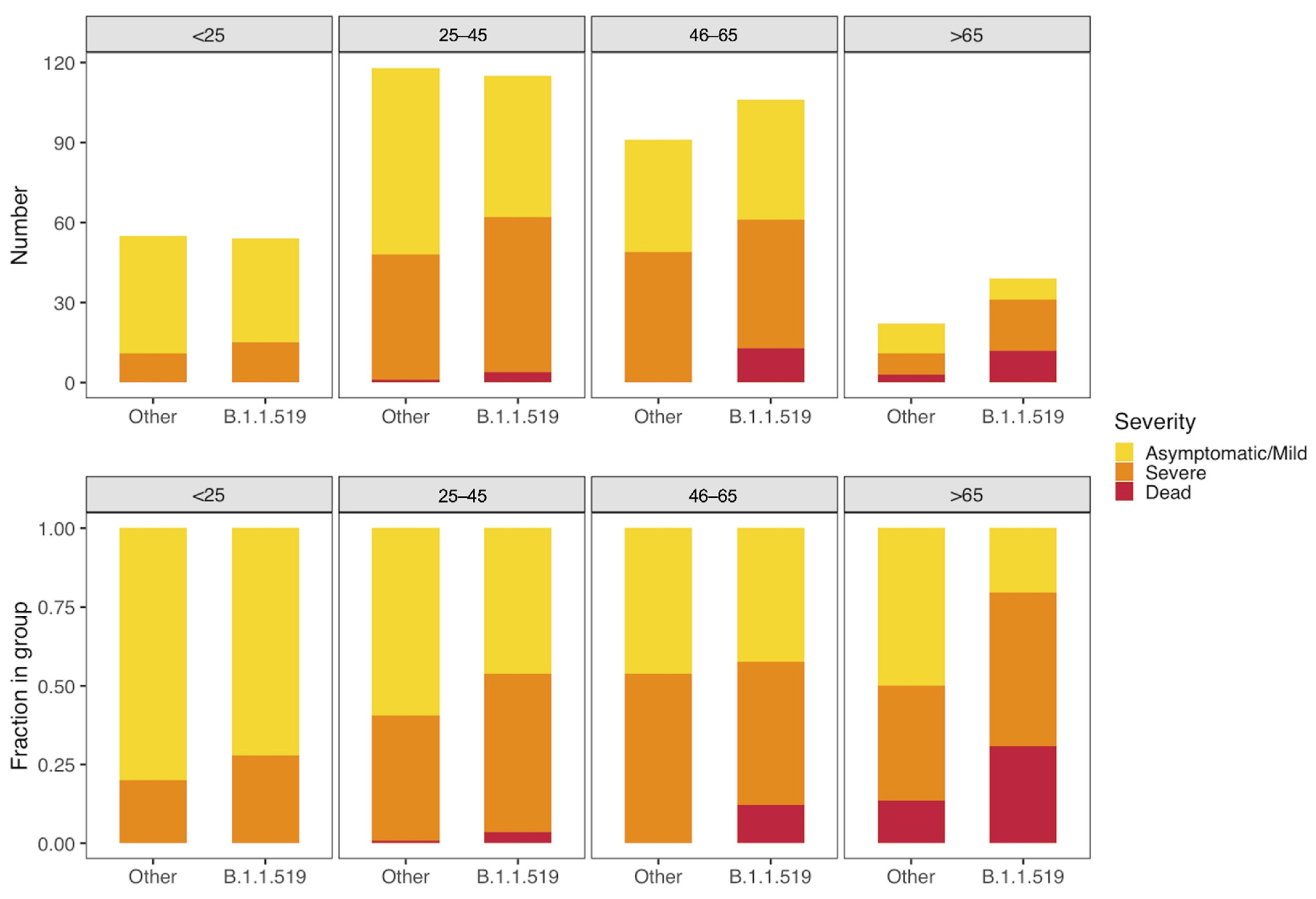
3.4. Clinical Association
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Coronavirus Disease-2019 (COVID-19): The Epidemic and the Challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Ng, W.H.; Tipih, T.; Makoah, N.A.; Vermeulen, J.-G.; Goedhals, D.; Sempa, J.B.; Burt, F.J.; Taylor, A.; Mahalingam, S. Comorbidities in SARS-CoV-2 Patients: A Systematic Review and Meta-Analysis. mBio 2021, 12, e03647-20. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Gargiulo, G.; Esposito, G.; Paolisso, G.; Marfella, R. Impact of Diabetes Mellitus on Clinical Outcomes in Patients Affected by Covid-19. Cardiovasc. Diabetol. 2020, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Matarese, A.; Gambardella, J.; Sardu, C.; Santulli, G. MiR-98 Regulates TMPRSS2 Expression in Human Endothelial Cells: Key Implications for COVID-19. Biomedicines 2020, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health: Data, Disease and Diplomacy. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention Real-Time RT-PCR Primers and Probes. 2020. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 1 November 2020).
- Mirtaleb, M.S.; Mirtaleb, A.H.; Nosrati, H.; Heshmatnia, J.; Falak, R.; Zolfaghari Emameh, R. Potential Therapeutic Agents to COVID-19: An Update Review on Antiviral Therapy, Immunotherapy, and Cell Therapy. Biomed. Pharmacother. 2021, 138, 111518. [Google Scholar] [CrossRef]
- Maurya, V.K.; Kumar, S.; Bhatt, M.L.B.; Saxena, S.K. Therapeutic Development and Drugs for the Treatment of COVID-19. In Coronavirus Disease 2019 (COVID-19); Saxena, S.K., Ed.; Medical Virology: From Pathogenesis to Disease Control; Springer: Singapore, 2020; pp. 109–126. ISBN 9789811548130. [Google Scholar]
- World Health Organization Tracking SARS-CoV-2 Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 1 November 2020).
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant Under Investigation B.1.617.1 With Sera of BBV152 Vaccinees. Clin. Infect. Dis. 2021, ciab411. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 Variant B.1.1.7 Is Susceptible to Neutralizing Antibodies Elicited by Ancestral Spike Vaccines. Cell Host Microbe 2021, 29, 529–539.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased Resistance of SARS-CoV-2 Variant P.1 to Antibody Neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Horby, P.; Huntley, C.; Davies, N.; Edmund, J.; Ferguson, N.; Medley, G.; Semple, C. NERVTAG Paper on COVID-19 Variant of Concern B.1.1.7; NERVTAG: London, UK, 2021. [Google Scholar]
- Loman, N.; Rambaut, A.; Rowe, W. NCoV-2019 Novel Coronavirus Bioinformatics Protocol. 2020. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 1 November 2020).
- Cori, A.; Ferguson, N.M.; Fraser, C.; Cauchemez, S. A New Framework and Software to Estimate Time-Varying Reproduction Numbers During Epidemics. Am. J. Epidemiol. 2013, 178, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiura, H.; Linton, N.M.; Akhmetzhanov, A.R. Serial Interval of Novel Coronavirus (COVID-19) Infections. Int. J. Infect. Dis. 2020, 93, 284–286. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E. Pegas: An R Package for Population Genetics with an Integrated-Modular Approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E. Analysis of Haplotype Networks: The Randomized Minimum Spanning Tree Method. Methods Ecol. Evol. 2018, 9, 1308–1317. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Bezerra, J.F.; Teixeira Vasconcelos, R.H.; Arantes, I.; Appolinario, L.; Mendonça, A.C.; Paixao, A.C.; Duarte, A.C.; Silva, T.; Rocha, A.S.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 P.2 Lineage Associated with Reinfection Case, Brazil, June–October 2020. Emerg. Infect. Dis. 2021, 27, 1789–1794. [Google Scholar] [CrossRef]
- Laiton-Donato, K.; Franco-Muñoz, C.; Álvarez-Díaz, D.A.; Ruiz-Moreno, H.A.; Usme-Ciro, J.A.; Prada, D.A.; Reales-González, J.; Corchuelo, S.; Herrera-Sepúlveda, M.T.; Naizaque, J.; et al. Characterization of the Emerging B.1.621 Variant of Interest of SARS-CoV-2. Infect. Genet. Evol. 2021, 95, 105038. [Google Scholar] [CrossRef]
- Tegally, H.; Ramuth, M.; Amoaka, D.; Scheepers, C.; Wilkinson, E.; Giovanetti, M.; Lessells, R.J.; Giandhari, J.; Ismail, A.; Martin, D.; et al. Genomic Epidemiology of SARS-CoV-2 in Mauritius Reveals a New Wave of Infections Dominated by the B.1.1.318, a Variant under Investigation. medRxiv 2021. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary Report on Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Spike Mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Rezaei, S.; Sefidbakht, Y.; Uskoković, V. Comparative Molecular Dynamics Study of the Receptor-Binding Domains in SARS-CoV-2 and SARS-CoV and the Effects of Mutations on the Binding Affinity. J. Biomol. Struct. Dyn. 2020, 1–20. [Google Scholar] [CrossRef]
- Maison, D.P.; Ching, L.L.; Shikuma, C.M.; Nerurkar, V.R. Genetic Characteristics and Phylogeny of 969-Bp S Gene Sequence of SARS-CoV-2 from Hawai’i Reveals the Worldwide Emerging P681H Mutation. Hawaii J. Health Soc. Welf 2021, 80, 52–61. [Google Scholar]
- Lubinski, B.; Tang, T.; Daniel, S.; Jaimes, J.A.; Whittaker, G.R. Functional Evaluation of Proteolytic Activation for the SARS-CoV-2 Variant B.1.1.7: Role of the P681H Mutation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Mejía, M.; Costa, Á.; Duarte, D.; Pessoa, K.; et al. Emergence and Spread of SARS-CoV-2 P.1 (Gamma) Lineage Variants Carrying Spike Mutations Δ141–144, N679K or P681H during Persistent Viral Circulation in Amazonas, Brazil. MedRxiv 2021. [Google Scholar] [CrossRef]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020–January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from Linking Epidemiological and Genetic Data. medRxiv 2021. [Google Scholar] [CrossRef]
- Morel, B.; Barbera, P.; Czech, L.; Bettisworth, B.; Hübner, L.; Lutteropp, S.; Serdari, D.; Kostaki, E.-G.; Mamais, I.; Kozlov, A.M.; et al. Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult. Mol. Biol. Evol. 2021, 38, 1777–1791. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dumaidi, K.; Qaraqe, H.; Al-Jawabreh, A.; Abu-Helu, R.; Samarah, F.; Al-Jawabreh, H. Genetic Diversity, Haplotype Analysis, and Risk Factor Assessment of Hepatitis A Virus Isolates from the West Bank, Palestine during the Period between 2014 and 2016. PLoS ONE 2020, 15, e0240339. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Pereson, M.J.; Flichman, D.M.; Martínez, A.P.; Baré, P.; Garcia, G.H.; Di Lello, F.A. Evolutionary Analysis of SARS-CoV-2 Spike Protein for Its Different Clades. J. Med. Virol. 2021, 93, 3000–3006. [Google Scholar] [CrossRef] [PubMed]
- Shishir, T.A.; Naser, I.B.; Faruque, S.M. In Silico Comparative Genomics of SARS-CoV-2 to Determine the Source and Diversity of the Pathogen in Bangladesh. PLoS ONE 2021, 16, e0245584. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Prates, E.T.; Pavicic, M.; Jones, P.; Amos, B.K.; Geiger, A.; Shah, M.B.; Streich, J.; Felipe Machado Gazolla, J.G.; Kainer, D.; et al. Potentially Adaptive SARS-CoV-2 Mutations Discovered with Novel Spatiotemporal and Explainable AI Models. Genome Biol. 2020, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Bhattacharjee, S.; Bhattacharjee, B. Association of Clade-G SARS-CoV-2 Viruses and Age with Increased Mortality Rates across 57 Countries and India. Infect. Genet. Evol. 2021, 90, 104734. [Google Scholar] [CrossRef]
- Song, S.; Ma, L.; Zou, D.; Tian, D.; Li, C.; Zhu, J.; Chen, M.; Wang, A.; Ma, Y.; Li, M.; et al. The Global Landscape of SARS-CoV-2 Genomes, Variants, and Haplotypes in 2019nCoVR. Genom. Proteom. Bioinform. 2020, 18, 749–759. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY. RECOVERY Trial Finds Regeneron’s Monoclonal Antibody Combination Reduces Deaths for Hospitalised COVID-19 Patients Who Have Not Mounted Their Own Immune Response; Nuffield Department of Population Health: Oxford, UK, 2021. [Google Scholar]
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Wohlfahrt, J.; Fonager, J.; Rasmussen, M.; Albertsen, M.; Michaelsen, T.Y.; Møller, C.H.; Ethelberg, S.; Legarth, R.; Button, M.S.F.; et al. Risk of Hospitalisation Associated with Infection with SARS-CoV-2 Lineage B.1.1.7 in Denmark: An Observational Cohort Study. Lancet Infect. Dis. 2021, 21, 1507–1517. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; CMMID COVID-19 Working Group; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic Characteristics and Clinical Effect of the Emergent SARS-CoV-2 B.1.1.7 Lineage in London, UK: A Whole-Genome Sequencing and Hospital-Based Cohort Study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Graham, M.S.; Sudre, C.H.; May, A.; Antonelli, M.; Murray, B.; Varsavsky, T.; Kläser, K.; Canas, L.S.; Molteni, E.; Modat, M.; et al. Changes in Symptomatology, Reinfection, and Transmissibility Associated with the SARS-CoV-2 Variant B.1.1.7: An Ecological Study. Lancet Public Health 2021, 6, e335–e345. [Google Scholar] [CrossRef]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C. SARS-CoV-2 Delta VOC in Scotland: Demographics, Risk of Hospital Admission, and Vaccine Effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.-M.; Cui, L.; Toh, M.P.H.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and Virological Features of SARS-CoV-2 Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta). Clin. Infec. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.; Pharris, A.; Spiteri, G.; Bundle, N.; Melidou, A.; Carr, M.; Gonzalez, G.; Garcia-Leon, A.; Crispie, F.; O’Connor, L.; et al. Characteristics of SARS-CoV-2 Variants of Concern B.1.1.7, B.1.351 or P.1: Data from Seven EU/EEA Countries, Weeks 38/2020 to 10/2021. Eurosurveillance 2021, 26, 1–10. [Google Scholar] [CrossRef]
- Freitas, A.R.R.; Lemos, D.R.Q.; Beckedorff, O.A.; de Góes Cavalcanti, L.P.; Siqueira, A.M.; de Mello, R.C.S.; Barros, E.N.C. The Increase in the Risk of Severity and Fatality Rate of Covid-19 in Southern Brazil after the Emergence of the Variant of Concern (VOC) SARS-CoV-2 P.1 Was Greater among Young Adults without Pre-Existing Risk Conditions. medRxiv 2021. [Google Scholar] [CrossRef]
- Jassat, W.; Mudara, C.; Ozougwu, L.; Tempia, S.; Blumberg, L.; Davies, M.-A.; Pillay, Y.; Carter, T.; Morewane, R.; Wolmarans, M.; et al. Increased Mortality among Individuals Hospitalised with COVID-19 during the Second Wave in South Africa. medRxiv 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symptom | B.1.1.519 n (%) | Other n (%) | aOR | 95% CI | p-Value |
|---|---|---|---|---|---|
| Dyspnea | 154 (49.0%) | 103 (36.0%) | 1.786 | (0.202–0.964) | 0.0028 |
| Chest Pain | 162 (51.6%) | 117 (40.9%) | 1.489 | (0.029–0.769) | 0.0350 |
| Cyanosis | 20 (6.4%) | 9 (3.1%) | 3.665 | (0.159–2.793) | 0.0456 |
| Diarrhea | 113 (36.0%) | 91 (31.8%) | 1.464 | (−0.007–0.777) | 0.0565 |
| Polipnea | 40 (12.7%) | 46 (16.1%) | 1.721 | (−0.067–1.200) | 0.0909 |
| Myalgia | 211 (67.2%) | 181 (63.3%) | 1.317 | (−0.101–0.652) | 0.1507 |
| Rhinorrhea | 98 (31.2%) | 103 (36.0%) | 0.757 | (−0.661–0.106) | 0.1549 |
| Anosmia | 173 (55.1%) | 183 (64.0%) | 0.769 | (−0.636–0.107) | 0.1647 |
| Conjuntivitis | 69 (22.0%) | 89 (31.1%) | 0.744 | (−0.714–0.125) | 0.1660 |
| Odynophalgia | 144 (45.9%) | 143 (50.0%) | 0.807 | (−0.581–0.151) | 0.2508 |
| Arthralgia | 196 (62.4%) | 172 (60.1%) | 1.235 | (−0.159–0.581) | 0.2628 |
| Cough | 204 (65.0%) | 166 (58.0%) | 1.233 | (−0.167–0.584) | 0.2740 |
| Vomit | 31 (9.9%) | 26 (9.1%) | 1.345 | (−0.336–0.970) | 0.3702 |
| Persistant.Fever | 47 (15.0%) | 52 (18.2%) | 0.800 | (−0.719–0.279) | 0.3780 |
| Cephalea | 213 (67.8%) | 201 (70.3%) | 0.861 | (−0.553–0.249) | 0.4651 |
| Fever | 185 (58.9%) | 173 (60.5%) | 0.925 | (−0.456–0.298) | 0.6868 |
| Abdominal.Pain | 31 (9.9%) | 37 (12.9%) | 1.011 | (−0.591–0.636) | 0.9714 |
| Summary | Ordinal Multivariable LR Model (Severity) | Binary Multivariable LR Model (Hospitalization) | |||||
|---|---|---|---|---|---|---|---|
| Characteristic | n = 600 1 | aOR 2 | 95% CI 2 | p-Value | aOR 2 | 95% CI 2 | p-Value |
| Severity | |||||||
| Asymptomatic/Mild | 312 (52%) | ||||||
| Severe | 255 (42%) | ||||||
| Dead | 33 (5.5%) | ||||||
| Hospitalized | 69 (12%) | ||||||
| Age | 42 (29, 54) | 1.04 | 1.03, 1.05 | <0.001 | 1.06 | 1.04, 1.08 | <0.001 |
| Sex | |||||||
| Female | 302 (50%) | — | — | — | — | ||
| Male | 298 (50%) | 1.21 | 0.87, 1.70 | 0.3 | 1.69 | 0.95, 3.04 | 0.075 |
| Ct | 19.13 (17.90, 20.40) | 0.99 | 0.93, 1.06 | 0.8 | 1.05 | 0.95, 1.17 | 0.3 |
| ImmunoSuppressed | 18 (3.0%) | 2.86 | 1.12, 7.42 | 0.029 | 2.43 | 0.59, 8.23 | 0.2 |
| HD_Hypertension | 107 (18%) | 1.18 | 0.73, 1.90 | 0.5 | 1.55 | 0.82, 2.91 | 0.2 |
| Diabetes | 73 (12%) | 0.91 | 0.53, 1.56 | 0.7 | 1.09 | 0.53, 2.14 | 0.8 |
| Obesity | 236 (39%) | 1.42 | 1.01, 1.99 | 0.044 | 1.61 | 0.91, 2.87 | 0.10 |
| Asthma | 20 (3.3%) | 1.53 | 0.62, 3.73 | 0.3 | 1.11 | 0.21, 4.43 | 0.9 |
| Smoker | 164 (27%) | 1.21 | 0.83, 1.75 | 0.3 | 0.84 | 0.43, 1.57 | 0.6 |
| Variant | |||||||
| Other | 286 (48%) | — | — | — | — | ||
| B.1.1.519 | 314 (52%) | 1.85 | 1.33, 2.60 | <0.001 | 2.35 | 1.32, 4.34 | 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cedro-Tanda, A.; Gómez-Romero, L.; Alcaraz, N.; de Anda-Jauregui, G.; Peñaloza, F.; Moreno, B.; Escobar-Arrazola, M.A.; Ramirez-Vega, O.A.; Munguia-Garza, P.; Garcia-Cardenas, F.; et al. The Evolutionary Landscape of SARS-CoV-2 Variant B.1.1.519 and Its Clinical Impact in Mexico City. Viruses 2021, 13, 2182. https://doi.org/10.3390/v13112182
Cedro-Tanda A, Gómez-Romero L, Alcaraz N, de Anda-Jauregui G, Peñaloza F, Moreno B, Escobar-Arrazola MA, Ramirez-Vega OA, Munguia-Garza P, Garcia-Cardenas F, et al. The Evolutionary Landscape of SARS-CoV-2 Variant B.1.1.519 and Its Clinical Impact in Mexico City. Viruses. 2021; 13(11):2182. https://doi.org/10.3390/v13112182
Chicago/Turabian StyleCedro-Tanda, Alberto, Laura Gómez-Romero, Nicolás Alcaraz, Guillermo de Anda-Jauregui, Fernando Peñaloza, Bernardo Moreno, Marco A. Escobar-Arrazola, Oscar A. Ramirez-Vega, Paulina Munguia-Garza, Francisco Garcia-Cardenas, and et al. 2021. "The Evolutionary Landscape of SARS-CoV-2 Variant B.1.1.519 and Its Clinical Impact in Mexico City" Viruses 13, no. 11: 2182. https://doi.org/10.3390/v13112182