
Early-Onset Ovarian Cancer <30 Years: What Do We Know about Its Genetic Predisposition?

 , , , and
, , , and
Abstract
:1. Introduction
2. Characteristics of Early-Onset and Late-Onset OC
3. OC Risk Factors and Predisposition
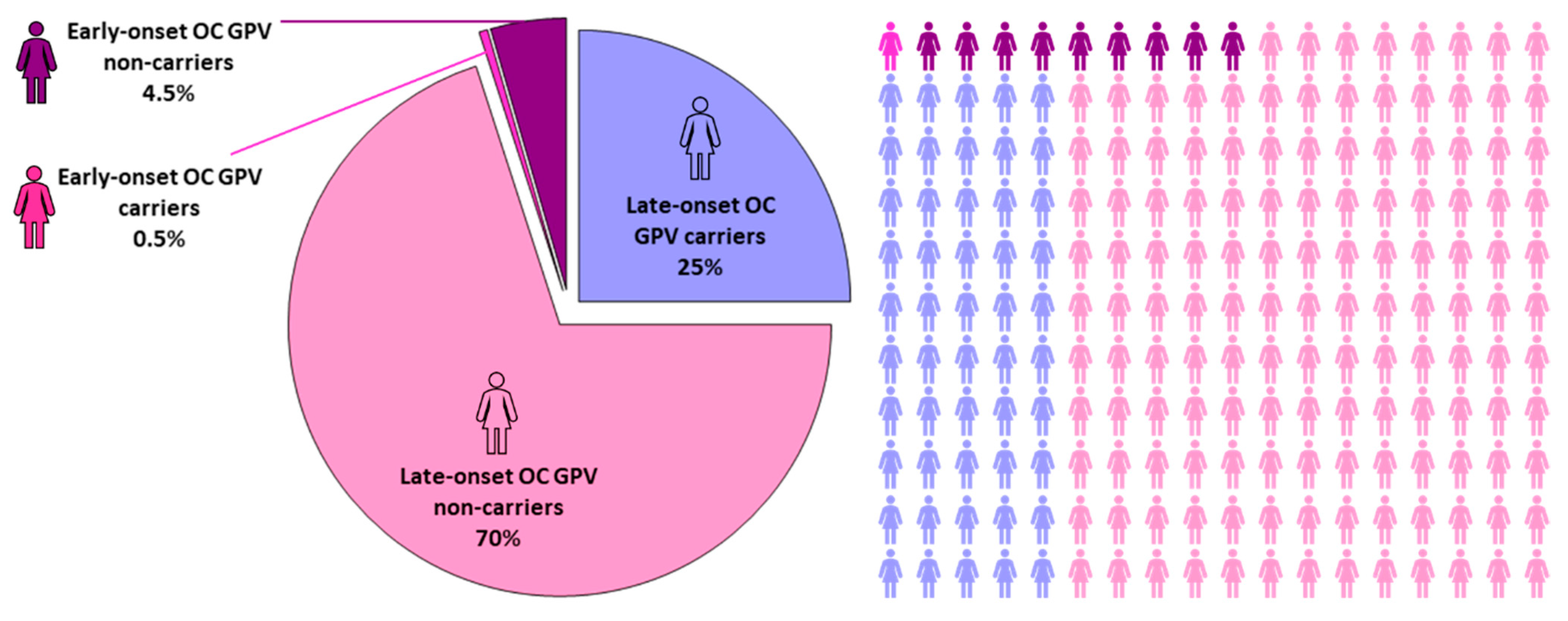
3.1. Established OC Predisposition Genes

{kind=link}
{kind=link}
| Gene | Heterozygote | Homozygote/ Compound Heterozygote [54] | |||
|---|---|---|---|---|---|
| Associated OC Histotype | Absolute Risk for OC [35] | GPV Identified in Early-Onset OC | Other Associated Cancer Types [35] | ||
| High penetrance | |||||
| BRCA1 | Epithelial [41] | 39–58% | Yes [4,9] | BC, PaC, PrC | FA-S |
| BRCA2 | Epithelial [41] | 13–29% | Yes [4] | BC, PaC, PrC, MM | FA-D1 |
| BRIP1 | Epithelial [41] | 5–15% | Yes [4,9] | BC, CrC, EC | FA-J |
| DICER1 | Sex cord-stromal [47] | NA | Yes [47,48] | DICER1 sy | - |
| MLH1 | Epithelial [37,41] | 4–20% | No | Lynch sy—CrC, EC, PaC | CMMRD |
| MSH2 | Epithelial [37] | 8–38% | Yes [40,52] | Lynch sy—CrC, EC, PaC | CMMRD |
| RAD51C | Epithelial [41] | 10–15% | Yes [4] | BC | FA-O |
| RAD51D | Epithelial [41] | 10–20% | No | BC | - |
| SMARCA4 | SCCOHT [50] | NA | Yes [50,51] | Rhabdoid tumor predisposition sy | - |
| STK11 | Non-epithelial [45] | >10% | Yes [46] | Peutz–Jeghers sy, BC, PaC, CrC | - |
| Moderate penetrance/Insufficient evidence | |||||
| ATM | Epithelial [41] | 2–3% | Yes [4,9] | PaC | AT |
| MSH6 | Epithelial [37] | 1–13% | No | Lynch sy—CrC, EC, PaC | CMMRD |
| PMS2 | Epithelial [37] | 1–3% | Yes [40,52] | Lynch sy—CrC, EC | CMMRD |
| PALB2 | Epithelial [42] | 3–5% | No | BC, PaC | FA-N |
3.2. Candidate OC Predisposition Genes
| Gene | GPVs Identified in Early-Onset OC | Associated Disease (Inheritance Mode) [54] | OC-Association Reported in |
|---|---|---|---|
| ABRAXAS1 (FAM175A) | No | - | [56,86] |
| ATR | No | Cutaneous telangiectasia and cancer sy (AD) | [78] |
| APC | Yes (early 30s) [63] | Familial adenomatous polyposis (AD) | [62,63] |
| BAP1 | No | Melanoma (AD) | [68] |
| BARD1 | Yes [6,25,55,59,60] | BC (AD) | [71] |
| BLM | No | Bloom sy (AR) | [5,76] |
| BMPR1A | Yes [64] | Juvenile polyposis sy, primary ovarian insufficiency (AD) | [64] |
| BRAT | Yes (in their 30s) [85] | Neurodevelopmental disorder (AR) | [85] |
| CNKSR1 | No | - | [87] |
| CDKN2A | No | MM, MM-PaC sy (AD) | [5,72] |
| CHEK2 | Yes [6,25,55,59,60] | BC (AD) | [5,73] |
| ERCC3 | No | Trichothiodystrophy, xeroderma pigmentosum (AR) | [88,89] |
| FANCA | No | FA (AR) | [77,78] |
| FANCC | No | FA (AR) | [77] |
| FANCL | No | FA (AR) | [77] |
| FANCM | Yes [51] | - | [77,79] |
| FH | No | Leiomyomatosis and renal cell cancer (AD), fumarase deficiency (AR) | [68] |
| MEN1 | No | Multiple endocrine neoplasia (AD) | [65] |
| MRE11 | No | AT-like disorder (AR) | [81,84] |
| NBN | Yes [25] | Nijmegen breakage sy (AR) | [41] |
| NF1 | No | Neurofribromatosis (AD) | [5] |
| PIK3C2G | No | - | [87] |
| POLD1 | No | CrC, EC (AD) | [74] |
| POLE | No | CrC, EC (AD), IMAGE-I sy (AR) | [73] |
| POLK | No | - | [75] |
| PTEN | Yes [66] | Cowden sy (AD) | [66,67] |
| RAD50 | No | Nijmegen breakage syndrome-like disorder (AR) | [84] |
| RAD51B | No | - | [5,82] |
| RAD52 | No | - | [83] |
| RAD54B | No | - | [83] |
| RAD54L | No | - | [55] |
| RB1 | No | Retinoblastoma (AD) | [5] |
| RTEL1 | No | Dyskeratosis congenita (AD/AR), telomere-related pulmonary fibrosis, and/or bone marrow failure sy (AD) | [68] |
| SLX4 | No | FA (AR) | [75,81] |
| TP53 | Yes [51,69] | Li–Fraumeni sy (AD) | [5,69,70] |
| TSC2 | No | Tuberous sclerosis (AD) | [68] |
| VHL | No | von Hippel–Lindau sy, pheochromocytoma (AD) | [68] |
| WT1 | No | Wilms tumor (AD) | [68] |
| XRCC3 | No | - | [82] |
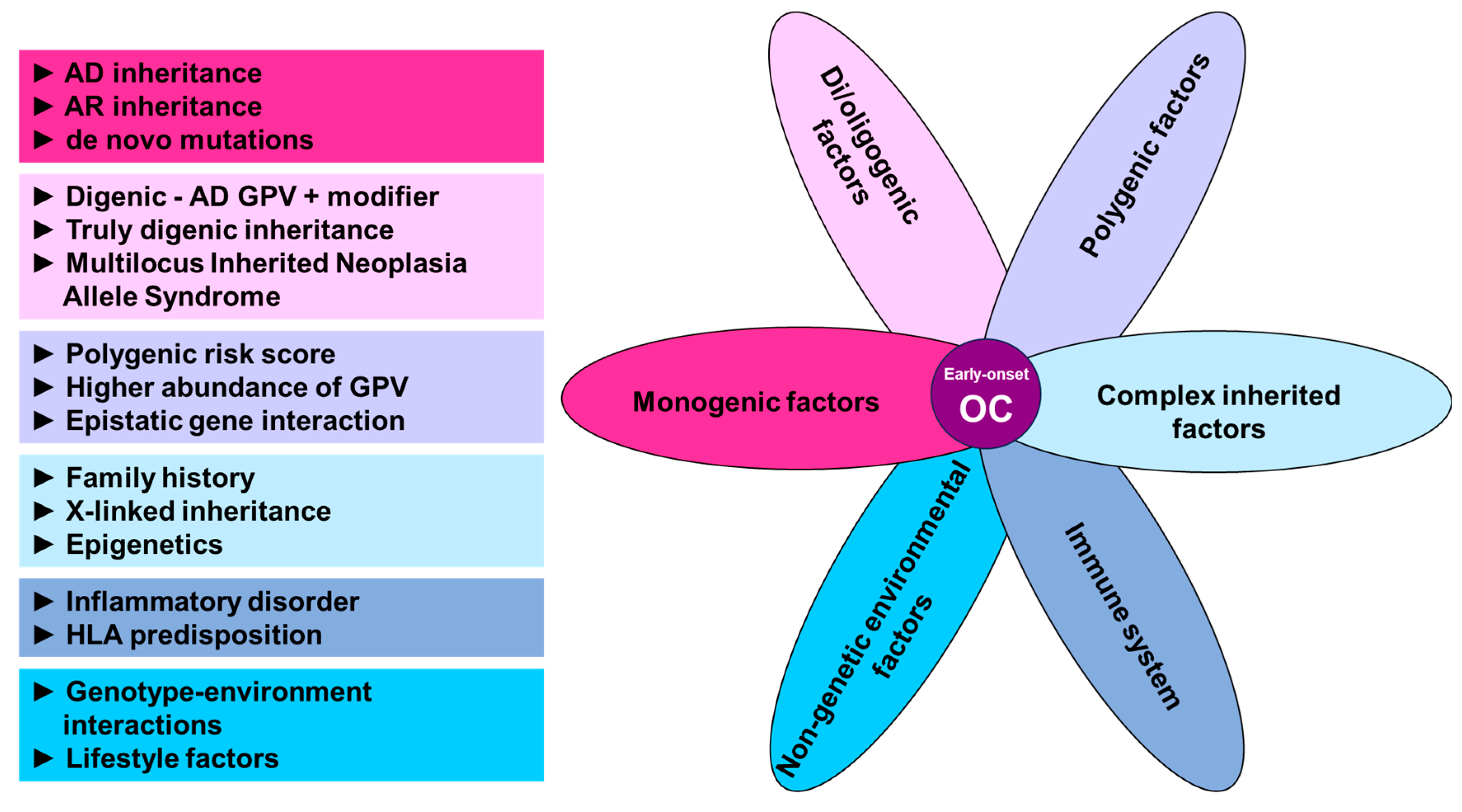
4. Alternative Approaches to Germline Genetic Testing in Early-Onset OC
4.1. Alternative Ways of Cancer Predisposition Inheritance
4.2. Family History and X-Linked Inheritance
4.3. Polygenic Inheritance
4.4. Di/Oligenic Inheritance
4.5. Immune-Related Modifiers of OC
4.6. Non-Genetic Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
Abbreviations
| AD | Autosomal Dominant |
| AR | Autosomal Recessive |
| AT | Ataxia-Telangiectasia |
| BC | Breast Cancer |
| CMMRD | Constitutional mismatch repair deficiency syndrome |
| CrC | Colorectal Cancer |
| Dg | Diagnosis |
| EC | Endometrial Cancer |
| FA | Fanconi Anemia |
| GPV | Germline Pathogenic/Likely Pathogenic Variant |
| HBOC | Hereditary Breast and Ovarian Cancer |
| HGSC | High-Grade Serous Carcinoma |
| HR | Homologous Recombination |
| IMAGE | Intrauterine growth restriction, Metaphyseal dysplasia, Adrenal hypoplasia congenita, and Genitourinary abnormalities |
| LGSC | Low-Grade Serous Carcinoma |
| LS | Lynch Syndrome |
| MINAS | Multilocus Inherited Neoplasia Allele Syndrome |
| MM | Malignant Melanoma |
| MMR | Mismatch Repair |
| NA | Not Available |
| NGS | Next-Generation Sequencing |
| No. | Number |
| OC | Ovarian cancer |
| OLIDA | Oligogenic Diseases Database |
| PaC | Pancreatic Cancer |
| PrC | Prostate Cancer |
| PRS | Polygenic Risk Score |
| SCCOHT | Small Cell Carcinoma of the Ovary Hypercalcemic Type |
| SNP | Single Nucleotide Polymorphism |
| Sy | Syndrome |
| WES | Whole Exome Sequencing |
| WGS | Whole Genome Sequencing |
| XCI | X Chromosome Inactivation |
| Yo | Years Old |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.; Bsc, M.F.B.; Me, J.F.; Soerjomataram, M.I.; et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-K.; Kim, K.; Kim, S.M.; Kim, J.W.; Park, N.-H.; Song, Y.-S.; Kang, S.-B. A hospital-based case-control study of identifying ovarian cancer using symptom index. J. Gynecol. Oncol. 2009, 20, 238–242. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Available online: www.seer.cancer.gov/ (accessed on 1 September 2023).
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. S4), iv1–iv18. [Google Scholar] [CrossRef] [PubMed]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; McLellan, M.D.; Leiserson, M.D.M.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Xie, M.; Kandoth, C.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 2001, 68, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.F.; Thompson, D.; Bobrow, L.; Dalal, N.; Gore, M.; Bishop, D.; Scott, I.; Evans, G.; Daly, P.; Easton, D.F.; et al. The genetic epidemiology of early-onset epithelial ovarian cancer: A population-based study. Am. J. Hum. Genet. 1999, 65, 1725–1732. [Google Scholar] [CrossRef]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline pathogenic variants identified in women with ovarian tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef]
- Prat, J. New insights into ovarian cancer pathology. Ann. Oncol. 2012, 23 (Suppl. S10), x111–x117. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Morden, C.R.; Farrell, A.C.; Sliwowski, M.; Lichtensztejn, Z.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. Chromosome instability is prevalent and dynamic in high-grade serous ovarian cancer patient samples. Gynecol. Oncol. 2021, 161, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Matz, M.; Coleman, M.P.; Sant, M.; Chirlaque, M.D.; Visser, O.; Gore, M.; Allemani, C.; Bouzbid, S.; Hamdi-Chérif, M.; Zaidi, Z.; et al. The histology of ovarian cancer: Worldwide distribution and implications for international survival comparisons (CONCORD-2). Gynecol. Oncol. 2017, 144, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- GLOBOCAN 2020. Available online: https://gco.iarc.fr/ (accessed on 1 September 2023).
- Lockley, M.; Stoneham, S.J.; Olson, T.A. Ovarian cancer in adolescents and young adults. Pediatr. Blood Cancer 2019, 66, e27512. [Google Scholar] [CrossRef]
- Chan, J.K.; Urban, R.; Cheung, M.K.; Osann, K.; Husain, A.; Teng, N.N.; Kapp, D.S.; Berek, J.S.; Leiserowitz, G.S. Ovarian cancer in younger vs older women: A population-based analysis. Br. J. Cancer. 2006, 95, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Grimley, P.M.; Matsuno, R.K.; Rosenberg, P.S.; Henson, D.E.; Schwartz, A.M.; Anderson, W.F. Qualitative Age Interactions between Low-grade and High-grade Serous Ovarian Carcinomas. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2256–2261. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Susini, T.; Savino, L.; Boddi, V.; Amunni, G.; Colafranceschi, M. Epithelial ovarian tumors in the reproductive age group: Age is not an independent prognostic factor. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 77, 1131–1136. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Fukuda, T.; Uemura, R.; Matsubara, H.; Wada, T.; Kawanishi, M.; Tasaka, R.; Kasai, M.; Hashiguchi, Y.; Ichimura, T.; et al. Age-related differences in prognosis and prognostic factors among patients with epithelial ovarian cancer. Mol. Clin. Oncol. 2018, 9, 329–334. [Google Scholar] [CrossRef]
- Schildkraut, J.M.; Halabi, S.; Bastos, E.; Marchbanks, P.A.; McDonald, J.A.; Berchuck, A. Prognostic factors in early-onset epithelial ovarian cancer: A population-based study. Obstet. Gynecol. 2000, 95, 119–127. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Bodurka, D.C.; Lu, K.H.; Nathan, L.C.; Milojevic, L.; Wong, K.K.; Malpica, A.; Sun, C.C. Impact of Age and Primary Disease Site on Outcome in Women With Low-Grade Serous Carcinoma of the Ovary or Peritoneum: Results of a Large Single-Institution Registry of a Rare Tumor. J. Clin. Oncol. 2015, 33, 2675–2682. [Google Scholar] [CrossRef]
- Lalrinpuii, E.; Bhageerathy, P.S.; Sebastian, A.; Jeyaseelan, L.; Thomas, V.; Thomas, A.; Chandy, R.; Peedicayil, A. Ovarian Cancer in Young Women. Indian J. Surg. Oncol. 2017, 8, 540–547. [Google Scholar] [CrossRef]
- Zhang, J.; Ugnat, A.-M.; Clarke, K.; Mao, Y. Ovarian cancer histology-specific incidence trends in Canada 1969–1993: Age-period-cohort analyses. Br. J. Cancer 1999, 81, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.T.; Terenziani, M.; Cecchetto, G.; Olson, T.A.; Schneider, D.T.; Terenziani, M.; Olson, T.A.; Schneider, D.T.; Cecchetto, G.; Olson, T.A.; et al. Gonadal and Extragonadal Germ Cell Tumors, Sex Cord Stromal and Rare Gonadal Tumors. In Rare Tumors in Children and Adolescents; Springer International Publishing: Cham, Switzerland, 2012; pp. 327–402. [Google Scholar]
- Huang, Y.; Ming, X.; Li, B.; Li, Z. Histological Characteristics and Early-Stage Diagnosis Are Associated With Better Survival in Young Patients With Epithelial Ovarian Cancer: A Retrospective Analysis Based on Surveillance Epidemiology and End Results Database. Front. Oncol. 2020, 10, 595789. [Google Scholar] [CrossRef]
- Fu, Z.; Brooks, M.M.; Irvin, S.; Jordan, S.; Aben, K.K.H.; Anton-Culver, H.; Bandera, E.V.; Beckmann, M.W.; Berchuck, A.; Brooks-Wilson, A.; et al. Lifetime ovulatory years and risk of epithelial ovarian cancer: A multinational pooled analysis. JNCI J. Natl. Cancer Inst. 2023, 115, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, M.; Chan, I.H.; Ariff, A.; Pharoah, P.D.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef] [PubMed]
- Witjes, V.M.; van Bommel, M.H.; Ligtenberg, M.J.; Vos, J.R.; Mourits, M.J.; Ausems, M.G.; de Hullu, J.A.; Bosse, T.; Hoogerbrugge, N. Probability of detecting germline BRCA1/2 pathogenic variants in histological subtypes of ovarian carcinoma. A meta-analysis. Gynecol. Oncol. 2022, 164, 221–230. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Chen, J.; Bae, E.; Zhang, L.; Hughes, K.; Parmigiani, G.; Braun, D.; Rebbeck, T.R. Penetrance of Breast and Ovarian Cancer in Women Who Carry a BRCA1/2 Mutation and Do Not Use Risk-Reducing Salpingo-Oophorectomy: An Updated Meta-Analysis. JNCI Cancer Spectr. 2020, 4, pkaa029. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.-K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 7. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 1.2023). 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 1 September 2023).
- Cummings, S.; Roman, S.S.; Saam, J.; Bernhisel, R.; Brown, K.; Lancaster, J.M.; Usha, L. Age of ovarian cancer diagnosis among BRIP1, RAD51C, and RAD51D mutation carriers identified through multi-gene panel testing. J. Ovarian Res. 2021, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Bernards, S.S.; Norquist, B.M.; Harrell, M.I.; Agnew, K.J.; Lee, M.K.; Walsh, T.; Swisher, E.M. Genetic characterization of early onset ovarian carcinoma. Gynecol. Oncol. 2016, 140, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Gupta, S.; Boland, C.R. Review article: Lynch Syndrome-a mechanistic and clinical management update. Aliment. Pharmacol. Ther. 2022, 55, 960–977. [Google Scholar] [CrossRef]
- Flaum, N.; Crosbie, E.J.; Woodward, E.R.; Lalloo, F.; Morgan, R.; Ryan, N.; Evans, D.G. MSH2 is the very young onset ovarian cancer predisposition gene, not BRCA1. J. Med. Genet. 2023, 60, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Narayan, P.; Ahsan, M.D.; Webster, E.M.; Perez, L.; Levi, S.R.; Harvey, B.; Wolfe, I.; Beaumont, S.; Brewer, J.T.; Siegel, D.; et al. Partner and localizer of BRCA2 (PALB2) pathogenic variants and ovarian cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2023, 177, 72–85. [Google Scholar] [CrossRef]
- Hearle, N.; Schumacher, V.; Menko, F.H.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin. Cancer Res. 2006, 12, 3209–3215. [Google Scholar] [CrossRef]
- Klimkowski, S.; Ibrahim, M.; Ibarra Rovira, J.J.; Elshikh, M.; Javadi, S.; Klekers, A.R.; Abusaif, A.A.; Moawad, A.W.; Ali, K.; Elsayes, K.M. Peutz-Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers 2021, 13, 5121. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz–Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- De Paolis, E.; Paragliola, R.M.; Concolino, P. Spectrum of DICER1 Germline Pathogenic Variants in Ovarian Sertoli-Leydig Cell Tumor. J. Clin. Med. 2021, 10, 1845. [Google Scholar] [CrossRef]
- Frio, T.R.; Bahubeshi, A.; Kanellopoulou, C.; Hamel, N.; Niedziela, M.; Sabbaghian, N.; Pouchet, C.; Gilbert, L.; O’Brien, P.K.; Serfas, K.; et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA 2011, 305, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Harris, A.; Doros, L.A.; Young, R.H.; Dehner, L.P.; Frazier, A.L.; Hill, D.A.; Messinger, Y.H. Clinical and genetic aspects of ovarian stromal tumors: A report from the International Ovarian and Testicular Stromal Tumor Registry. J. Clin. Oncol. 2014, 32 (Suppl. S15), 5520. [Google Scholar] [CrossRef]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Schmolling, J.; Ernst, C.; Ataseven, B.; Blümcke, B.; Schömig-Markiefka, B.; Heikaus, S.; Göhring, U.; Engel, C.; Lampe, B.; et al. Pathogenic germline variants in SMARCA4 and further cancer predisposition genes in early onset ovarian cancer. Cancer Med. 2023, 12, 15256–15260. [Google Scholar] [CrossRef]
- Ryan, N.; Evans, D.; Green, K.; Crosbie, E. Pathological features and clinical behavior of Lynch syndrome-associated ovarian cancer. Gynecol. Oncol. 2017, 144, 491–495. [Google Scholar] [CrossRef]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- McKusick-Nathans Institute of Genetic Medicine Johns Hopkins University; Baltimore, M. Online Mendelian Inheritance in Man, OMIM®. 2022. Available online: https://omim.org/ (accessed on 1 September 2023).
- Felicio, P.S.; Grasel, R.S.; Campacci, N.; de Paula, A.E.; Galvão, H.C.; Torrezan, G.T.; Sabato, C.S.; Fernandes, G.C.; Souza, C.P.; Michelli, R.D.; et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021, 42, 290–299. [Google Scholar] [CrossRef]
- da Costa e Silva Carvalho, S.; Cury, N.M.; Brotto, D.B.; De Araujo, L.F.; Rosa, R.C.A.; Texeira, L.A.; Plaça, J.R.; Marques, A.A.; Peronni, K.C.; Ruy, P.D.C.; et al. Germline variants in DNA repair genes associated with hereditary breast and ovarian cancer syndrome: Analysis of a 21 gene panel in the Brazilian population. BMC Med. Genom. 2020, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.; Sonoda, Y.; Federici, M.G.; Bogomolniy, F.; Rhei, E.; Maresco, D.L.; Saigo, P.E.; Almadrones, L.A.; Barakat, R.R.; Brown, C.L.; et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA 2000, 283, 2260–2265. [Google Scholar] [CrossRef] [PubMed]
- Cibula, D.; Laco, J.; Dundr, P.; Hájková, N.; Tichá, I.; Hojný, J.; Němejcová, K.; Bártů, M.; Michálková, R.; Zikán, M.; et al. Synchronous endometrioid endometrial and ovarian carcinomas are biologically related: A clinico-pathological and molecular (next generation sequencing) study of 22 cases. Oncol. Lett. 2019, 17, 2207–2214. [Google Scholar]
- Jarhelle, E.; Stensland, H.M.F.R.; Hansen, G.M.; Skarsfjord, S.; Jonsrud, C.; Ingebrigtsen, M.; Strømsvik, N.; Van Ghelue, M. Identifying sequence variants contributing to hereditary breast and ovarian cancer in BRCA1 and BRCA2 negative breast and ovarian cancer patients. Sci. Rep. 2019, 9, 19986. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Anand, L.; Padmavathi, V.; Dhivya, V.; Mahalaxmi, I.; Balachandar, V. De novo germ-line mutation of APC gene in periampullary carcinoma with familial adenomatous polyps—A novel familial case report in South India. Karbala Int. J. Mod. Sci. 2016, 2, 266–270. [Google Scholar] [CrossRef]
- Vibert, R.; Le Gall, J.; Buecher, B.; Mouret-Fourme, E.; Bataillon, G.; Becette, V.; Trabelsi-Grati, O.; Moncoutier, V.; Dehainault, C.; Carriere, J.; et al. APC germline pathogenic variants and epithelial ovarian cancer: Causal or coincidental findings? J. Med. Genet. 2023, 60, 460–463. [Google Scholar] [CrossRef]
- Babovic, N.; Simmons, P.S.; Moir, C.; Thorland, E.C.; Scheithauer, B.; Gliem, T.J.; Babovic-Vuksanovic, D. Mucinous cystadenoma of ovary in a patient with juvenile polyposis due to 10q23 microdeletion: Expansion of phenotype. Am. J. Med. Genet. A 2010, 152A, 2623–2627. [Google Scholar] [CrossRef]
- Lou, L.; Zhou, L.; Wang, W.; Li, H.; Li, Y. Atypical ovarian carcinoid tumor with widespread skeletal metastases: A case report of multiple endocrine neoplasia type 1 in a young woman. BMC Cancer 2019, 19, 1107. [Google Scholar] [CrossRef]
- Yauy, K.; Imbert-Bouteille, M.; Bubien, V.; Lindet-Bourgeois, C.; Rathat, G.; Perrochia, H.; MacGrogan, G.; Longy, M.; Bessis, D.; Tinat, J.; et al. Ovarian Clear Cell Carcinoma in Cowden Syndrome. J. Natl. Compr. Cancer Netw. 2019, 17, 7–11. [Google Scholar] [CrossRef]
- Cho, M.-Y.; Kim, H.S.; Eng, C.; Kim, D.S.; Kang, S.J.; Eom, M.; Yi, S.Y.; Bronner, M.P. First report of ovarian dysgerminoma in Cowden syndrome with germline PTEN mutation and PTEN-related 10q loss of tumor heterozygosity. Am. J. Surg. Pathol. 2008, 32, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Sia, T.Y.; Maio, A.; Kemel, Y.M.; Arora, K.S.; Gordhandas, S.B.; Kahn, R.M.; Salo-Mullen, E.E.; Sheehan, M.A.; Tejada, P.R.; Bandlamudi, C.; et al. Germline Pathogenic Variants and Genetic Counseling by Ancestry in Patients With Epithelial Ovarian Cancer. JCO Precis. Oncol. 2023, 7, e2300137. [Google Scholar] [CrossRef] [PubMed]
- Janavičius, R.; Andrėkutė, K.; Mickys, U.; Rudaitis, V.; Brasiūnienė, B.; Griškevičius, L. Apparently “BRCA-related” breast and ovarian cancer patient with germline TP53 mutation. Breast J. 2011, 17, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Grana, B.; Fachal, L.; Santamarina, M.; Cameselle-Teijeiro, J.; Ruíz-Ponte, C.; Carracedo, A.; Vega, A. Beyond BRCA1 and BRCA2 wild-type breast and/or ovarian cancer families: Germline mutations in TP53 and PTEN. Clin. Genet. 2010, 77, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Dace, P.; Olita, H.; Ludmila, E.; Ingrida, D. Tumour suppressor gene CDKN2A/p16 germline mutations in melanoma patients with additional cancer and cancer in their family history. Acta Univ. Latv. 2003, 662, 25–32. [Google Scholar]
- Eoh, K.J.; Kim, J.E.; Park, H.S.; Lee, S.T.; Park, J.S.; Han, J.W.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, J.H.; et al. Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res. Treat. 2018, 50, 917–925. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.M.; Tyrer, J.; Intermaggio, M.; Chenevix-Trench, G.; Bowtell, D.D.; Traficante, N.; AOCS Group; Brenton, J.; Goranova, T.; et al. Population-based targeted sequencing of 54 candidate genes identifies PALB2 as a susceptibility gene for high-grade serous ovarian cancer. J. Med Genet. 2020, 58, 305–313. [Google Scholar] [CrossRef]
- Schubert, S.; van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer. 2019, 144, 2683–2694. [Google Scholar] [CrossRef]
- del Valle, J.; Rofes, P.; Moreno-Cabrera, J.M.; López-Dóriga, A.; Belhadj, S.; Vargas-Parra, G.; Teulé, À.; Cuesta, R.; Muñoz, X.; Campos, O.; et al. Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients. Cancers 2020, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, B.; Tuxen, I.V.; Yde, C.W.; Gabrielaite, M.; Torp, M.H.; Kinalis, S.; Oestrup, O.; Rohrberg, K.; Spangaard, I.; Santoni-Rugiu, E.; et al. High frequency of pathogenic germline variants within homologous recombination repair in patients with advanced cancer. NPJ Genom. Med. 2019, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Cavaillé, M.; Uhrhammer, N.; Privat, M.; Ponelle-Chachuat, F.; Gay-Bellile, M.; Lepage, M.; Molnar, I.; Viala, S.; Bidet, Y.; Bignon, Y. Analysis of 11 candidate genes in 849 adult patients with suspected hereditary cancer predisposition. Genes Chromosomes Cancer 2021, 60, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Kostantopoulou, I.; Apostolou, P.; Papamentzelopoulou, M.S.; Papadimitriou, C.; Faliakou, E.; Christodoulou, C.; Boukovinas, I.; Razis, E.; Tryfonopoulos, D.; et al. One in three highly selected Greek patients with breast cancer carries a loss-of-function variant in a cancer susceptibility gene. J. Med Genet. 2019, 57, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.; Harrell, M.; Walsh, T.; Lee, M.; King, M.; Davidson, S.; Mannel, R.; DiSilvestro, P.; Swisher, E.; Birrer, M. Germline mutations in DNA repair genes in women with ovarian, peritoneal, or fallopian tube cancer treated on GOG protocols 218 and 262. Gynecol. Oncol. 2014, 133, 6. [Google Scholar] [CrossRef]
- Golmard, L.; Castéra, L.; Krieger, S.; Moncoutier, V.; Abidallah, K.; Tenreiro, H.; Laugé, A.; Tarabeux, J.; Millot, G.A.; Nicolas, A.; et al. Contribution of germline deleterious variants in the RAD51 paralogs to breast and ovarian cancers. Eur. J. Hum. Genet. 2017, 25, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yang, J.; Li, L.; Cao, D.; Yu, M.; Shen, K. Germline and somatic mutations in homologous recombination genes among Chinese ovarian cancer patients detected using next-generation sequencing. J. Gynecol. Oncol. 2017, 28, e39. [Google Scholar] [CrossRef]
- Subramanian, D.N.; Zethoven, M.; McInerny, S.; Morgan, J.A.; Rowley, S.M.; Lee, J.E.A.; Li, N.; Gorringe, K.L.; James, P.A.; Campbell, I.G. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat. Commun. 2020, 11, 1640. [Google Scholar] [CrossRef]
- Srivastava, S.; Olson, H.E.; Cohen, J.S.; Gubbels, C.S.; Lincoln, S.; Davis, B.T.; Shahmirzadi, L.; Gupta, S.; Picker, J.; Yu, T.W.; et al. BRAT1 mutations present with a spectrum of clinical severity. Am. J. Med. Genet. A 2016, 170, 2265–2273. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.M.; Huang, K.-L.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef]
- Stradella, A.; Del Valle, J.; Rofes, P.; Vargas-Parra, G.; Salinas, M.; González, S.; Montes, E.; López-Doriga, A.; Gómez, C.; de Cid, R.; et al. ERCC3, a new ovarian cancer susceptibility gene? Eur. J. Cancer 2020, 141, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Zemankova, P.; Nehasil, P.; Kleibl, Z.; Kleibl, Z.; Soukupová, J.; Janatová, M.; Zemánková, P.; Černá, M.; Jelínková, S.; et al. Re: ERCC3, a new ovarian cancer susceptibility gene? Eur. J. Cancer 2021, 150, 278–280. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.; Pozzi, E.; Gatti, R.A.; Brusco, A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur. J. Hum. Genet. 2013, 21, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5’ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Rusch, M.; Nakitandwe, J.; Shurtleff, S.; Newman, S.; Zhang, Z.; Edmonson, M.N.; Parker, M.; Jiao, Y.; Ma, X.; Liu, Y.; et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018, 9, 3962. [Google Scholar] [CrossRef] [PubMed]
- Funingana, I.; Trotman, J.; Ambrose, J.; Roberts, T.; Watkins, J.; Ridley, M.; Gilson, B.; Freeman, S.; Jimenez-Linan, M.; Sosinsky, A.; et al. 7P Integration of whole genome sequencing (WGS) into NHS pathways for high-grade ovarian cancer (HGOC): A single-centre prospective experience. ESMO Open 2023, 8, 100861. [Google Scholar] [CrossRef]
- Guan, Z.; Begg, C.B.; Shen, R. Predicting Cancer Risk from Germline Whole-exome Sequencing Data Using a Novel Context-based Variant Aggregation Approach. Cancer Res. Commun. 2023, 3, 483–488. [Google Scholar] [CrossRef]
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.A.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Scherz, A.; Stoll, S.; Rothlisberger, B.; Rabaglio, M. A New de novo BRCA1 Mutation in a Young Breast Cancer Patient: A Case Report. Appl. Clin. Genet. 2023, 16, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Tenedini, E.; Piana, S.; Toss, A.; Marino, M.; Barbieri, E.; Artuso, L.; Venturelli, M.; Gasparini, E.; Mandato, V.D.; Marchi, I.; et al. Constitutional Mosaicism: A Critical Issue in the Definition of BRCA-Inherited Cancer Risk. JCO Precis. Oncol. 2022, 6, e2200138. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Lalonde, E.; Zhang, J.; Albrecht, S.; Hamel, N.; Cavallone, L.; May, S.T.; Nicholson, J.C.; Coleman, N.; Murray, M.J.; et al. Familial rhabdoid tumour ‘avant la lettre’—From pathology review to exome sequencing and back again. J. Pathol. 2013, 231, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Golmard, L.; Delnatte, C.; Laugé, A.; Moncoutier, V.; Lefol, C.; Abidallah, K.; Tenreiro, H.; Copigny, F.; Giraudeau, M.; Guy, C.; et al. Breast and ovarian cancer predisposition due to de novo BRCA1 and BRCA2 mutations. Oncogene 2016, 35, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- von Hardenberg, S.; Wallaschek, H.; Du, C.; Schmidt, G.; Auber, B. A holistic approach to maximise diagnostic output in trio exome sequencing. Front. Pediatr. 2023, 11, 1183891. [Google Scholar] [CrossRef] [PubMed]
- Speight, B.; Colvin, E.; Epurescu, E.D.; Drummond, J.; Verhoef, S.; Pereira, M.; Evans, D.G.; Tischkowitz, M. Low-level constitutional mosaicism of BRCA1 in two women with young onset ovarian cancer. Hered. Cancer Clin. Pract. 2022, 20, 32. [Google Scholar] [CrossRef]
- Alhopuro, P.; Vainionpää, R.; Anttonen, A.-K.; Aittomäki, K.; Nevanlinna, H.; Pöyhönen, M. Constitutional mosaicism for a BRCA2 mutation as a cause of early-onset breast cancer. Fam. Cancer 2020, 19, 307–310. [Google Scholar] [CrossRef]
- Schwartz, M.; Ibadioune, S.; Chansavang, A.; Vacher, S.; Caputo, S.M.; Delhomelle, H.; Wong, J.; Abidallah, K.; Moncoutier, V.; Becette, V.; et al. Mosaic BRCA1 promoter methylation contribution in hereditary breast/ovarian cancer pedigrees. J. Med. Genet. 2023. [Google Scholar] [CrossRef]
- Pinto, D.; Pinto, C.; Guerra, J.; Pinheiro, M.; Santos, R.; Vedeld, H.M.; Yohannes, Z.; Peixoto, A.; Santos, C.; Pinto, P.; et al. Contribution of MLH1 constitutional methylation for Lynch syndrome diagnosis in patients with tumor MLH1 downregulation. Cancer Med. 2018, 7, 433–444. [Google Scholar] [CrossRef]
- Rantala, J.N.J.; Heikkinen, S.M.M.; Hirvonen, E.M.; Tanskanen, T.; Malila, N.K.; Pitkäniemi, J.M. Familial aggregation of early-onset cancers in early-onset breast cancer families. Int. J. Cancer 2023, 153, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Imbert-Bouteille, M.; Corsini, C.; Picot, M.-C.; Mizrahy, L.; Akouete, S.; Huguet, H.; Thomas, F.; Geneviève, D.; Taourel, P.; Ychou, M.; et al. No Association of Early-Onset Breast or Ovarian Cancer with Early-Onset Cancer in Relatives in BRCA1 or BRCA2 Mutation Families. Genes 2021, 12, 1100. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.F.; Pharoah, P.; Smith, S.K.; Easton, D.; Ponder, B.A.J. A systematic review and meta-analysis of family history and risk of ovarian cancer. BJOG Int. J. Obstet. Gynaecol. 1998, 105, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Eng, K.H.; Szender, J.B.; Etter, J.L.; Kaur, J.; Poblete, S.; Huang, R.Y.; Zhu, Q.; Grzesik, K.A.; Battaglia, S.; Cannioto, R.; et al. Paternal lineage early onset hereditary ovarian cancers: A Familial Ovarian Cancer Registry study. PLoS Genet. 2018, 14, e1007194. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.E.; Sood, A.K.; Lallas, T.; Buekers, T.; Skilling, J.S. Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. J. Natl. Cancer Inst. 1999, 91, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Kain, M.; Wang, L. Inactivation of X-linked tumor suppressor genes in human cancer. Future Oncol. 2012, 8, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Lose, F.; Duffy, D.L.; Kay, G.F.; Kedda, M.A.; Spurdle, A.B. Skewed X chromosome inactivation and breast and ovarian cancer status: Evidence for X-linked modifiers of BRCA1. J. Natl. Cancer Inst. 2008, 100, 1519–1529. [Google Scholar] [CrossRef]
- Winham, S.J.; Larson, N.B.; Armasu, S.M.; Fogarty, Z.C.; Larson, M.C.; McCauley, B.M.; Wang, C.; Lawrenson, K.; Gayther, S.; Cunningham, J.M.; et al. Molecular signatures of X chromosome inactivation and associations with clinical outcomes in epithelial ovarian cancer. Hum. Mol. Genet. 2019, 28, 1331–1342. [Google Scholar] [CrossRef]
- Qing, T.; Mohsen, H.; Marczyk, M.; Ye, Y.; O’meara, T.; Zhao, H.; Townsend, J.P.; Gerstein, M.; Hatzis, C.; Kluger, Y.; et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 2020, 11, 2438. [Google Scholar] [CrossRef]
- Jia, G.; Lu, Y.; Wen, W.; Long, J.; Liu, Y.; Tao, R.; Li, B.; Denny, J.C.; Shu, X.-O.; Zheng, W. Evaluating the Utility of Polygenic Risk Scores in Identifying High-Risk Individuals for Eight Common Cancers. JNCI Cancer Spectr. 2020, 4, pkaa021. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Gentry-Maharaj, A.; Ryan, A.; Intermaggio, M.; Lee, A.; Kalsi, J.K.; Tyrer, J.; Gaba, F.; Manchanda, R.; et al. Evaluation of polygenic risk scores for ovarian cancer risk prediction in a prospective cohort study. J. Med Genet. 2018, 55, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Dareng, E.O.; Tyrer, J.P.; Barnes, D.R.; Jones, M.R.; Yang, X.; Aben, K.K.H.; Adank, M.A.; Agata, S.; Andrulis, I.L.; Anton-Culver, H.; et al. Polygenic risk modeling for prediction of epithelial ovarian cancer risk. Eur. J. Hum. Genet. 2022, 30, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Borde, J.; Laitman, Y.; Blümcke, B.; Niederacher, D.; Weber-Lassalle, K.; Sutter, C.; Rump, A.; Arnold, N.; Wang-Gohrke, S.; Horváth, J.; et al. Polygenic risk scores indicate extreme ages at onset of breast cancer in female BRCA1/2 pathogenic variant carriers. BMC Cancer 2022, 22, 706. [Google Scholar] [CrossRef] [PubMed]
- Fatapour, Y.; Brody, J.P. Genetic Risk Scores and Missing Heritability in Ovarian Cancer. Genes 2023, 14, 762. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Mitra, N.; Wan, F.; Chen, S.; Andrulis, I.L.; Apostolou, P.; Arnold, N.; Arun, B.K.; Barrowdale, D.; et al. Inheritance of deleterious mutations at both BRCA1 and BRCA2 in an international sample of 32,295 women. Breast Cancer Res. 2016, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, J.; Yu, H.; Hu, Q.; Bateman, N.W.; Long, M.; Rosario, S.; Schultz, E.; Dalgard, C.L.; Wilkerson, M.D.; et al. Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers. Cancers 2022, 14, 2350. [Google Scholar] [CrossRef] [PubMed]
- Laitman, Y.; Michaelson-Cohen, R.; Chen-Shtoyerman, R.; Goldberg, Y.; Reish, O.; Bernstein-Molho, R.; Levy-Lahad, E.; Ben Baruch, N.E.; Kedar, I.; Evans, D.G.; et al. Age at diagnosis of cancer in 185delAG BRCA1 mutation carriers of diverse ethnicities: Tentative evidence for modifier factors. Fam. Cancer 2021, 20, 189–194. [Google Scholar] [CrossRef]
- Lee, C.H.; Subramanian, S.; Beck, A.H.; Espinosa, I.; Senz, J.; Zhu, S.X.; Huntsman, D.; van de Rijn, M.; Gilks, C.B. MicroRNA profiling of BRCA1/2 mutation-carrying and non-mutation-carrying high-grade serous carcinomas of ovary. PLoS ONE 2009, 4, e7314. [Google Scholar] [CrossRef]
- Pastrello, C.; Polesel, J.; Della Puppa, L.; Viel, A.; Maestro, R. Association between hsa-mir-146a genotype and tumor age-of-onset in BRCA1/BRCA2-negative familial breast and ovarian cancer patients. Carcinogenesis 2010, 31, 2124–2126. [Google Scholar] [CrossRef]
- Zhang, K.; Chandrakasan, S.; Chapman, H.; Valencia, C.A.; Husami, A.; Kissell, D.; Johnson, J.A.; Filipovich, A.H. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood 2014, 124, 1331–1334. [Google Scholar] [CrossRef]
- Moreno-Ruiz, N.; Ambrose, J.C.; Arumugam, P.; Baple, E.L.; Bleda, M.; Boardman-Pretty, F.; Boissiere, J.M.; Boustred, C.R.; Brittain, H.; Caulfield, M.J.; et al. Assessing the digenic model in rare disorders using population sequencing data. Eur. J. Hum. Genet. 2022, 30, 1439–1443. [Google Scholar] [CrossRef]
- Kamar, A.; Khalil, A.; Nemer, G. The Digenic Causality in Familial Hypercholesterolemia: Revising the Genotype-Phenotype Correlations of the Disease. Front. Genet. 2020, 11, 572045. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, R.; Woon, S.-T.; Bryant, V.L.; Steele, R.; Slade, C.; Leung, E.Y.; Lehnert, K. Clinical Implications of Digenic Inheritance and Epistasis in Primary Immunodeficiency Disorders. Front. Immunol. 2017, 8, 1965. [Google Scholar] [CrossRef]
- Nachtegael, C.; Gravel, B.; Dillen, A.; Smits, G.; Nowé, A.; Papadimitriou, S.; Lenaerts, T. Scaling up oligogenic diseases research with OLIDA: The Oligogenic Diseases Database. Database 2022, 2022, baac023. [Google Scholar] [CrossRef] [PubMed]
- Fijneman, R.J.; de Vries, S.S.; Jansen, R.C.; Demant, P. Complex interactions of new quantitative trait loci, Sluc1, Sluc2, Sluc3, and Sluc4, that influence the susceptibility to lung cancer in the mouse. Nat. Genet. 1996, 14, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Tansel, A.; Katz, L.H.; El-Serag, H.B.; Thrift, A.P.; Parepally, M.; Shakhatreh, M.H.; Kanwal, F. Incidence and Determinants of Hepatocellular Carcinoma in Autoimmune Hepatitis: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2017, 15, 1207–1217.e4. [Google Scholar] [CrossRef]
- Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001, 49, 347. [Google Scholar] [CrossRef]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O’Connell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Early-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 2752–2759.e2. [Google Scholar] [CrossRef]
- Bae, E.; Lim, S.; Han, K.-D.; Jung, J.-H.; Choi, H.; Kim, C.; Ma, S.; Kim, S. Systemic lupus erythematosus is a risk factor for cancer: A nationwide population-based study in Korea. Lupus 2019, 28, 317–323. [Google Scholar] [CrossRef]
- Kübler, K.; Arndt, P.F.; Wardelmann, E.; Krebs, D.; Kuhn, W.; van der Ven, K. HLA-class II haplotype associations with ovarian cancer. Int. J. Cancer. 2006, 119, 2980–2985. [Google Scholar] [CrossRef]
- Carbone, M.; Arron, S.T.; Beutler, B.; Bononi, A.; Cavenee, W.; Cleaver, J.E.; Croce, C.M.; D’andrea, A.; Foulkes, W.D.; Gaudino, G.; et al. Tumour predisposition and cancer syndromes as models to study gene-environment interactions. Nat. Rev. Cancer 2020, 20, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Novelli, F.; Bononi, A.; Wang, Q.; Bai, F.; Patergnani, S.; Kricek, F.; Haglund, E.; Suarez, J.S.; Tanji, M.; Xu, R.; et al. BAP1 forms a trimer with HMGB1 and HDAC1 that modulates gene x environment interaction with asbestos. Proc. Natl. Acad. Sci. USA 2021, 118, e2111946118. [Google Scholar] [CrossRef] [PubMed]
- Kurzynska-Kokorniak, A.; Koralewska, N.; Pokornowska, M.; Urbanowicz, A.; Tworak, A.; Mickiewicz, A.; Figlerowicz, M. The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015, 43, 4365–4380. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef] [PubMed]
- L’espérance, K.; Grundy, A.; Abrahamowicz, M.; Arseneau, J.; Gilbert, L.; Gotlieb, W.H.; Provencher, D.; Koushik, A. Alcohol intake and the risk of epithelial ovarian cancer. Cancer Causes Control 2023, 34, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Townsend, M.K.; Vinci, C.; Jake-Schoffman, D.E.; Tworoger, S.S. Early life exposure to tobacco smoke and ovarian cancer risk in adulthood. Int. J. Epidemiol. 2021, 50, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian cancer and smoking: Individual participant meta-analysis including 28,114 women with ovarian cancer from 51 epidemiological studies. Lancet Oncol. 2012, 13, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Zhai, T.; Hatanaka, K.C.; Hatanaka, Y.; Amano, T.; Wang, L.; Tanaka, S.; Watari, H. Germline PRDM1 Variant rs2185379 in Long-Term Recurrence-Free Survivors of Advanced Ovarian Cancer. Pharmacogenomics Pers. Med. 2022, 15, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Jiang, R.; Wang, P.; Xu, Y.; Yin, S.; Cheng, X.; Zang, R. Significant association of the EXO1 rs851797 polymorphism with clinical outcome of ovarian cancer. Onco Targets Ther. 2017, 10, 4841–4851. [Google Scholar] [CrossRef]


| Patients | Early-Onset OC | Late-Onset OC |
|---|---|---|
| Incidence in females | 1.6/100,000 [3,15] | 22.0/100,000 [3,15] |
| 5-year relative survival rate | 58–87% [17,19,20,23] -lower in LGSC [22] | Approx. 50% [3,17] |
| Clinicopathology | ||
| Histology | ~40% epithelial—LGSC prevails [16,17] ~50% germ-cell [16,24] ~10% sex cord-stromal [24,25] | ~90% epithelial—HGSC prevails (70%) [13,26] ~6% sex cord-stromal [6] ~3% germ-cell [6] |
| Dominant tumor stage | Localized disease [3] | Distant disease [3] |
| Genetic predispositions | ||
| GPV | Low <10% [7,8,9] | High >20% [4,5,9] |
| Study | Population | Study Details | No. of Tested Genes * | No. of All OC Patients | No. of Early-Onset OC Patients | Range of Early-Onset Patients’ Age at Dg. | Early-Onset OC Patients | ||
|---|---|---|---|---|---|---|---|---|---|
| No. of High-Penetrance GPV Carriers | GPVs in Established High-Penetrance OC Predisposition Genes | GPVs in Candidate OC Predisposition Genes | |||||||
| Stratton (1999) [8] | UK | Early-onset epithelial OC | 4 ** | 169 | 169 | 13–30 | 0 *** | 0 | 0 |
| Carter (2018) [9] | US | OC | 15 | 4439 | 147 | 6–30 | 2 (1.4%) | 1×BRCA1; 1×BRIP1 | 3×ATM; 1×BARD1; 5×CHEK2 |
| Lhotova (2020) [4] | CZ | OC | 219 | 1333 | 84 | 15–30 | 6 (7.1%) | 2×BRCA1; 1×BRCA2; 2×RAD51C; 1×STK11 | 1×ATM; 1×BARD1; 4×CHEK2; 1×NBN; |
| Herold (2023) [51] | GER | OC | 25 | 206 | 83 | 13–30 | 3 (3.6%) | 1× BRIP1; 2×SMARCA4 | 1×FANCM; 1×MUTYH het; 1×PMS2; 1×TP53 |
| Flaum (2023) [40] | UK | Early-onset OC | 15 | 77 | 77 | 15–30 | 4 (5.2%) | 4×MSH2 | 1×PMS2 |
| Bernards (2015) [38] | US | Early-onset OC | 18 | 47 | 5 | 27–30 | 0 | 0 | 0 |
| Felicio (2020) [55] | BRA | BRCA neg., TP53 neg. HBOC patients | WES | 11 | 3 | 20–21 | 0 | 0 | 1×CHEK2 |
| Da Costa (2020) [56] | BRA | HBOC | 21 | 6 | 2 | 22–30 | 0 | 0 | 0 |
| Boyd (2000) [57] | Jew | OC | 2 | 189 | 1 | 25 | 0 | 0 | 0 |
| Hajkova (2019) [58] | CZ | Synchronous EC and OC | 73 | 22 | 1 | 29 | 0 | 0 | 1×BARD1 |
| Jarhelle (2019) [59] | NOR | HBOC BRCA1/BRCA2 neg. | 94 | 20 | 1 | 27 | 0 | 0 | 1×CHEK2 |
| Risch (2001) [7] | US | OC | 2 | 649 | NA (96 <40 yo) | 20-30 | 0 | 0 | 0 |
| Koczkowska (2018) [60] | PL | OC | 25 | 333 | NA | NA | 0 | 0 | 1×CHEK2 |
| Pal (2005) [61] | US | epithelial OC | 2 | 209 | NA (11 <40 yo) | 18-30 | 0 | 0 | 0 |
| Ryan (2017) [52] | UK | LS assoc. preselected OC positive for LS-GPV | 4 | 53 | NA | 24-30 | 2 | 2×MSH2 | 1×PMS2 biallelic |
| Hajkova (2019) [58] | CZ | Synchronous EC and OC | 73 | 22 | 1 | 29 | 0 | 0 | 1×BARD1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horackova, K.; Janatova, M.; Kleiblova, P.; Kleibl, Z.; Soukupova, J. Early-Onset Ovarian Cancer <30 Years: What Do We Know about Its Genetic Predisposition? Int. J. Mol. Sci. 2023, 24, 17020. https://doi.org/10.3390/ijms242317020
Horackova K, Janatova M, Kleiblova P, Kleibl Z, Soukupova J. Early-Onset Ovarian Cancer <30 Years: What Do We Know about Its Genetic Predisposition? International Journal of Molecular Sciences. 2023; 24(23):17020. https://doi.org/10.3390/ijms242317020
Chicago/Turabian StyleHorackova, Klara, Marketa Janatova, Petra Kleiblova, Zdenek Kleibl, and Jana Soukupova. 2023. "Early-Onset Ovarian Cancer <30 Years: What Do We Know about Its Genetic Predisposition?" International Journal of Molecular Sciences 24, no. 23: 17020. https://doi.org/10.3390/ijms242317020