
Novel Phthalic-Based Anticancer Tyrosine Kinase Inhibitors: Design, Synthesis and Biological Activity
Abstract
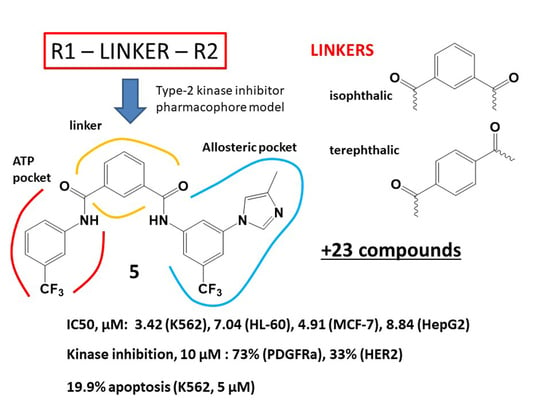
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
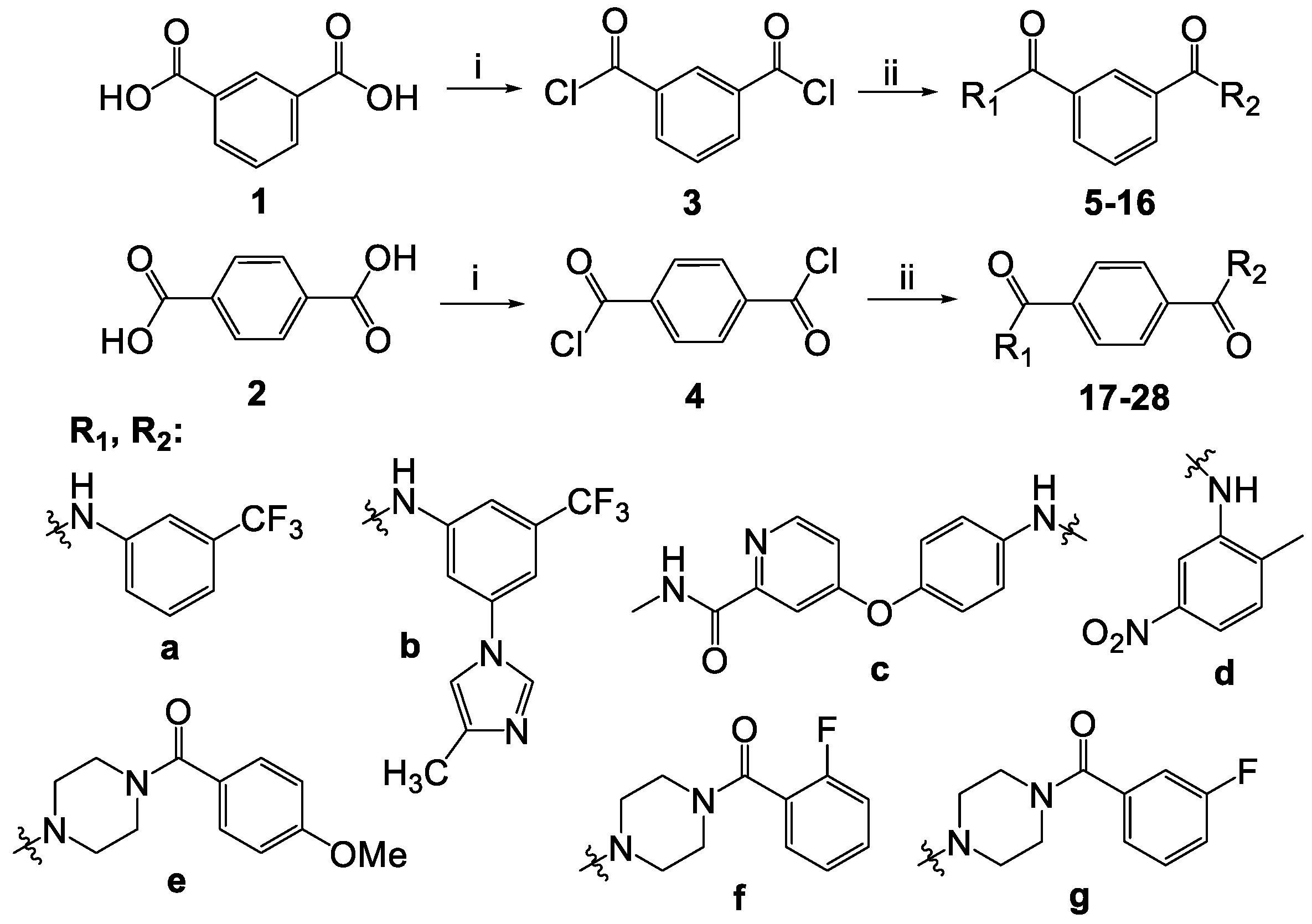
2.1.1. Synthesis of Isophthaloyl and Terephthaloyl Dichlorides 3 and 4
2.1.2. General Method for the Synthesis of Compounds 5–28
2.2. Cell Proliferation Assay
2.3. Kinase Inhibitory Assay
2.4. Cell Apoptosis Assay
2.5. In Vitro Cell Cycle Effects
2.6. ROS Determination
2.7. Molecular Docking and Molecular Dynamics
3. Results and Discussion
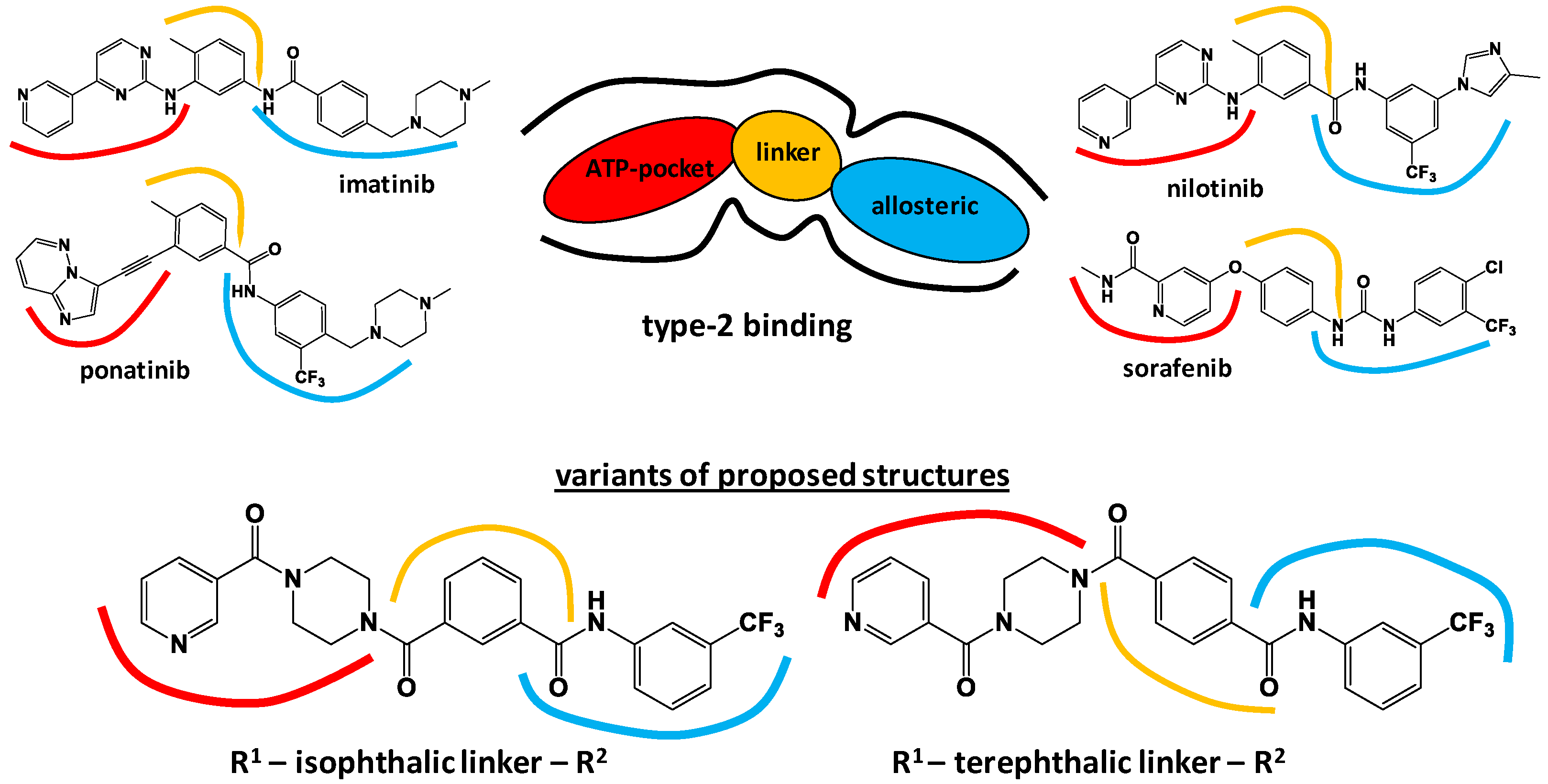
3.1. Molecular Design
3.2. Chemistry
3.3. Biological Studies
3.3.1. Antiproliferative Activity
3.3.2. Kinase Inhibitory Assays
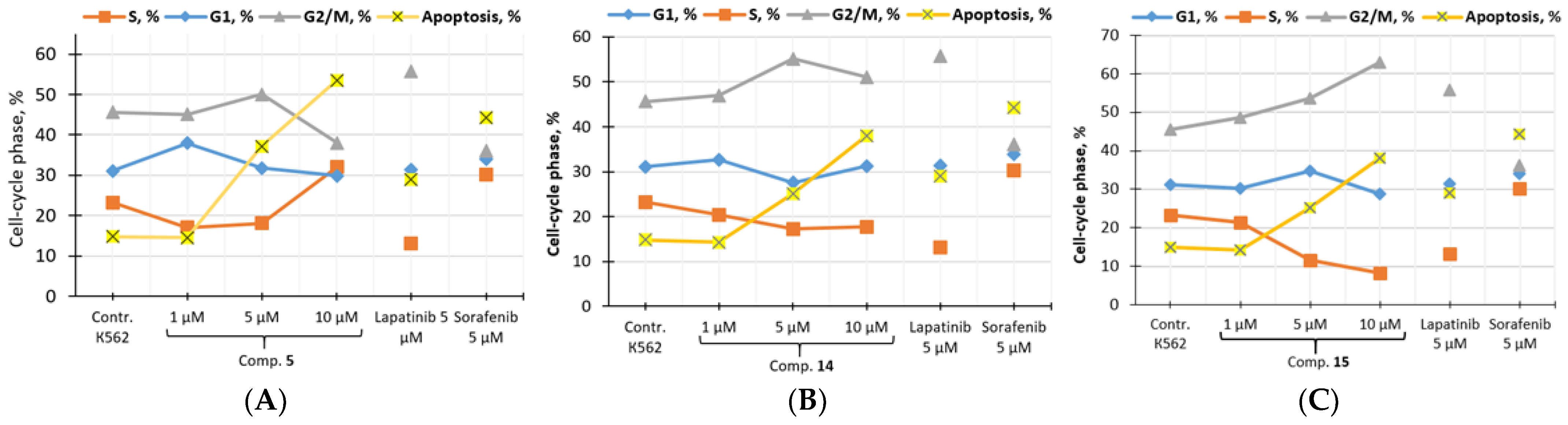
3.3.3. In Vitro Cell Cycle Effects of Compounds 5, 14 and 15
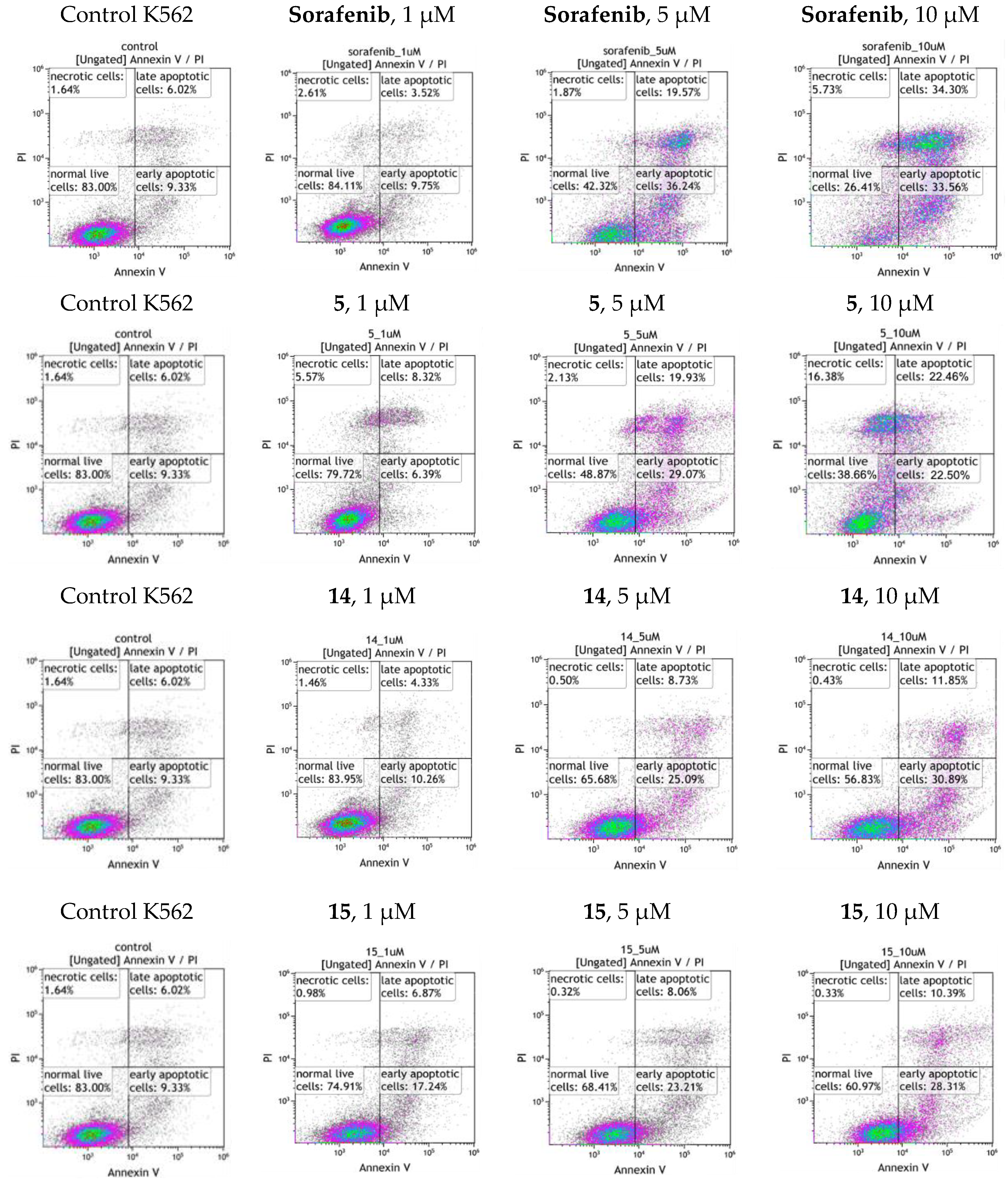
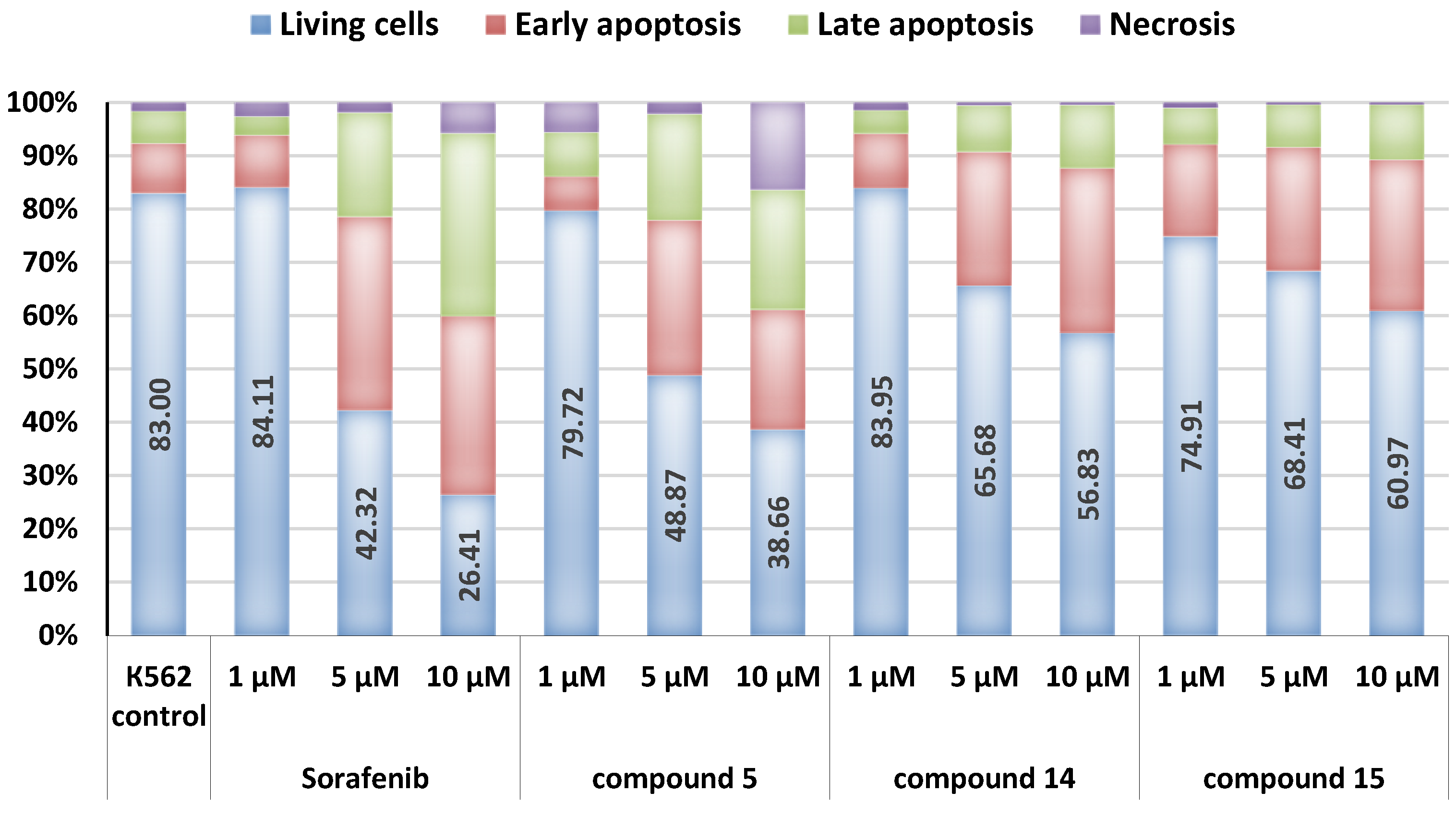
3.3.4. Apoptosis Assay
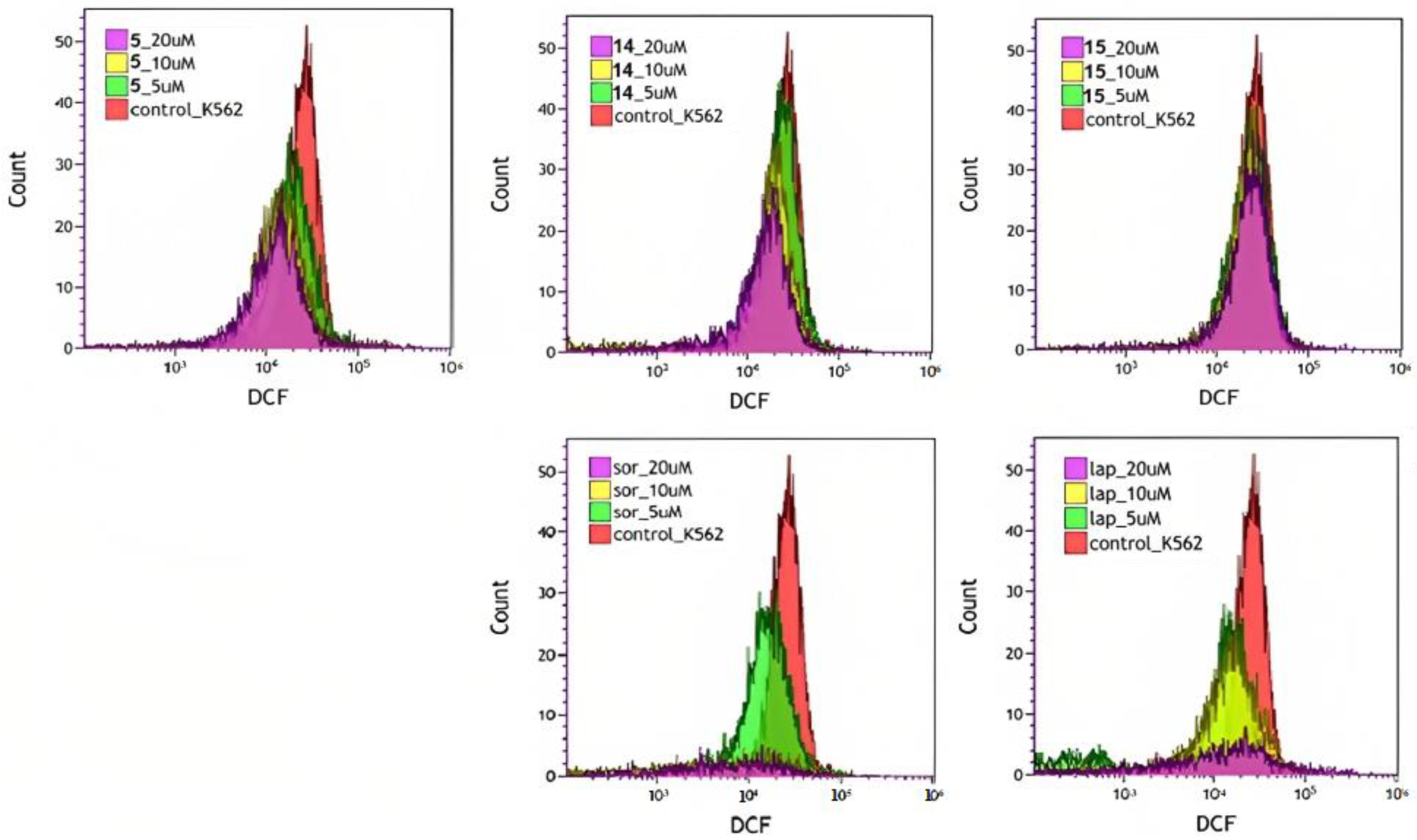
3.3.5. Reactive Oxygen Species (ROS)
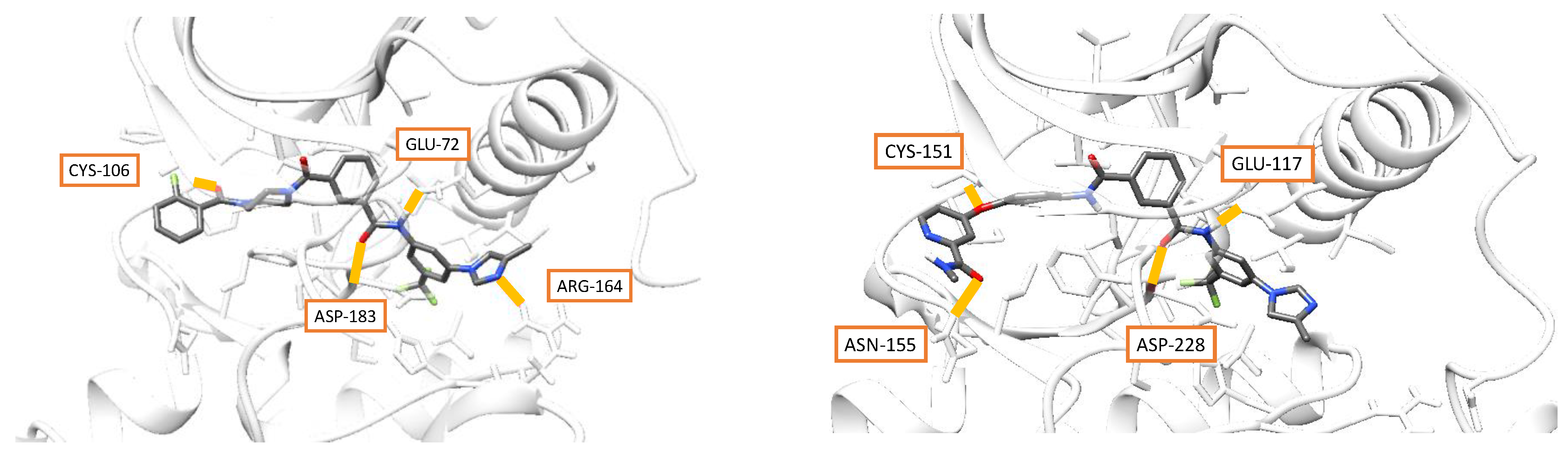
3.4. Docking and Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moen, M.D.; McKeage, K.; Plosker, G.L.; Siddiqui, M.A.A. Imatinib: A review of its use in chronic myeloid leukaemia. Drugs 2007, 67, 299–320. [Google Scholar] [CrossRef]
- Riegel, K.; Vijayarangakannan, P.; Kechagioglou, P.; Bogucka, K.; Rajalingam, K. Recent advances in targeting protein kinases and pseudokinases in cancer biology. Front. Cell Dev. Biol. 2022, 10, 942500. [Google Scholar] [CrossRef]
- Theivendren, P.; Kunjiappan, S.; Hegde, Y.M.; Vellaichamy, S.; Gopal, M.; Dhramalingam, S.R.; Kumar, S. Importance of Protein Kinase and Its Inhibitor: A Review. In Protein Kinases; Singh, R.K., Ed.; IntechOpen: Rijeka, Croatia, 2021; Chapter 5. [Google Scholar] [CrossRef]
- Bhatia, R.; Singh, R.K. Introductory chapter: Protein kinases as promising targets for drug design against cancer. In Protein Kinases; Singh, R.K., Ed.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Bajinting, A.; Ng, H.L. Chapter Nine—Structural studies of full-length receptor tyrosine kinases and their implications for drug design. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 124, pp. 311–336. [Google Scholar] [CrossRef]
- Barouch-Bentov, R.; Sauer, K. Mechanisms of drug resistance in kinases. Expert. Opin. Investig. Drugs 2011, 20, 153–208. [Google Scholar] [CrossRef]
- Soverini, S.; Rosti, G.; Iacobucci, I.; Baccarani, M.; Martinelli, G. Choosing the best second-line tyrosine kinase inhibitor in imatinib-resistant chronic myeloid leukemia patients harboring Bcr-Abl kinase domain mutations: How reliable is the IC50? Oncologist 2011, 16, 868–876. [Google Scholar] [CrossRef]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, D.; Yokota, K.; Gouda, M.; Narumi, Y.; Ohmoto, H.; Nishiwaki, E.; Akita, K.; Kirii, Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells 2013, 18, 110–122. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [CrossRef]
- Papaetis, G.S.; Syrigos, K.N. Sunitinib: A multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 2009, 23, 377–389. [Google Scholar] [CrossRef]
- Escudier, B.; Worden, F.; Kudo, M. Sorafenib: Key lessons from over 10 years of experience. Expert Rev. Anticancer Ther. 2019, 19, 177–189. [Google Scholar] [CrossRef]
- Takeda, T.; Tsubaki, M.; Kato, N.; Genno, S.; Ichimura, E.; Enomoto, A.; Imano, M.; Satou, T.; Nishida, S. Sorafenib treatment of metastatic melanoma with c Kit aberration reduces tumor growth and promotes survival. Oncol. Lett. 2021, 22, 827. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Gallego, G.; Blanco, M.; Figueroa, A.; García-Campelo, R.; Valladares-Ayerbes, M.; Grande-Pulido, E.; Antón-Aparicio, L. New insights into molecular mechanisms of sunitinib-associated side effects. Mol. Cancer Ther. 2011, 10, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schult, C.; Dahlhaus, M.; Ruck, S.; Sawitzky, M.; Amoroso, F.; Lange, S.; Etro, D.; Glass, A.; Fuellen, G.; Boldt, S.; et al. The multikinase inhibitor Sorafenib displays significant antiproliferative effects and induces apoptosis via caspase 3, 7 and PARP in B- and T-lymphoblastic cells. BMC Cancer 2010, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Björklund, A.C.; Zhivotovsky, B.; Grandér, D.; et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Gao, L.; Huang, X.; Weng, J.; Chen, Y.; Lin, S.; Yin, Q. Sorafenib prevents the proliferation and induces the apoptosis of liver cancer cells by regulating autophagy and hypoxia-inducible factor-1. Exp. Ther. Med. 2021, 22, 980. [Google Scholar] [CrossRef]
- Blackhall, F.; Cappuzzo, F. Crizotinib: From discovery to accelerated development to front-line treatment. Ann Oncol. 2016, 27, iii35–iii41. [Google Scholar] [CrossRef]
- Elnair, R.; Galal, A. Finding the right BCR-ABL1 tyrosine kinase inhibitor: A case report of successful treatment of a patient with chronic myeloid leukemia and a V299L mutation using nilotinib. BMC Cancer 2018, 18, 1097. [Google Scholar] [CrossRef]
- Tadesse, F.; Asres, G.; Abubeker, A.; Gebremedhin, A.; Radich, J. Spectrum of BCR-ABL Mutations and Treatment Outcomes in Ethiopian Imatinib-Resistant Patients with Chronic Myeloid Leukemia. JCO Glob. Oncol. 2021, 7, 1187–1193. [Google Scholar] [CrossRef]
- Li, W.; Feng, C.; Di, W.; Hong, S.; Chen, H.; Ejaz, M.; Yang, Y.; Xu, T.R. Clinical use of vascular endothelial growth factor receptor inhibitors for the treatment of renal cell carcinoma. Eur. J. Med. Chem. 2020, 200, 112482. [Google Scholar] [CrossRef]
- Epaillard, N.; Simonaggio, A.; Elaidi, R.; Azzouz, F.; Braychenko, E.; Thibault, C.; Sun, C.M.; Moreira, M.; Oudard, S.; Vano, Y.A. BIONIKK: A phase 2 biomarker driven trial with nivolumab and ipilimumab or VEGFR tyrosine kinase inhibitor (TKI) in naïve metastatic kidney cancer. Bull. Cancer 2020, 107, eS22–eS27. [Google Scholar] [CrossRef]
- Killock, D. Biomarkers 101—Personalizing therapy for RCC. Nat. Rev. Clin. Oncol. 2020, 17, 653. [Google Scholar] [CrossRef] [PubMed]
- D’Avella, C.; Abbosh, P.; Pal, S.K.; Geynisman, D.M. Mutations in renal cell carcinoma. Urol. Oncol. 2020, 38, 763–773. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Yang, X.; Duan, Y.; Han, J.; Liao, C. Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. J. Med. Chem. 2021, 64, 1283–1345. [Google Scholar] [CrossRef]
- Faryna, A.V.; Kalinichenko, E.N. Computer-aided molecular design of new potential inhibitors of protein kinases using of 4-methyl-benzoic acid as a linker. J. Comput. Chem. Mol. Model. 2019, 3, 285–293. [Google Scholar] [CrossRef]
- Kalinichenko, E.; Faryna, A.; Kondrateva, V.; Vlasova, A.; Shevchenko, V.; Melnik, A.; Avdoshko, O.; Belko, A. Synthesis, biological activities and docking studies of novel 4-(arylaminomethyl)benzamide derivatives as potential tyrosine kinase inhibitors. Molecules 2019, 24, 3543. [Google Scholar] [CrossRef] [Green Version]
- Kalinichenko, E.; Faryna, A.; Bozhok, T.; Panibrat, A. Synthesis, In Vitro and In Silico Anticancer Activity of New 4-Methylbenzamide Derivatives Containing 2,6-Substituted Purines as Potential Protein Kinases Inhibitors. Int. J. Mol. Sci. 2021, 22, 12738. [Google Scholar] [CrossRef]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [Green Version]
- NCI Chemical Identifier Resolver. Available online: http://cactus.nci.nih.gov/chemical/structure (accessed on 31 October 2022).
- Huang, W.; Shakespeare, W.C. An Efficient Synthesis of Nilotinib (AMN107). Synthesis 2007, 14, 2121–2124. [Google Scholar] [CrossRef]
- Wei, Y.; To, K.K.W.; Au-Yeng, S.C.F. Synergistic cytotoxicity from combination of imatinib and platinum-based anticancer drugs specifically in Bcr-Abl positive leukemia cells. J. Pharmacol. Sci. 2015, 129, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabarius, A.; Giehl, M.; Rebacz, B.; Krämer, A.; Frank, O.; Haferlach, C.; Duesberg, P.; Hehlmann, R.; Seifarth, W.; Hochhaus, A. Centrosome aberrations and G1 phase arrest after in vitro and in vivo treatment with the SRC/ABL inhibitor dasatinib. Haematologica 2008, 93, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauzay, C.; Louandre, C.; Bodeau, S.; Anglade, F.; Godin, C.; Saidak, Z.; Fontaine, L.X.; Usureau, C.; Martin, N.; Molinie, R.; et al. Protein biosynthesis, a target of sorafenib, interferes with the unfolded protein response (UPR) and ferroptosis in hepatocellular carcinoma cells. Oncotarget 2018, 9, 8400–8414. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Yao, M.; Shang, Y.Y.; Zhou, Z.W.; Yang, Y.X.; Wu, Y.S.; Guan, L.F.; Wang, X.Y.; Zhou, S.F.; Wei, X. The research on lapatinib in autophagy, cell cycle arrest and epithelial to mesenchymal transition via Wnt/ErK/PI3K-AKT signaling pathway in human cutaneous squamous cell carcinoma. J. Cancer 2017, 8, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 50, 2597. [Google Scholar] [CrossRef] [Green Version]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Matés, J.M.; Sánchez-Jiménez, F.M. Role of reactive oxygen species in apoptosis: Implications for cancer therapy. Int. J. Biochem. Cell Biol. 2000, 32, 157–170. [Google Scholar] [CrossRef]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The Tumorigenic Roles of the Cellular REDOX Regulatory Systems. Oxid. Med. Cell Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_mmpbsa: A new tool to perform end-state free energy calculations with gromacs. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faryna, A.; Kalinichenko, E. N1-(3-(Trifluoromethyl)Phenyl) Isophthalamide Derivatives as Promising Inhibitors of Vascular Endothelial Growth Factor Receptor: Pharmacophore-Based Design, Docking, and MM-PBSA/MM-GBSA Binding Energy Estimation. In Molecular Docking-Recent Advances; Erman Salih, I., Ed.; IntechOpen: Rijeka, Croatia, 2022; Chapter 8. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | Isophthalic Derivatives | Terephthalic Derivatives | ||||
|---|---|---|---|---|---|---|---|
 |  | ||||||
| No. | Structure | Yield, % | No. | Structure | Yield, % | ||
| a | b | 5 |  | 15 | 17 |  | 20 |
| a | c | 6 |  | 23 | 18 |  | 10 |
| a | e | 7 |  | 15 | 19 |  | 16 |
| a | f | 8 |  | 57 | 20 |  | 35 |
| a | g | 9 |  | 44 | 21 |  | 50 |
| b | b | 10 |  | 14 | 22 |  | 27 |
| b | c | 11 |  | 11 | 23 |  | 12 |
| b | d | 12 |  | 13 | 24 |  | 20 |
| b | e | 13 |  | 10 | 25 |  | 14 |
| b | f | 14 |  | 25 | 26 |  | 18 |
| b | g | 15 |  | 17 | 27 |  | 12 |
| c | d | 16 |  | 9 | 28 |  | 10 |
| Comp. | Acid | R1 | R2 | Anti-Proliferative Activities in Deferent Cell Lines (IC50 µM) 1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K562 | HL-60 | MCF-7 | HepG2 | A549 | OKP-GS | RPMI 1788 | ||||
| 5 | Iso | a | b | 3.42 ± 0.78 | 7.04 ± 0.29 | 4.91 ± 1.27 | 7.84 ± 2.61 | >50 | 17.76 ± 2.13 | 2.86 ± 0.09 |
| 6 | Iso | a | c | >50 | >50 | 41.15 ± 5.06 | >50 | >50 | >50 | 12.98 ± 2.99 |
| 8 | Iso | a | f | 18.33 ± 1.15 | 18.19 ± 1.02 | 24.71 ± 5.95 | 35.49 ± 0.34 | >50 | >50 | 24.91 ± 1.17 |
| 9 | Iso | a | g | 20.99 ± 0.59 | 21.90 ± 1.59 | 26.29 ± 3.71 | 33.29 ± 3.45 | >50 | >50 | 26.26 ± 1.88 |
| 10 | Iso | b | b | 25.96 ± 2.38 | 35.75 ± 3.19 | 38.47 ± 1.95 | 23.21 ± 1.81 | >50 | 13.23 ± 1.30 | 13.00 ± 0.30 |
| 11 | Iso | b | c | >50 | >50 | >50 | >50 | >50 | 26.51 ± 3.65 | 21.89 ± 0.44 |
| 12 | Iso | b | d | >50 | >50 | >50 | >50 | >50 | 27.47 ± 2.58 | 2.36 ± 1.13 |
| 14 | Iso | b | f | 7.71 ± 2.84 | 32.50 ± 1.50 | 27.30 ± 0.16 | 44.09 ± 4.19 | >50 | >50 | 26.63 ± 2.31 |
| 15 | Iso | b | g | 10.51 ± 2.54 | 34.76 ± 4.70 | 27.38 ± 0.26 | 45.81 ± 1.76 | >50 | >50 | 29.40 ± 1.79 |
| 17 | Tere | a | b | 15.62 ± 0.00 | >50 | >50 | >50 | >50 | >50 | >50 |
| 18 | Tere | a | c | 26.93 ± 0.00 | >50 | >50 | 45.85 ± 1.83 | >50 | 34.43 ± 2.52 | 46.02 ± 1.76 |
| 19 | Tere | a | e | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 20 | Tere | a | f | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 21 | Tere | a | g | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 22 | Tere | b | b | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 24 | Tere | b | d | 16.98 ± 0.20 | 26.42 ± 3.35 | 15.48 ± 1.16 | >50 | >50 | >50 | 36.49 ± 0.86 |
| 26 | Tere | b | f | 18.53 ± 0.73 | 26.41 ± 1.73 | 48.83 ± 1.17 | 44.28 ± 4.79 | >50 | >50 | 23.97 ± 0.03 |
| 27 | Tere | b | g | 22.25 ± 2.12 | 26.04 ± 4.15 | 49.36 ± 0.67 | 38.63 ± 1.11 | >50 | >50 | 24.08 ± 0.07 |
| Reference Compounds | ||||||||||
| Sorafenib | 2.44 ± 0.21 | 3.37 ± 0.42 | 8.62 ± 2.24 | 5.01 ± 0.14 | 12.52 ± 1.64 | 7.17 ± 0.50 | 3.48 ± 0.39 | |||
| Nilotinib | <0.1 | 22.67 ± 4.76 | 4.20 ± 0.14 | 4.81 ± 0.21 | >50 | 2.76 ± 0.06 | 19.32 ± 0.09 | |||
| Lapatinib | 9.62 ± 1.56 | 10.58 ± 1.02 | 10.93 ± 0.34 | 27.10 ± 0.08 | 18.55 ± 2.65 | 7.32 ± 2.15 | 9.31 ± 0.90 | |||
| Kinase | % Inhibition at 10 µM | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lapat. | Soraf. | Sunit. | 5 | 8 | 9 | 10 | 14 | 17 | 19 | 24 | |
| Tyrosine Kinases | |||||||||||
| EGFR | 100 | 0 | - | 0 | 14 | 90 | 1 | 21 | 0 | 4 | 2 |
| HER2 | 76 | 0 | - | 33 | 0 | 64 | 19 | 14 | 13 | 39 | 49 |
| HER4 | 100 | 0 | - | 0 | 0 | 37 | 0 | 0 | 0 | 2 | 3 |
| IGF1R | 0 | 0 | - | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| InsR | 0 | 0 | - | 0 | 6 | 0 | 0 | 0 | 0 | 17 | 0 |
| KDR | 24 | 100 | - | 18 | 34 | 33 | 33 | 26 | 0 | 16 | 0 |
| PDGFRα | 2 | 100 | - | 73 | 0 | 20 | 17 | 24 | 20 | 19 | 0 |
| PDGFRβ | 10 | 100 | - | 18 | 0 | 24 | 0 | 23 | 15 | 20 | 0 |
| Serine/Threonine Kinases | |||||||||||
| Aurora A | - | - | 68 | 0 | 0 | 0 | 0 | 0 | 0 | - | 0 |
| Aurora B | - | - | 90 | 0 | 0 | 0 | 0 | 7 | 0 | - | 3 |
| Comp. | Concentration, µM | G1, % | S, % | G2/M, % | Apoptosis, % |
|---|---|---|---|---|---|
| K562 (control) | 31.11 | 23.27 | 45.62 | 14.80 | |
| 5 | 1 | 37.92 | 17.01 | 45.08 | 14.56 |
| 5 | 31.77 | 18.14 | 50.09 | 37.20 | |
| 10 | 29.86 | 32.05 | 38.08 | 53.50 | |
| 14 | 1 | 32.65 | 20.41 | 46.95 | 14.28 |
| 5 | 27.58 | 17.25 | 55.16 | 25.10 | |
| 10 | 31.21 | 17.71 | 51.08 | 38.02 | |
| 15 | 1 | 30.15 | 21.31 | 48.54 | 17.90 |
| 5 | 34.76 | 11.59 | 53.65 | 20.54 | |
| 10 | 28.85 | 8.16 | 62.99 | 32.72 | |
| Lap. | 1 | 34.47 | 13.31 | 52.23 | 14.74 |
| 5 | 31.31 | 13.02 | 55.68 | 28.92 | |
| Sor. | 1 | 36.50 | 7.72 | 55.78 | 22.60 |
| 5 | 33.87 | 30.14 | 35.99 | 44.20 | |
| Concentration, µM | Control | ROS Level, % of Control | ||||
|---|---|---|---|---|---|---|
| Compound | ||||||
| 5 | 14 | 15 | Sor. | Lap. | ||
| 5 | 100% | 76.76 | 92.84 | 92.89 | 69.20 | 64.12 |
| 10 | 64.66 | 76.96 | 87.87 | 79.03 | 72.23 | |
| 20 | 62.90 | 71.90 | 94.07 | 71.59 | 89.07 | |
| Protein Family | PDB Code | Linker | N Comp. | Gmx_Mmpbsa, kcal/mol | RF- Score | g_mmpbsa, kJ/mol | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| v.d.w. | Electr. | Entr. | Total | v.d.w. | Electr. | Total | |||||
| ABL | 3cs9 | iso | 7 | −68 | −21.6 | 13.8 | −46 | 6.1 | −284.4 | −43.96 | −141.8 |
| ABL | 3cs9 | iso | 8 | −62.6 | −14 | 13.2 | −41 | 6.2 | −262 | −29.21 | −128.6 |
| ABL | 3cs9 | iso | 11 | −77.1 | −37.1 | 9.1 | −47.9 | 6.3 | −322.3 | −77.15 | −144.4 |
| ABL | 3cs9 | iso | 14 | −72.6 | −13.5 | 6 | −53.3 | 6.3 | −303.5 | −28.01 | −144.9 |
| TRKC | 6kzd | iso | 8 | −59.3 | −2.4 | 13.3 | −34.5 | 6.0 | −248 | −5.111 | −123.5 |
| TRKC | 6kzd | iso | 9 | −48.5 | −30.7 | 14.4 | −21.5 | 6.0 | −202.8 | −64.39 | −76.8 |
| TRKC | 6kzd | iso | 13 | −65.8 | −40.7 | 17.8 | −31.4 | 6.3 | −275.3 | −85.14 | −95.59 |
| TRKC | 6kzd | iso | 14 | −62.7 | −4.4 | 16.2 | −31.4 | 6.1 | −262.3 | −9.205 | −111.5 |
| TRKC | 6kzd | iso | 15 | −70.1 | −20 | 9.9 | −45.6 | 6.2 | −293.4 | −41.89 | −137.9 |
| TRKC | 6kzd | tere | 21 | −58.1 | −29.8 | 8.6 | −39.3 | 6.0 | −243.1 | −62.39 | −128.4 |
| TRKC | 6kzd | tere | 25 | −69.8 | −22.4 | 14.2 | −37 | 6.2 | −291.8 | −46.92 | −149 |
| TRKC | 6kzd | tere | 26 | −69.6 | −19 | 18.7 | −38.8 | 6.2 | −291.3 | −39.37 | −141.1 |
| TRKC | 6kzd | tere | 27 | −63.8 | −5.5 | 15.2 | −27.4 | 6.2 | −266.7 | −11.69 | −125.9 |
| VEGFR1 | 3hng | iso | 5 | −64.9 | −22.6 | 16 | −36.8 | 6.3 | −271.3 | −47.31 | −119.6 |
| VEGFR2 | 4ag8 | iso | 5 | −65.4 | −23.2 | 11.2 | −44 | 6.2 | −273.6 | −49.12 | −138.8 |
| VEGFR2 | 4ag8 | iso | 7 | −65.2 | −33.1 | 7.7 | −58 | 6.2 | −272.9 | −69.09 | −148.4 |
| VEGFR2 | 4ag8 | iso | 11 | −76.4 | −34.4 | 9.3 | −53.5 | 6.5 | −319.5 | −71.87 | −150.8 |
| VEGFR2 | 3wze | iso | 11 | −71.1 | −26.2 | 9.9 | −48.8 | 6.5 | −297.4 | −54.7 | −150.5 |
| VEGFR2 | 4asd | iso | 11 | −79.9 | −33.8 | 10.1 | −53.6 | 6.5 | −334.2 | −70.75 | −155.3 |
| VEGFR2 | 3wze | iso | 14 | −74 | −30.8 | 7.4 | −64 | 6.5 | −309.6 | −64.57 | −157.0 |
| VEGFR2 | 3wze | tere | 21 | −51.9 | −30.3 | 8 | −34.7 | 6.0 | −217 | −63.19 | −85.65 |
| BRAF | 5hi2 | iso | 8 | −57.4 | −27.4 | 7 | −46.6 | 6.1 | −240.2 | −57.45 | −118.9 |
| ERBB2 | 3pp0 | iso | 9 | −55.6 | −9.1 | 7.7 | −34.1 | 6.0 | −232.6 | −19.04 | −96.72 |
| ERBB2 | 3pp0 | tere | 20 | −50.8 | −15.6 | 11.5 | −26.7 | 6.0 | −212.6 | −32.59 | −86.55 |
| ERBB4 | 3bbt | iso | 14 | −50.4 | −22.4 | 8.6 | −30.7 | 6.0 | −211.0 | −46.8 | −79.0 |
| Reference ligands | |||||||||||
| ABL | 2hyy | imatinib | −67.4 | −20.1 | 5.3 | −50.5 | 6.2 | −282.1 | −42.4 | −154.7 | |
| ABL | 3cs9 | nilotinib | −72.8 | −28.1 | 6.1 | −56.1 | 6.6 | −304.6 | −57.8 | −183.1 | |
| VEGFR2 | 3wze | sorafenib | −60.8 | −35.3 | 7.4 | −58.5 | 6.2 | −254.3 | −73.9 | −170.0 | |
| VEGFR2 | 4asd | sorafenib | −60.3 | −37.9 | 5.9 | −59.8 | 6.2 | −252.2 | −79.3 | −153.0 | |
| ERBB4 | 3bbt | lapatinib | −58.7 | −26.5 | 6.2 | −54.4 | 6.1 | −245.6 | −55.2 | −179.8 | |
| TRKC | 6kzd | not approved | −82.6 | −31.7 | 8.2 | −61.0 | 6.4 | −345.5 | −66.2 | −206.9 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinichenko, E.; Faryna, A.; Bozhok, T.; Golyakovich, A.; Panibrat, A. Novel Phthalic-Based Anticancer Tyrosine Kinase Inhibitors: Design, Synthesis and Biological Activity. Curr. Issues Mol. Biol. 2023, 45, 1820-1842. https://doi.org/10.3390/cimb45030117
Kalinichenko E, Faryna A, Bozhok T, Golyakovich A, Panibrat A. Novel Phthalic-Based Anticancer Tyrosine Kinase Inhibitors: Design, Synthesis and Biological Activity. Current Issues in Molecular Biology. 2023; 45(3):1820-1842. https://doi.org/10.3390/cimb45030117
Chicago/Turabian StyleKalinichenko, Elena, Aliaksandr Faryna, Tatyana Bozhok, Anna Golyakovich, and Alesya Panibrat. 2023. "Novel Phthalic-Based Anticancer Tyrosine Kinase Inhibitors: Design, Synthesis and Biological Activity" Current Issues in Molecular Biology 45, no. 3: 1820-1842. https://doi.org/10.3390/cimb45030117