
Multi-Omics Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Poplar under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Treatment
2.2. Construction and Sequencing of Transcriptome, sRNA, and Degradome Libraries
2.3. Analysis of Transcriptome Sequencing Data
2.4. Analysis of Small RNA Sequencing and miRNA Identification
2.5. Analysis of Degradome Sequencing Data and miRNA Targets Identification
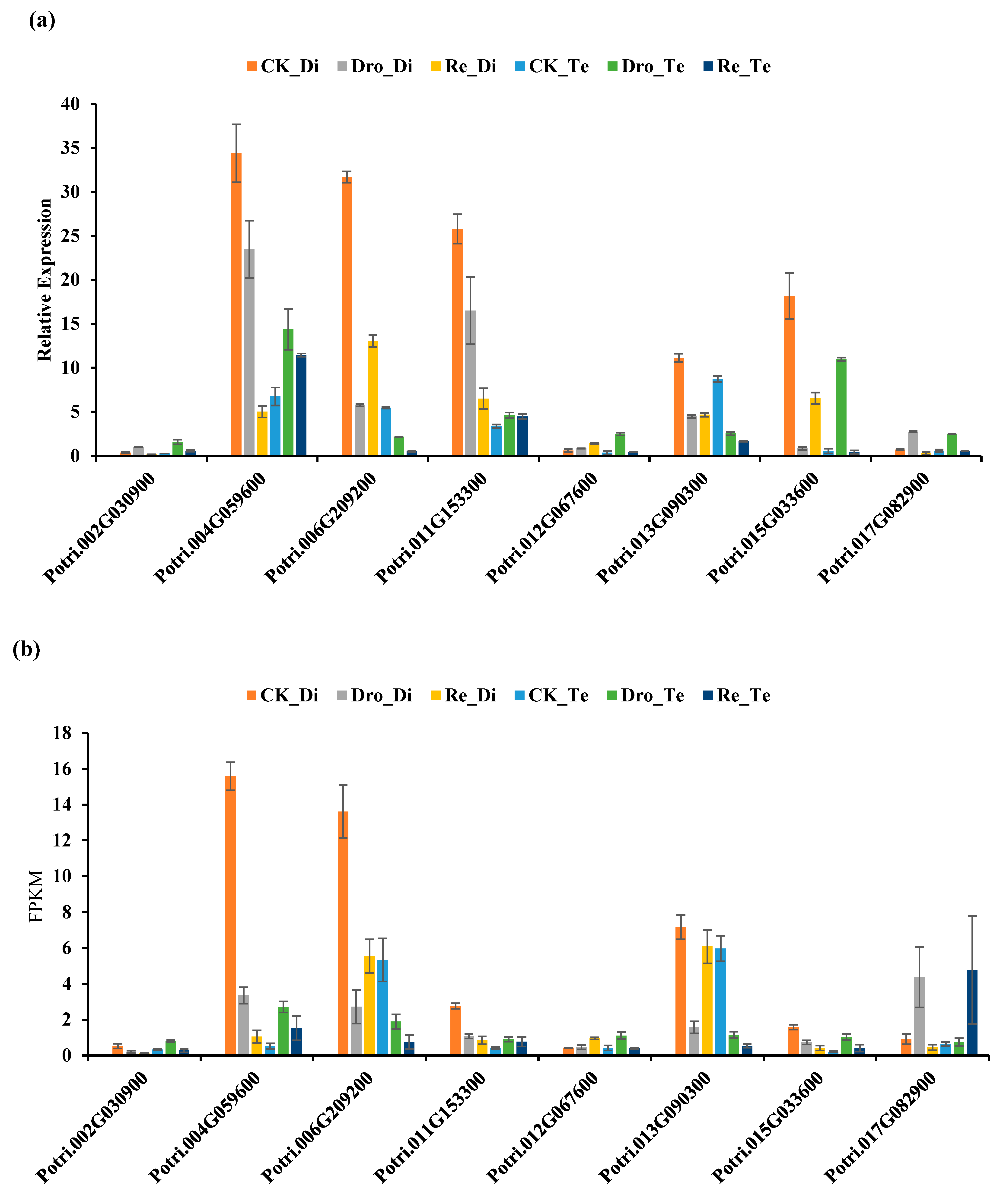
2.6. Verification of RNA-seq Data by qRT-PCR Analysis
3. Results
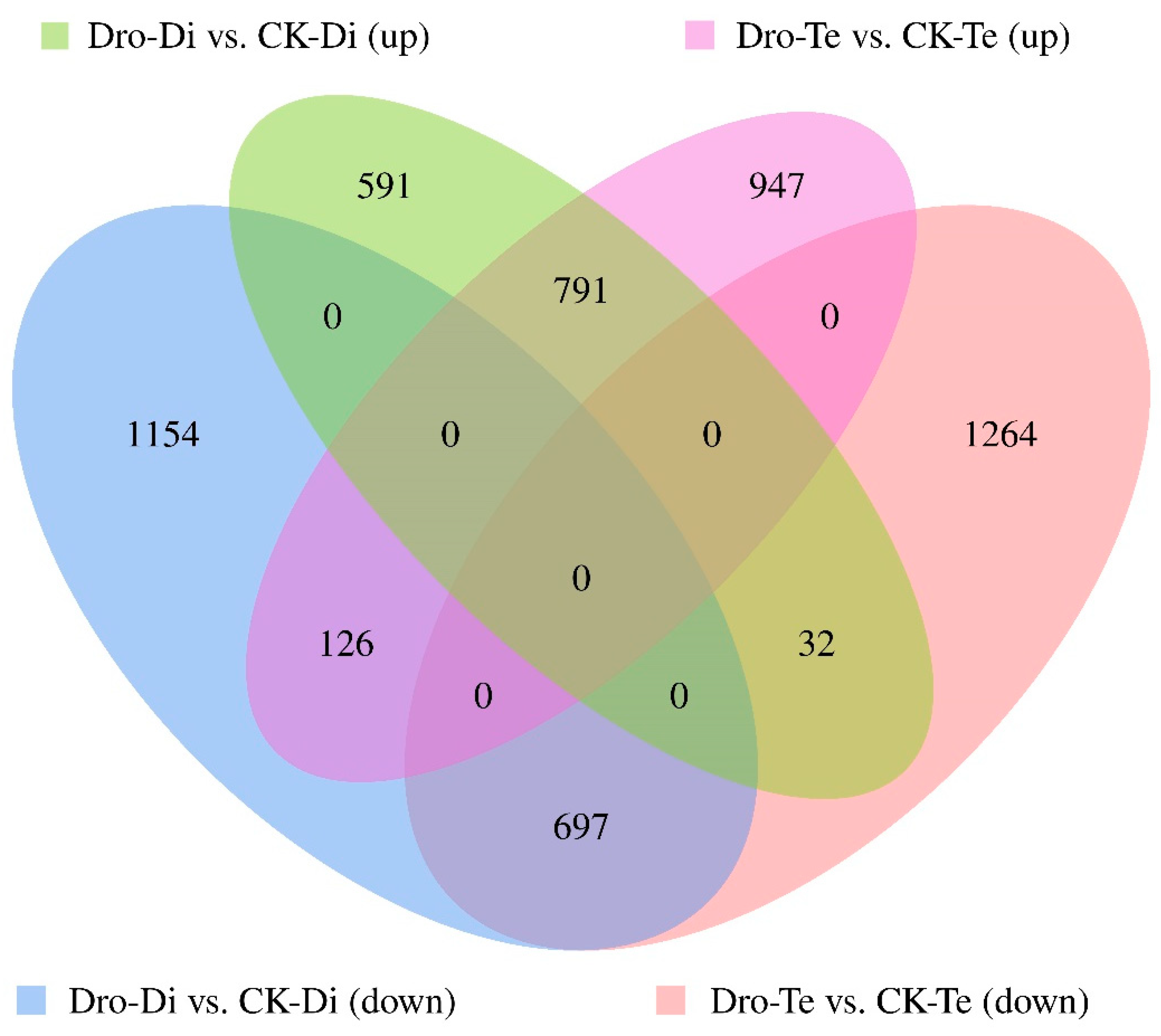
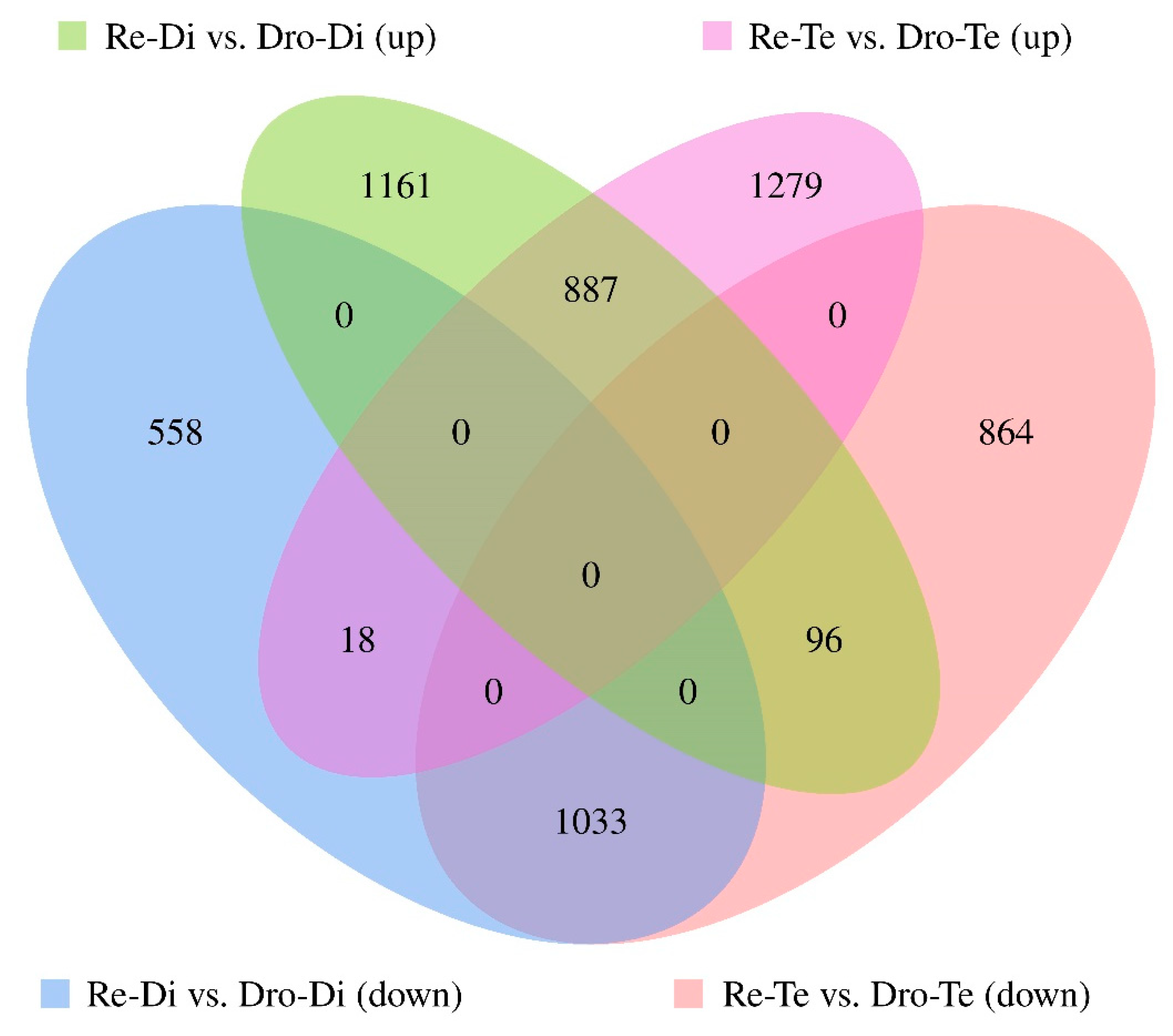
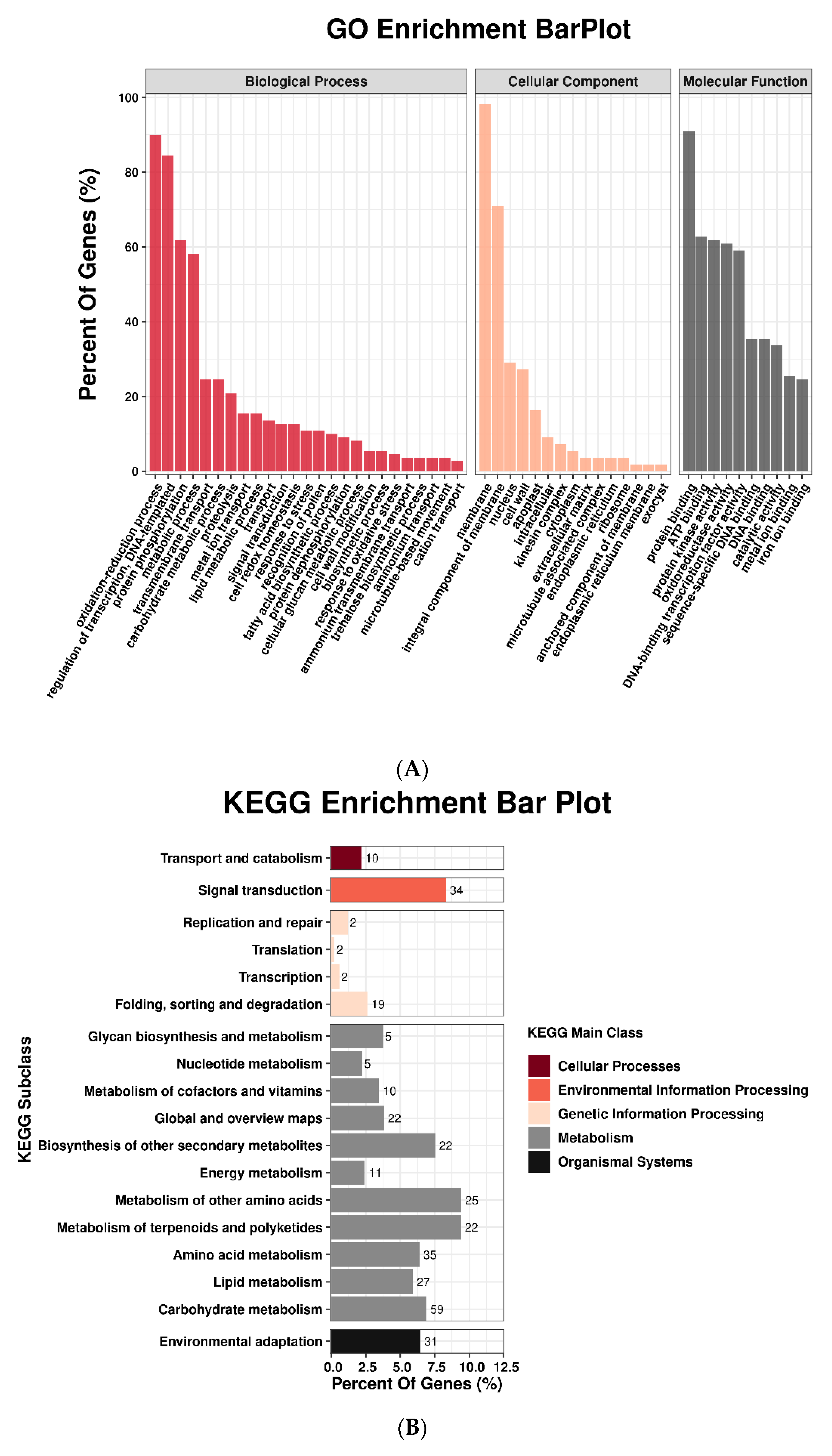
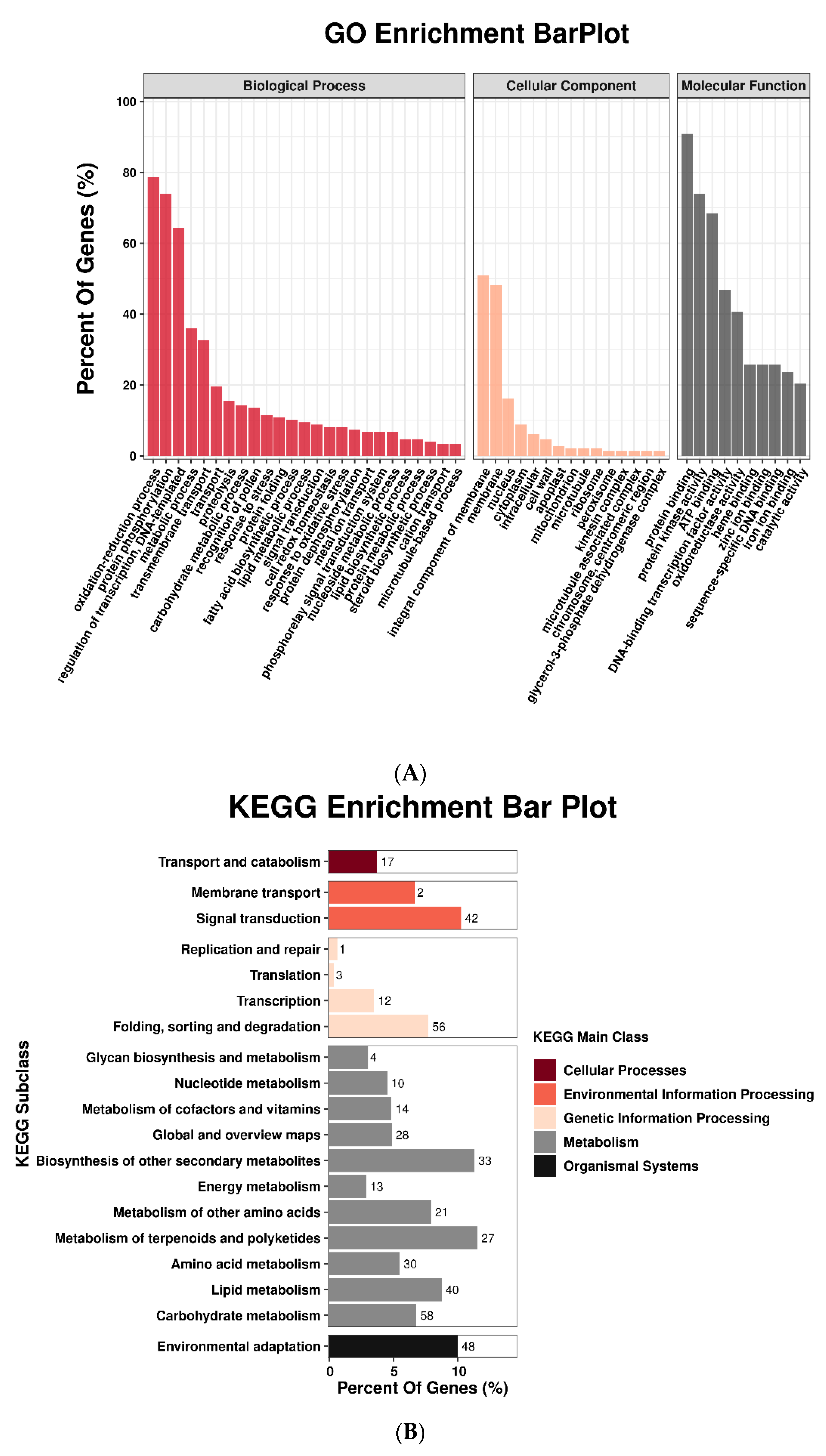
3.1. mRNA Expression Profiling
3.2. MicroRNA Sequencing Analysis
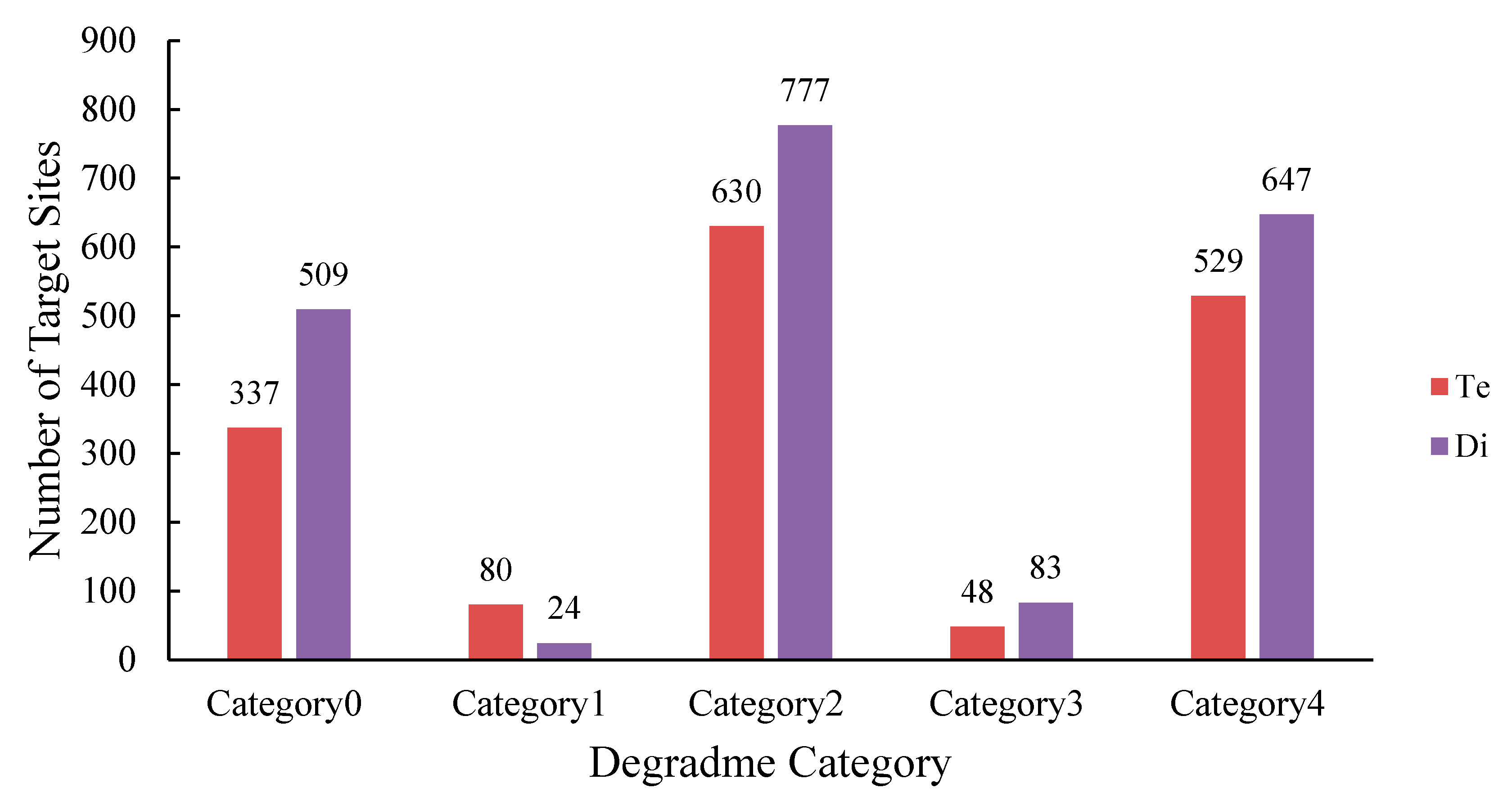
3.3. Determination of miRNAs-Target Genes by Degradome Analysis
4. Discussion
4.1. The Role of Transcription Factors in the Adaptive Responses to Drought Stress and Rewatering
4.2. miRNA Involved in the Responses to Drought Stress and Rewatering
4.3. Protein Phosphorylation Involved in the Responses to Drought Stress and Rewatering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hammond, W.M.; Williams, A.P.; Abatzoglou, J.T.; Adams, H.D.; Klein, T.; López, R.; Sáenz-Romero, C.; Hartmann, H.; Breshears, D.D.; Allen, C.D. Global field observations of tree die-off reveal hotter-drought fingerprint for Earth’s forests. Nat. Commun. 2022, 13, 1761. [Google Scholar] [CrossRef]
- Ma, T.; Liang, Y.; Li, Z.; Liu, Z.; Liu, B.; Wu, M.M.; Lau, M.K.; Fang, Y. Age-related patterns and climatic driving factors of drought-induced forest mortality in Northeast China. Agric. For. Meteorol. 2023, 332, 109360. [Google Scholar] [CrossRef]
- Estravis-Barcala, M.; Mattera, M.G.; Soliani, C.; Bellora, N.; Opgenoorth, L.; Heer, K.; Arana, M.V. Molecular bases of responses to abiotic stress in trees. J. Exp. Bot. 2020, 71, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, J.; Hui, W.; Zhao, F.; Wang, P.; Su, C.; Gong, W. Physiology of plant responses to water stress and related genes: A review. Forests 2022, 13, 324. [Google Scholar] [CrossRef]
- Greer, B.T.; Still, C.; Cullinan, G.L.; Brooks, J.R.; Meinzer, F.C. Polyploidy influences plant–environment interactions in quaking aspen (Populus tremuloides Michx.). Tree Physiol. 2018, 38, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Shang, X.H.; Cao, S.; Xie, X.Y.; Zeng, W.D.; Lu, L.Y.; Chen, S.B.; Yan, H.B. Comparative physiology and transcriptome analysis allows for identification of lncRNAs imparting tolerance to drought stress in autotetraploid cassava. BMC Genom. 2019, 20, 514. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Wang, Y.; Xie, Z.; Guo, D.; Chen, C.; Fan, Q.; Deng, X.; Liu, J.H. Enhanced ROS scavenging and sugar accumulation contribute to drought tolerance of naturally occurring autotetraploids in Poncirus trifoliata. Plant Biotechnol. J. 2019, 17, 1394–1407. [Google Scholar] [CrossRef]
- Abdolinejad, R.; Shekafandeh, A. Tetraploidy confers superior in vitro water-stress tolerance to the fig tree (Ficus carica) by reinforcing hormonal, physiological, and biochemical defensive systems. Front. Plant Sci. 2022, 12, 796215. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Compatible solute, transporter protein, transcription factor, and hormone-related gene expression provides an indicator of drought stress in Paulownia fortunei. Funct. Integr. Genom. 2014, 14, 479–491. [Google Scholar]
- Xu, J.; Jin, J.; Zhao, H.; Li, K. Drought stress tolerance analysis of Populus ussuriensis clones with different ploidies. J. For. Res. 2019, 30, 1267–1275. [Google Scholar] [CrossRef]
- Zhang, W.W.; Song, J.; Wang, M.; Liu, Y.Y.; Li, N.; Zhang, Y.J.; Holbrook, N.M.; Hao, G.Y. Divergences in hydraulic architecture form an important basis for niche differentiation between diploid and polyploid Betula species in NE China. Tree Physiol. 2017, 37, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, D.; Marat, M.; Marasek-Ciołakowska, A.; Klamkowski, K.; Buler, Z.; Podwyszyńska, M.; Tomczyk, P.P.; Wójcik, K.; Treder, W.; Filipczak, J. Apple autotetraploids—Phenotypic characterisation and response to drought stress. Agronomy 2022, 12, 161. [Google Scholar] [CrossRef]
- Sannigrahi, P.; Ragauskas, A.J.; Tuskan, G.A. Poplar as a feedstock for biofuels: A review of compositional characteristics. Biofuels Bioprod. Biorefin. 2010, 4, 209–226. [Google Scholar] [CrossRef]
- Zhang, A.P.; Liu, C.F.; Sun, R.C. Fractional isolation and characterization of lignin and hemicelluloses from Triploid of Populus tomentosa Carr. Ind. Crops Prod. 2010, 31, 357–362. [Google Scholar] [CrossRef]
- Han, Q.; Zhang, Y.; Geng, X.; Du, K.; Yang, J.; Kang, X. Response of tree growth, crown, and branch development to planting density in four Populus tomentosa clones. Austrian J. For. Sci. 2020, 137. [Google Scholar]
- Sun, S.; He, C.; Qiu, L.; Li, C.; Zhang, J.; Meng, P. Stable isotope analysis reveals prolonged drought stress in poplar plantation mortality of the Three-North Shelter Forest in Northern China. Agric. For. Meteorol. 2018, 252, 39–48. [Google Scholar] [CrossRef]
- Kang, X.; Wei, H. Breeding polyploid Populus: Progress and perspective. For. Res. 2022, 2, 4. [Google Scholar] [CrossRef]
- Chen, T.Y.; Wang, B.; Wu, Y.Y.; Wen, J.L.; Liu, C.F.; Yuan, T.Q.; Sun, R.C. Structural variations of lignin macromolecule from different growth years of Triploid of Populus tomentosa Carr. Int. J. Biol. Macromol. 2017, 101, 747–757. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, Y.; Han, Q.; Kang, X. Molecular mechanism of slow vegetative growth in Populus tetraploid. Genes 2020, 11, 1417. [Google Scholar] [CrossRef]
- Liu, B.; Sun, G. Transcriptome and miRNAs analyses enhance our understanding of the evolutionary advantages of polyploidy. Crit. Rev. Biotechnol. 2019, 39, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım, K.; Kaya, Z. Gene regulation network behind drought escape, avoidance and tolerance strategies in black poplar (Populus nigra L.). Plant Physiol. Biochem. 2017, 115, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Fox, H.; Ben-Dor, S.; Doron-Faigenboim, A.; Goldsmith, M.; Klein, T.; David-Schwartz, R. Carbohydrate dynamics in Populus trees under drought: An expression atlas of genes related to sensing, translocation, and metabolism across organs. Physiol. Plant. 2023, 175, e14001. [Google Scholar] [CrossRef]
- Kim, T.-L.; Lim, H.; Denison, M.I.J.; Oh, C. Transcriptomic and Physiological Analysis Reveals Genes Associated with Drought Stress Responses in Populus alba × Populus glandulosa. Plants 2023, 12, 3238. [Google Scholar] [CrossRef]
- Jiao, Z.; Lian, C.; Han, S.; Huang, M.; Shen, C.; Li, Q.; Niu, M.-X.; Yu, X.; Yin, W.; Xia, X. PtmiR169o plays a positive role in regulating drought tolerance and growth by targeting the PtNF-YA6 gene in poplar. Environ. Exp. Bot. 2021, 189, 104549. [Google Scholar] [CrossRef]
- Cai, H.; Yang, C.; Liu, S.; Qi, H.; Wu, L.; Xu, L.-A.; Xu, M. MiRNA-target pairs regulate adventitious rooting in Populus: A functional role for miR167a and its target Auxin response factor 8. Tree Physiol. 2019, 39, 1922–1936. [Google Scholar] [CrossRef]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef]
- German, M.A.; Luo, S.; Schroth, G.; Meyers, B.C.; Green, P.J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009, 4, 356–362. [Google Scholar] [CrossRef]
- Ma, Z.; Coruh, C.; Axtell, M.J. Arabidopsis lyrata small RNAs: Transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell 2010, 22, 1090–1103. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genom. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Jiang, S.; Chen, H.; Xia, Y.; Guo, R.; Ling, A.; Liao, T.; Wu, W.; Kang, X. Spatiotemporal miRNA and transcriptomic network dynamically regulate the developmental and senescence processes of poplar leaves. Hortic. Res. 2023, 10, uhad186. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Guo, J.; Zhang, S.; Zhang, G. Integration of small RNAs, degradome, and transcriptome sequencing in Populus× euramericana “Neva” provides insights into the allelopathic interference of para-hydroxybenzoic acid. Can. J. For. Res. 2020, 50, 422–437. [Google Scholar] [CrossRef]
- Tossi, V.E.; Martinez Tosar, L.J.; Laino, L.E.; Iannicelli, J.; Regalado, J.J.; Escandón, A.S.; Baroli, I.; Causin, H.F.; Pitta-Álvarez, S.I. Impact of polyploidy on plant tolerance to abiotic and biotic stresses. Front. Plant Sci. 2022, 13, 869423. [Google Scholar] [CrossRef]
- Mangena, P.; Mushadu, P.N. Colchicine-Induced Polyploidy in Leguminous Crops Enhances Morpho-Physiological Characteristics for Drought Stress Tolerance. Life 2023, 13, 1966. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Zhang, H.; Zhang, D. Genome-wide analysis of gene expression in response to drought stress in Populus simonii. Plant Mol. Biol. Rep. 2013, 31, 946–962. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef] [PubMed]
- Salvato, F.; Loziuk, P.; Kiyota, E.; Daneluzzi, G.S.; Araújo, P.; Muddiman, D.C.; Mazzafera, P. Label-free quantitative proteomics of enriched nuclei from sugarcane (Saccharum ssp) stems in response to drought stress. Proteomics 2019, 19, 1900004. [Google Scholar] [CrossRef]
- Tang, Y.; Bao, X.; Zhi, Y.; Wu, Q.; Guo, Y.; Yin, X.; Zeng, L.; Li, J.; Zhang, J.; He, W. Overexpression of a MYB family gene, OsMYB6, increases drought and salinity stress tolerance in transgenic rice. Front. Plant Sci. 2019, 10, 168. [Google Scholar] [CrossRef]
- Mun, B.G.; Lee, S.U.; Park, E.J.; Kim, H.H.; Hussain, A.; Imran, Q.M.; Lee, I.J.; Yun, B.W. Analysis of transcription factors among differentially expressed genes induced by drought stress in Populus davidiana. 3 Biotech 2017, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef]
- Khatun, K.; Robin, A.H.K.; Park, J.I.; Nath, U.K.; Kim, C.K.; Lim, K.B.; Nou, I.S.; Chung, M.Y. Molecular characterization and expression profiling of tomato GRF transcription factor family genes in response to abiotic stresses and phytohormones. Int. J. Mol. Sci. 2017, 18, 1056. [Google Scholar] [CrossRef]
- Wang, W.; Wu, P.; Li, Y.; Hou, X. Genome-wide analysis and expression patterns of ZF-HD transcription factors under different developmental tissues and abiotic stresses in Chinese cabbage. Mol. Genet. Genom. 2016, 291, 1451–1464. [Google Scholar] [CrossRef]
- Alam, I.; Wu, X.; Yu, Q.; Ge, L. Comprehensive genomic analysis of G2-like transcription factor genes and their role in development and abiotic stresses in Arabidopsis. Diversity 2022, 14, 228. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, B.; Lu, G.; Han, B. Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem. Biophys. Res. Commun. 2009, 379, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dane, F. NAC (NAM/ATAF/CUC) transcription factors in different stresses and their signaling pathway. Acta Physiol. Plant 2013, 35, 1397–1408. [Google Scholar] [CrossRef]
- Qu, X.; Zou, J.; Wang, J.; Yang, K.; Wang, X.; Le, J. A rice R2R3-type MYB transcription factor OsFLP positively regulates drought stress response via OsNAC. Int. J. Mol. Sci. 2022, 23, 5873. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, Z.; Lai, J.; Zhang, Y.; Yang, C.; Yin, B.; Zhao, Q.; Zhang, L.; Li, Y.; Yang, C. Dual function of Arabidopsis ATAF1 in abiotic and biotic stress responses. Cell Res. 2009, 19, 1279–1290. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhong, M.; He, L.; Wang, B.; Liu, Q.L.; Pan, Y.Z.; Jiang, B.B.; Zhang, L. Overexpression of a chrysanthemum transcription factor gene DgNAC1 improves drought tolerance in chrysanthemum. J. Plant Cell Tissue 2018, 135, 119–132. [Google Scholar] [CrossRef]
- Mao, X.; Zhang, H.; Qian, X.; Li, A.; Zhao, G.; Jing, R. TaNAC2, a NAC-type wheat transcription factor conferring enhanced multiple abiotic stress tolerances in Arabidopsis. J. Exp. Bot. 2012, 63, 2933–2946. [Google Scholar] [CrossRef]
- Sun, L.; Huang, L.; Hong, Y.; Zhang, H.; Song, F.; Li, D. Comprehensive analysis suggests overlapping expression of rice ONAC transcription factors in abiotic and biotic stress responses. Int. J. Mol. Sci. 2015, 16, 4306–4326. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Lei, G.; Tian, A.G.; Zhang, W.K. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 2011, 68, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [PubMed]
- He, J.F.; Wang, L.W.; Fang, Y.Y.; Wang, Y.H.; Wang, C.; Liu, H.K. Transcriptome Analysis on Transcription Factors of Haloxylon ammodendron under Drought Stress and Rehydration Treatment. Acta Agric. Bor. Sin. 2020, 35, 36–43. [Google Scholar]
- An, X.; Jin, G.; Luo, X.; Chen, C.; Li, W.; Zhu, G. Transcriptome analysis and transcription factors responsive to drought stress in Hibiscus cannabinus. PeerJ 2020, 8, e8470. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, L.; Liu, N.; Cao, H.; Li, H.; Gui, D.; Wang, J.; Zhang, C. Analysis of MYB genes in four plant species and the detection of genes associated with drought resistance. Botany 2022, 100, 519–532. [Google Scholar] [CrossRef]
- Gao, F.; Wang, N.; Li, H.; Liu, J.; Fu, C.; Xiao, Z.; Wei, C.; Lu, X.; Feng, J.; Zhou, Y. Identification of drought-responsive microRNAs and their targets in Ammopiptanthus mongolicus by using high-throughput sequencing. Sci. Rep. 2016, 6, 34601. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Xu, Y.X.; Ma, J.Q.; Wang, W.W.; Chen, W.; Huang, D.J.; Fang, J.; Li, X.J.; Chen, L. Small RNA and degradome profiling reveals important roles for microRNAs and their targets in tea plant response to drought stress. Physiol. Plant 2016, 158, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, S.; Zhu, C.; Chang, X.; Yue, C.; Wang, Z.; Lin, Y.; Lai, Z. Identification of drought-responsive miRNAs and physiological characterization of tea plant (Camellia sinensis L.) under drought stress. BMC Plant Biol. 2017, 17, 211. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Han, S.; Zhao, W.; Chen, T.; Zhou, J.; Li, L. Genome-wide identification of abiotic stress-regulated and novel microRNAs in mulberry leaf. Plant Physiol. Biochem. 2015, 95, 75–82. [Google Scholar] [CrossRef]
- Li, R.; Chen, D.; Wang, T.; Wan, Y.; Li, R.; Fang, R.; Wang, Y.; Hu, F.; Zhou, H.; Li, L. High throughput deep degradome sequencing reveals microRNAs and their targets in response to drought stress in mulberry (Morus alba). PLoS ONE 2017, 12, e0172883. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Fan, G.; Cao, L.; Deng, M.; Zhao, Z.; Niu, S.; Wang, Z.; Wang, Y. Drought stress-induced changes of microRNAs in diploid and autotetraploid Paulownia tomentosa. Genes Genom. 2017, 39, 77–86. [Google Scholar] [CrossRef]
- Deng, M.; Cao, Y.; Zhao, Z.; Yang, L.; Zhang, Y.; Dong, Y.; Fan, G. Discovery of microRNAs and their target genes related to drought in Paulownia “Yuza 1” by high-throughput sequencing. Int. J. Genom. 2017, 2017, 3674682. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Chen, L.; Zhang, Y.; Kang, X.; Zhang, Z.; Wang, Y. Identification of novel and conserved Populus tomentosa microRNA as components of a response to water stress. Funct. Integr. Genom. 2012, 12, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Su, Y.; Liang, D.; Zhang, Z.; Xia, X.; Yin, W. Identification of phasiRNAs and their drought-responsiveness in Populus trichocarpa. FEBS Lett. 2016, 590, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, C.G.; Yoshimoto, N.; Maruyama-Nakashita, A.; Tsuchiya, Y.N.; Saito, K.; Takahashi, H.; Dalmay, T. Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J. 2009, 57, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Ai, Q.; Yu, D. Uncovering miRNAs involved in crosstalk between nutrient deficiencies in Arabidopsis. Sci. Rep. 2015, 5, 11813. [Google Scholar] [CrossRef]
- Frazier, T.P.; Sun, G.; Burklew, C.E.; Zhang, B. Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol. Biotechnol. 2011, 49, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genomic. 2016, 16, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Cai, Y.; Yang, Z.; Zheng, Y. MeJA induces chilling tolerance in loquat fruit by regulating proline and γ-aminobutyric acid contents. Food Chem. 2012, 133, 1466–1470. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.E.; Repetti, P.P.; Adams, T.R.; Creelman, R.A.; Wu, J.; Warner, D.C.; Anstrom, D.C.; Bensen, R.J.; Castiglioni, P.P.; Donnarummo, M.G. Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres. Proc. Natl. Acad. Sci. USA 2007, 104, 16450–16455. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Bhaskara, G.B.; Wen, T.N.; Nguyen, T.T.; Verslues, P.E. Protein phosphatase 2Cs and microtubule-associated stress protein 1 control microtubule stability, plant growth, and drought response. Plant Cell 2017, 29, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Geiger, D.; Scherzer, S.; Mumm, P.; Stange, A.; Marten, I.; Bauer, H.; Ache, P.; Matschi, S.; Liese, A.; Al-Rasheid, K.A. Activity of guard cell anion channel SLAC1 is controlled by drought-stress signaling kinase-phosphatase pair. Proc. Natl. Acad. Sci. USA 2009, 106, 21425–21430. [Google Scholar] [CrossRef]
- Huang, K.; Peng, L.; Liu, Y.; Yao, R.; Liu, Z.; Li, X.; Yang, Y.; Wang, J. Arabidopsis calcium-dependent protein kinase AtCPK1 plays a positive role in salt/drought-stress response. Biochem. Biophys. Res. Commun. 2018, 498, 92–98. [Google Scholar] [CrossRef]
- Zhu, M.; Monroe, J.G.; Suhail, Y.; Villiers, F.; Mullen, J.; Pater, D.; Hauser, F.; Jeon, B.W.; Bader, J.S.; Kwak, J.M. Molecular and systems approaches towards drought-tolerant canola crops. New Phytol. 2016, 210, 1169–1189. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Han, G.; He, H.; Li, J. Differential regulation of proteins and phosphoproteins in rice under drought stress. Biochem. Biophys. Res. Commun. 2009, 379, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lv, J.; Ma, Z.; Dong, W. The mechanism of alfalfa (Medicago sativa L.) response to abiotic stress. Plant Growth Regul. 2019, 89, 239–249. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Known miRNA | Novel miRNA | Total | ||
|---|---|---|---|---|---|
| Up | Down | Up | Down | ||
| Dro-Di vs. CK-Di | 8 | 17 | 3 | 2 | 35 |
| Re-Di vs. Dro-Di | 11 | 52 | 2 | 4 | 69 |
| Dro-Te vs. CK-Te | 34 | 24 | 1 | 2 | 61 |
| Re-Te vs. Dro-Te | 17 | 17 | 7 | 2 | 43 |
| miRNA | Dro-Te vs. CK-Te | Re-Te vs. Dro-Te |
|---|---|---|
| ptc-MIR156j-p3 | up | down |
| rco-MIR156h-p3 | up | down |
| gra-MIR7486j-p5_2ss9TG17CG | up | down |
| gra-MIR7486j-p3_2ss9TG17CG | up | down |
| ptc-miR399i_L+1_1ss22AT | down | down |
| ptc-MIR481b-p5_1ss1CT | down | up |
| ptc-miR169ac_R-2_1ss16TA | up | down |
| ptc-MIR169k-p3 | up | down |
| ptc-MIR477c-p3_1ss16TC | up | up |
| PC-3p-19149_647 | up | down |
| Sample | Di | Te | ||
|---|---|---|---|---|
| Number | Ratio | Number | Ratio | |
| Raw Reads | 23,659,860 | / | 16,514,363 | / |
| reads < 15nt after removing 3 adaptor | 127,762 | 0.54% | 95,754 | 0.58% |
| Mappable Reads | 23,532,098 | 99.46% | 16,418,609 | 99.42% |
| Unique Raw Reads | 7,699,368 | / | 5,654,938 | / |
| Unique reads < 15 nt after removing 3 adaptor | 58,802 | 0.76% | 43,558 | 0.77% |
| Unique Mappable Reads | 7,640,566 | 99.24% | 5,611,380 | 99.23% |
| Transcript Mapped Reads | 14,573,489 | 61.60% | 10,863,582 | 65.78% |
| Unique Transcript Mapped Reads | 4,688,516 | 60.89% | 3,642,364 | 64.41% |
| Number of input Transcript | 63,498 | / | 63,498 | / |
| Number of Coverd Transcript | 52,596 | 82.83% | 51,046 | 80.39% |
| miRNA | Transcript | Annotation |
|---|---|---|
| gma-miR156a_R+1 | Potri.015G098900.3 | SPL13A |
| Potri.015G098900.1 | SPL13A | |
| Potri.015G098900.2 | SPL13A | |
| Potri.003G169400.2 | SPL13B | |
| Potri.003G169400.1 | SPL13B | |
| Potri.014G057800.1 | SPL10 | |
| Potri.001G058600.1 | SPL13B | |
| ptc-miR2111a | Potri.001G331500.4 | Galactose oxidase/kelch repeat superfamily protein |
| Potri.001G331500.3 | ||
| Potri.001G331500.1 | ||
| Potri.001G331500.2 | ||
| ptc-MIR395k-p5 | Potri.012G039000.2 | glutamate decarboxylase-like |
| Potri.012G039000.5 | ||
| Potri.012G039000.7 | ||
| PC-3p-19149_647 | Potri.018G100700.5 | amidase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Q.; Du, K.; Xia, Y.; Kang, X. Multi-Omics Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Poplar under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing. Forests 2023, 14, 2268. https://doi.org/10.3390/f14112268
Han Q, Du K, Xia Y, Kang X. Multi-Omics Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Poplar under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing. Forests. 2023; 14(11):2268. https://doi.org/10.3390/f14112268
Chicago/Turabian StyleHan, Qiang, Kang Du, Yufei Xia, and Xiangyang Kang. 2023. "Multi-Omics Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Poplar under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing" Forests 14, no. 11: 2268. https://doi.org/10.3390/f14112268