
Assessing the Phenotype of a Homologous Recombination Deficiency Using High Resolution Array-Based Comparative Genome Hybridization in Ovarian Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
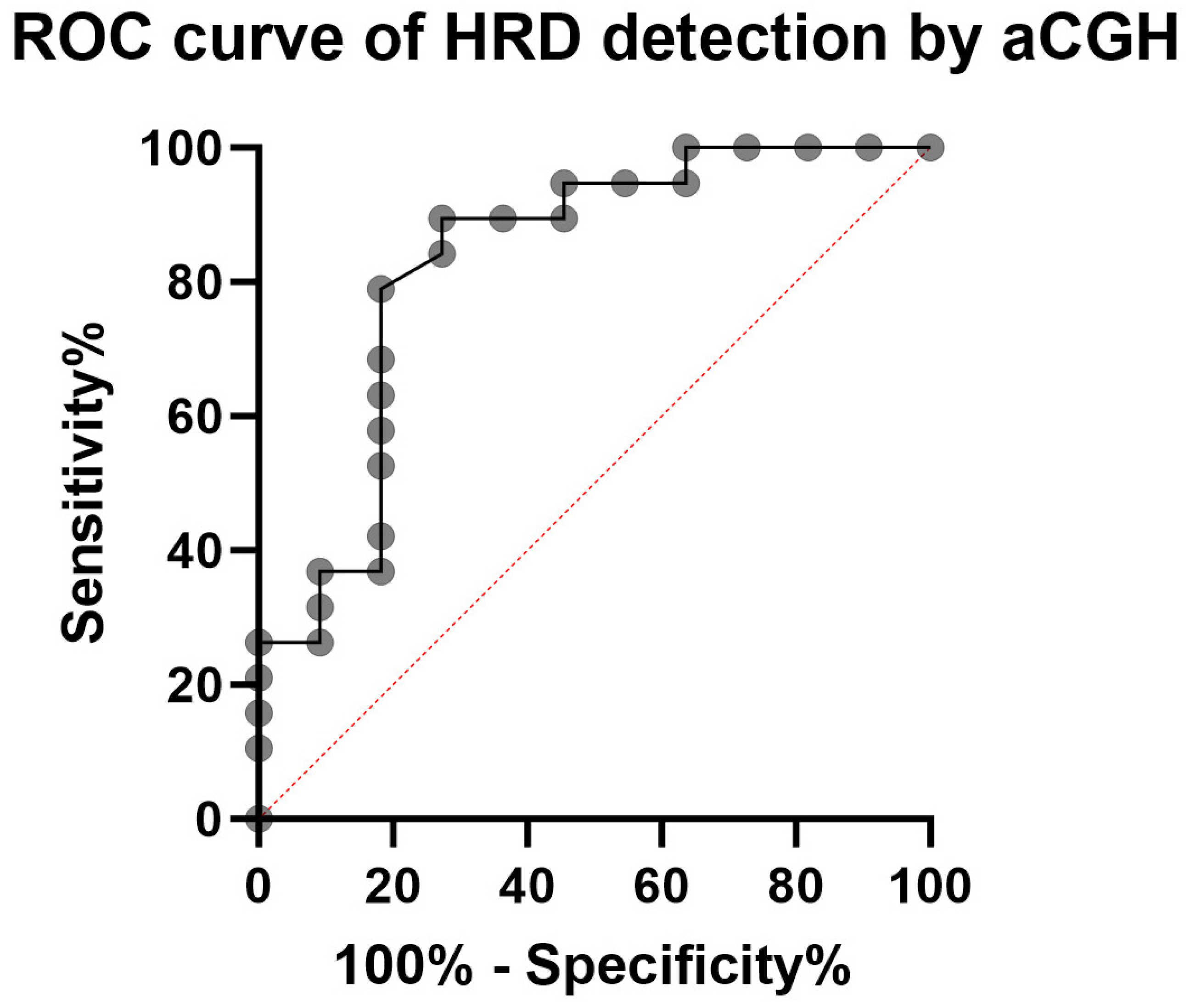
2.1. Determination of HRD Score Cut-Off
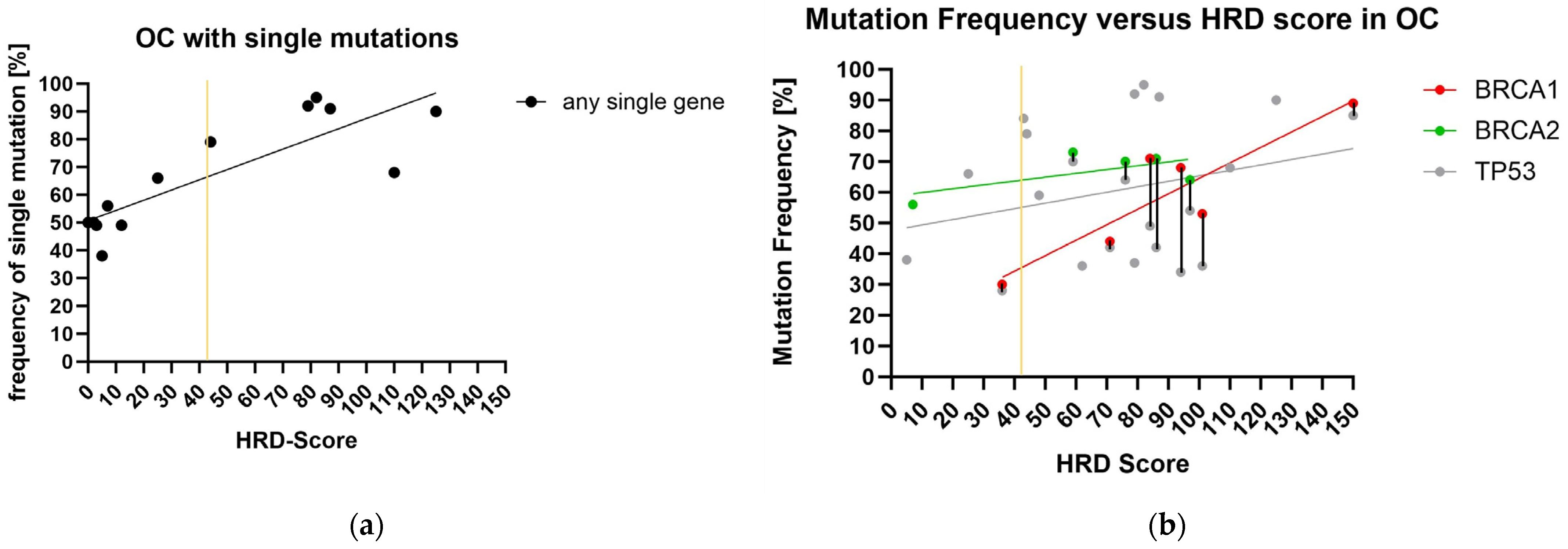
2.2. Panel Sequencing and HRD-Score
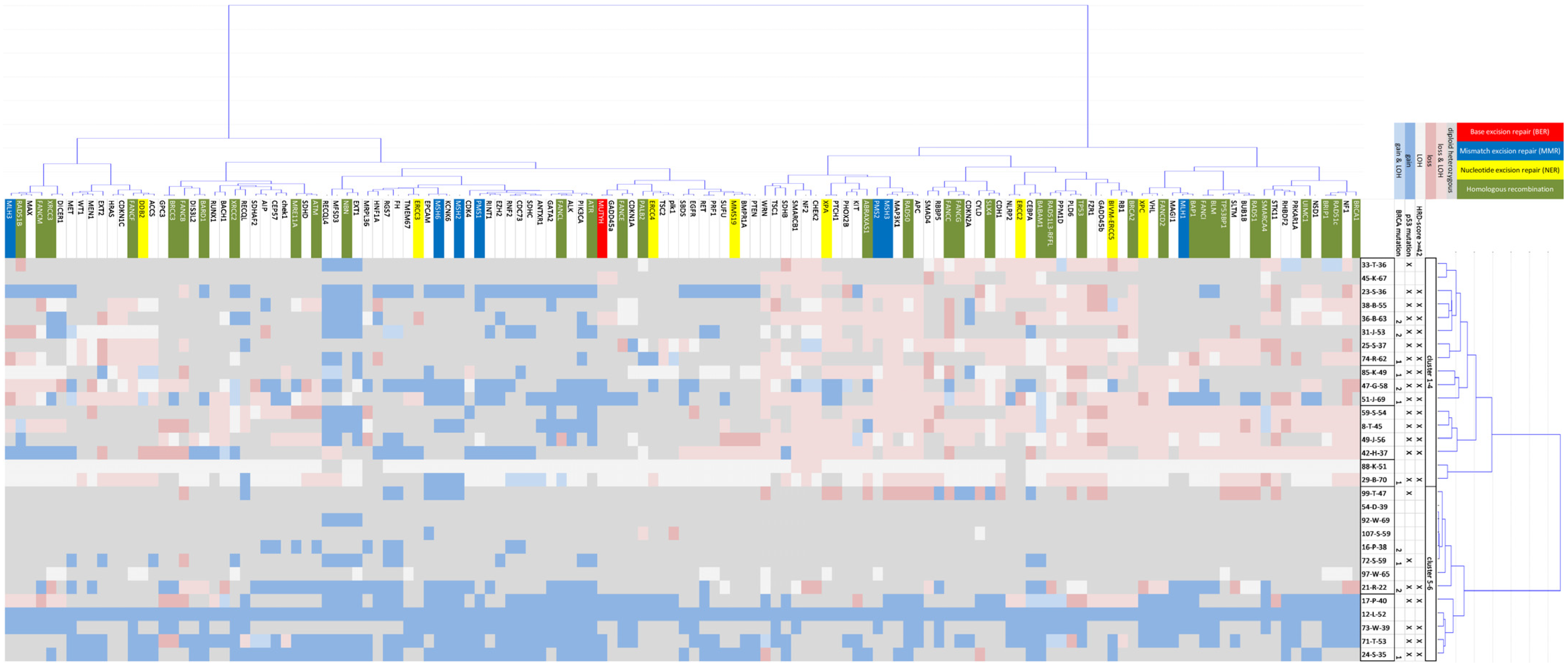
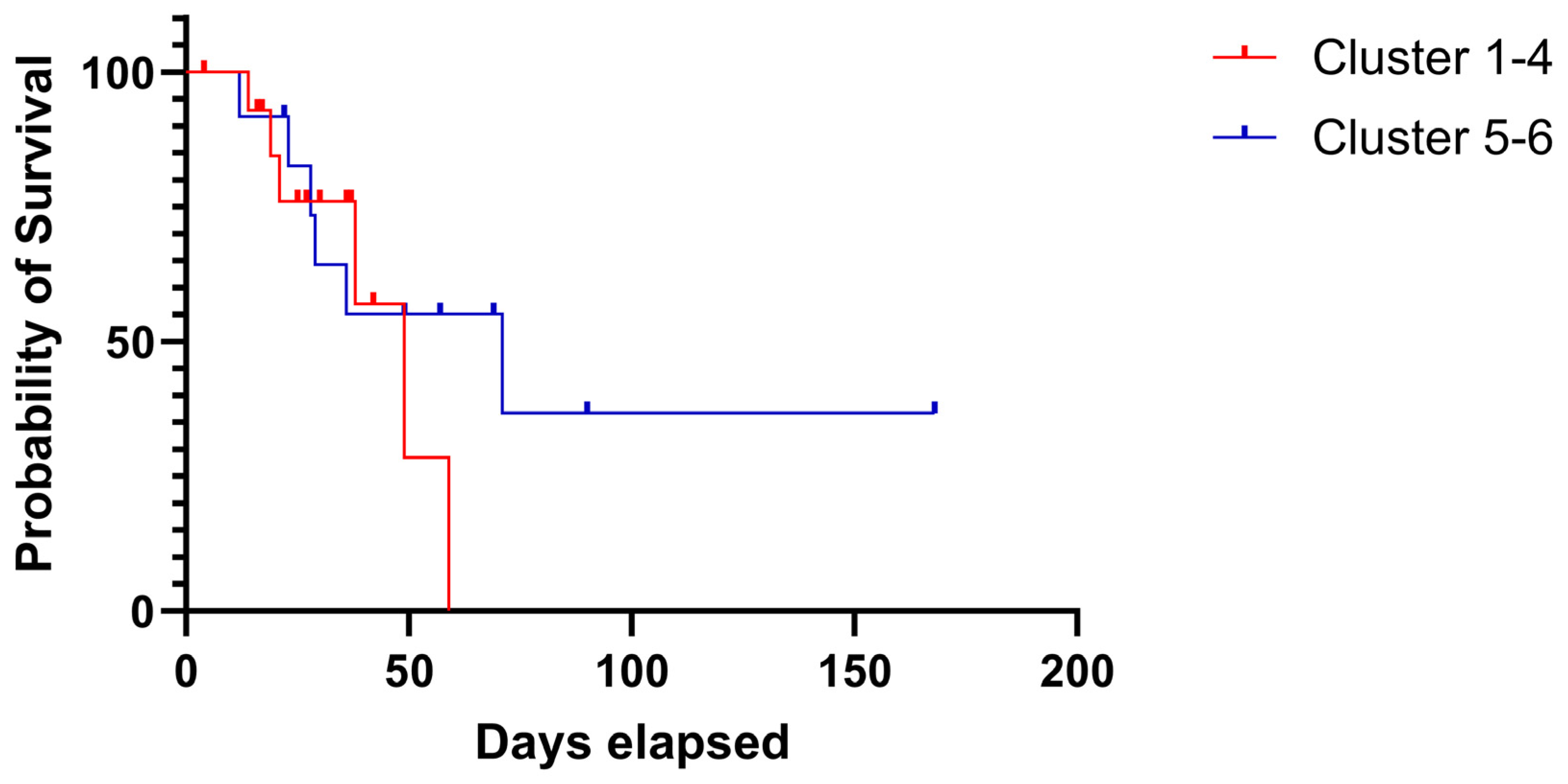
2.3. CNV Cluster Analysis in 132 Tumor Genes and 30 OC Cases
2.4. CNV Differences in 30 OC Cases with and without BRCA Mutation
2.5. Validation of HRD Detection Using aCGH on 19 OC
3. Discussion
4. Materials and Methods
4.1. Biomaterial
4.2. Panel Sequencing
4.3. aCGH
4.4. Determination of the HRD Score
4.5. Evaluation of the HRD Score
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chang, L.C.; Huang, C.F.; Lai, M.S.; Shen, L.J.; Wu, F.L.; Cheng, W.F. Prognostic factors in epithelial ovarian cancer: A population-based study. PLoS ONE 2018, 13, e0194993. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Shorokhova, M.; Nikolsky, N.; Grinchuk, T. Chromothripsis-Explosion in Genetic Science. Cells 2021, 10, 1102. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Marsden, C.G.; Dragon, J.A.; Wallace, S.S.; Sweasy, J.B. Base Excision Repair Variants in Cancer. Methods Enzym. 2017, 591, 119–157. [Google Scholar]
- Dantzer, F.; de La Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Mangerich, A.; Burkle, A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer J. Int. Du Cancer 2011, 128, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.; Martinez, A.; Szul, T.; Birrer, M.J. Biomarkers in ovarian cancer: To be or not to be. Cancer 2019, 125 (Suppl. 24), 4563–4572. [Google Scholar] [CrossRef] [PubMed]
- Futaki, M.; Liu, J.M. Chromosomal breakage syndromes and the BRCA1 genome surveillance complex. Trends Mol. Med. 2001, 7, 560–565. [Google Scholar] [CrossRef]
- Deng, C.X.; Brodie, S.G. Roles of BRCA1 and its interacting proteins. Bioessays 2000, 22, 728–737. [Google Scholar] [CrossRef]
- Kanaar, R.; Wyman, C. DNA repair by the MRN complex: Break it to make it. Cell 2008, 135, 14–16. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Pauty, J.; Maity, R.; Zhao, W.; Coulombe, Y.; Sung, P.; Masson, J.Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 6, 553–564. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Tufail, M. DNA repair pathways in breast cancer: From mechanisms to clinical applications. Breast Cancer Res. Treat. 2023, 200, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. BCR 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed]
- Esplen, H.P.; Yang, R.K.; Kalia, A.; Tang, Z.; Tang, G.; Medeiros, L.J.; Toruner, G.A. Recurrent Somatic Copy Number Alterations and Their Association with Oncogene Expression Levels in High-Grade Ovarian Serous Carcinoma. Life 2023, 13, 2192. [Google Scholar] [CrossRef]
- Ren, J.; Kong, P.; Wang, Y.; Guo, D.; Zhang, L. Copy number variations in esophageal squamous cell carcinoma: Emerging cancer drivers and biomarkers (Review). Oncol. Rep. 2024, 51, 8. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP Inhibitors in Ovarian Cancer: A Review. Target Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef]
- Pandya, K.; Scher, A.; Omene, C.; Ganesan, S.; Kumar, S.; Ohri, N.; Potdevin, L.; Haffty, B.; Toppmeyer, D.L.; George, M.A. Clinical efficacy of PARP inhibitors in breast cancer. Breast Cancer Res. Treat. 2023, 200, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.; George, A.; Banerjee, S. PARP inhibitors in ovarian cancer: An overview of the practice-changing trials. Genes Chromosomes Cancer 2021, 60, 385–397. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, W.; Xu, S.; Shang, H.; Li, J.; Guo, Y.; Tong, J. PARP inhibitors as maintenance therapy in newly diagnosed advanced ovarian cancer: A meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 485–493. [Google Scholar] [CrossRef]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Naiman, K.; Campillo-Funollet, E.; Watson, A.T.; Budden, A.; Miyabe, I.; Carr, A.M. Replication dynamics of recombination-dependent replication forks. Nat. Commun. 2021, 12, 923. [Google Scholar] [CrossRef] [PubMed]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell Dev. Biol. 2021, 113, 14–26. [Google Scholar] [CrossRef]
- Incorvaia, L.; Passiglia, F.; Rizzo, S.; Galvano, A.; Listi, A.; Barraco, N.; Maragliano, R.; Calo, V.; Natoli, C.; Ciaccio, M.; et al. “Back to a false normality”: New intriguing mechanisms of resistance to PARP inhibitors. Oncotarget 2017, 8, 23891–23904. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhu, J.; Yin, R.; Wang, J.; Pan, L.; Kong, B.; Zheng, H.; Liu, J.; Wu, X.; Wang, L.; et al. Treatment With Niraparib Maintenance Therapy in Patients with Newly Diagnosed Advanced Ovarian Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2023, 9, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- How, J.A.; Jazaeri, A.A.; Fellman, B.; Daniels, M.S.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.S.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef]
- Willing, E.M.; Vollbrecht, C.; Vossing, C.; Weist, P.; Schallenberg, S.; Herbst, J.M.; Schatz, S.; Jori, B.; Bataillon, G.; Harter, P.; et al. Development of the NOGGO GIS v1 Assay, a Comprehensive Hybrid-Capture-Based NGS Assay for Therapeutic Stratification of Homologous Repair Deficiency Driven Tumors and Clinical Validation. Cancers 2023, 15, 3445. [Google Scholar] [CrossRef]
- Asphaug, L.; Melberg, H.O. The Cost-Effectiveness of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer in Norway. MDM Policy Pract. 2019, 4, 2381468318821103. [Google Scholar] [CrossRef]
- Suratannon, N.; van Wijck, R.T.A.; Broer, L.; Xue, L.; van Meurs, J.B.J.; Barendregt, B.H.; van der Burg, M.; Dik, W.A.; Chatchatee, P.; Langerak, A.W.; et al. Rapid Low-Cost Microarray-Based Genotyping for Genetic Screening in Primary Immunodeficiency. Front. Immunol. 2020, 11, 614. [Google Scholar] [CrossRef]
- Pawlyn, C.; Loehr, A.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Sun, J.; Fedorchak, K.; Mughal, T.; Davies, F.E.; Walker, B.A.; et al. Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: A role for PARP inhibition? Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2018, 32, 1561–1566. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef]
- Kim, J.; Jeong, K.; Jun, H.; Kim, K.; Bae, J.M.; Song, M.G.; Yi, H.; Park, S.; Woo, G.U.; Lee, D.W.; et al. Mutations of TP53 and genes related to homologous recombination repair in breast cancer with germline BRCA1/2 mutations. Hum Genom. 2023, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A.; Bottani, S.; Birchler, J.A. Gene dosage effects: Nonlinearities, genetic interactions, and dosage compensation. Trends Genet. 2013, 29, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhang, J. Gene dosage and gene duplicability. Genetics 2008, 179, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. BCR 2014, 16, 211. [Google Scholar] [CrossRef]
- Kyo, S.; Kanno, K.; Takakura, M.; Yamashita, H.; Ishikawa, M.; Ishibashi, T.; Sato, S.; Nakayama, K. Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance. Cancers 2022, 14, 2504. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient-ID | Mutation BRCA1/2 | LOH | TAI | LST-Break Points | HRD-Score |
|---|---|---|---|---|---|
| 51-J-69 | 1 | 24 | 28 | 98 | 150 |
| 31-J-53 | 2 | 13 | 10 | 74 | 97 |
| 85-K-49 | 1 | 22 | 15 | 64 | 101 |
| 21-R-22 | 2 | 9 | 19 | 58 | 86 |
| 24-S-35 | 1 | 1 | 17 | 53 | 71 |
| 74-R-62 | 1 | 17 | 20 | 47 | 84 |
| 47-G-58 | 2 | 15 | 17 | 44 | 76 |
| 29-B-70 | 1 | 37 | 14 | 43 | 94 |
| 36-B-63 | 2 | 9 | 18 | 32 | 59 |
| 72-S-59 | 1 | 0 | 7 | 29 | 36 |
| 16-P-38 | 2 | 0 | 0 | 7 | 7 |
| Mean | 13 | 15 | 50 | ||
| Std. dev. | 11 | 7 | 24 | ||
| Mean–std. dev. | 2 | 8 | 26 | Sum 35 |
| Patient-Code | BRCA1 | BRCA2 | TP53 | Further Genes | LOH | TAI | LST-Break Points | HRD- Score |
|---|---|---|---|---|---|---|---|---|
| 51-J-69 | c.5266_5267 insC (89%) | c.520A>T (85%) | 24 | 28 | 98 | 150 | ||
| 71-T-53 | c.375G>A (31%), c.764T>G (59%) | 3 | 25 | 97 | 125 | |||
| 49-J-56 | c.268_269delTC (68%) | 24 | 20 | 66 | 110 | |||
| 85-K-49 | c.1166delG (53%) | c.463A>C (36%) | CHEK2 (42%) | 22 | 15 | 64 | 101 | |
| 31-J-53 | c.5466_5467insA (64%) | c.659A>G (54%) | 13 | 10 | 74 | 97 | ||
| 29-B-702 | c.1687C>T (68%) | c.375+2T>A (34%) | MLH1 (48%) | 37 | 14 | 43 | 94 | |
| 23-S-36 | c.452C>A (91%) | 16 | 20 | 51 | 87 | |||
| 21-R-22 | c.657_658delTG (71%) | c.1013_1014insT (42%) | MUTYH (25%), SLX4 (49%) | 9 | 19 | 58 | 86 | |
| 74-R-62 | c.4035delA (71%) | c.659A>G (49%) | 17 | 20 | 47 | 84 | ||
| 8-T-45 | c.824G>T (95%) | 15 | 21 | 46 | 82 | |||
| 38-B-55 | c.402T>A (37%) | RECQL (50%), RAD51D (67%), RAD51C (31%) | 20 | 16 | 43 | 79 | ||
| 42-H-37 | c.524G>A (92%) | 16 | 17 | 46 | 79 | |||
| 47-G-58 | c.8933C>G (70%) | c.638G>T (64%) | NF1 (30%) | 15 | 17 | 44 | 76 | |
| 24-S-35 | c.2908A>T (44%) | c.224delC (44%) | Fam175A (62%) | 1 | 17 | 53 | 71 | |
| 25-S-37 | c.814G>A (36%) | FANCM (43%) | 21 | 12 | 9 | 62 | ||
| 36-B-63 | c.4363G>T (73%) | c.949C>T (70%) | 9 | 18 | 32 | 59 | ||
| 73-W-39 | c.559+1G>C (59%) | MAP3K1 (44%), RAD50 (75%), SMARCA4 (54%) | 1 | 17 | 30 | 48 | ||
| 59-S-54 | c.764_766delTCA (79%) | 12 | 13 | 19 | 44 | |||
| 17-P-40 | c.517G>A (84%) | CDH1 (36%) | 3 | 12 | 28 | 43 | ||
| 72-S-59 | c.906delT (30%) | c.626_627delGA (28%) | PMS2 (47%) | 0 | 7 | 29 | 36 | |
| 33-T-36 | c.536A>G (66%) | 10 | 9 | 6 | 25 | |||
| 45-K-67 | MSH2 (49%) | 4 | 4 | 4 | 12 | |||
| 97-W-65 | 9 | 0 | 0 | 9 | ||||
| 16-P-38 | c.5164_5165delAG (56%) | 0 | 0 | 7 | 7 | |||
| 99-T-47 | c.626_627delGA (38%) | 0 | 2 | 3 | 5 | |||
| 107-S-59 | FANCM (49%) | 1 | 0 | 2 | 3 | |||
| 92-W-69 | CDH1 (50%) | 0 | 1 | 1 | 2 | |||
| 88-K-51 | MSH6 (50%) | 0 | 0 | 0 | 0 | |||
| 12-L-52 | 0 | 0 | 0 | 0 | ||||
| 54-D-39 | 0 | 0 | 0 | 0 | ||||
| Mutation (Frequency) |
| Cases with BRCA 1/2 Mutation | Cases without BRCA 1/2 Mutation | |||||||
|---|---|---|---|---|---|---|---|---|
| Chr. Region (bp-Position) | Incl. Tumor Gene | No. Gains | No. Balance | No. Losses | No. Gains | No. Balance | No. Losses | Chi2 |
| 10p15.3 (171262–1306517) | no | 8 | 3 | 0 | 3 | 15 | 1 | 0.0073 |
| 10p15.3 (1306517–3793806) | no | 7 | 4 | 0 | 3 | 15 | 1 | 0.0252 |
| 11p15.4 (2976948–5785900) | no | 0 | 5 | 6 | 1 | 16 | 2 | 0.0282 |
| 11p15.4 (5785900–5809417) | no | 0 | 4 | 7 | 1 | 16 | 2 | 0.0086 |
| 11p15.4 (5809417–9463851) | no | 0 | 5 | 6 | 1 | 16 | 2 | 0.0282 |
| 11p15.4 (9463851–9516249) | no | 0 | 5 | 6 | 2 | 15 | 2 | 0.0248 |
| 11p15.4–11p15.3 (9516249–12042992) | no | 0 | 5 | 6 | 1 | 16 | 2 | 0.0282 |
| Patient-Code | Extern HRD Examination | LOH | TAI | LST-Break Points | HRD- Score | HRD |
|---|---|---|---|---|---|---|
| Valid-01 | HRD detected | 18 | 11 | 63 | 92 | yes |
| Valid-02 | mutated BRCA1 | 5 | 14 | 61 | 80 | yes |
| Valid-03 | mutated BRCA2 | 15 | 18 | 36 | 69 | yes |
| Valid-04 | mutated BRCA1 | 18 | 9 | 32 | 59 | yes |
| Valid-05 | mutated BRCA1 | 9 | 8 | 38 | 55 | yes |
| Valid-06 | mutated BRCA1 | 0 | 12 | 43 | 55 | yes |
| Valid-07 | BRCA1/2 wt | 5 | 14 | 32 | 51 | yes |
| Valid-08 | BRCA1/2 wt | 12 | 9 | 22 | 43 | yes |
| Valid-09 | mutated BRCA2 | 10 | 17 | 15 | 42 | yes |
| Valid-10 | BRCA1/2 wt | 13 | 14 | 14 | 41 | no |
| Valid-11 | mutated BRCA1 | 11 | 16 | 14 | 41 | no |
| Valid-12 | BRCA1/2 wt; HRD negative | 0 | 14 | 26 | 40 | no |
| Valid-13 | BRCA1/2 wt | 0 | 1 | 23 | 24 | no |
| Valid-14 | mutated BRCA2 | 0 | 4 | 6 | 10 | no |
| Valid-15 | BRCA1/2 wt | 4 | 0 | 0 | 4 | no |
| Valid-16 | HRD negative | 0 | 1 | 2 | 3 | no |
| Valid-17 | BRCA1/2 wt | 0 | 0 | 2 | 2 | no |
| Valid-18 | BRCA1/2 wt | 0 | 0 | 0 | 0 | no |
| Valid-19 | BRCA1/2 wt | 0 | 0 | 0 | 0 | no |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magadeeva, S.; Qian, X.; Korff, N.; Flörkemeier, I.; Hedemann, N.; Rogmans, C.; Forster, M.; Arnold, N.; Maass, N.; Bauerschlag, D.O.; et al. Assessing the Phenotype of a Homologous Recombination Deficiency Using High Resolution Array-Based Comparative Genome Hybridization in Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 17467. https://doi.org/10.3390/ijms242417467
Magadeeva S, Qian X, Korff N, Flörkemeier I, Hedemann N, Rogmans C, Forster M, Arnold N, Maass N, Bauerschlag DO, et al. Assessing the Phenotype of a Homologous Recombination Deficiency Using High Resolution Array-Based Comparative Genome Hybridization in Ovarian Cancer. International Journal of Molecular Sciences. 2023; 24(24):17467. https://doi.org/10.3390/ijms242417467
Chicago/Turabian StyleMagadeeva, Svetlana, Xueqian Qian, Nadine Korff, Inken Flörkemeier, Nina Hedemann, Christoph Rogmans, Michael Forster, Norbert Arnold, Nicolai Maass, Dirk O. Bauerschlag, and et al. 2023. "Assessing the Phenotype of a Homologous Recombination Deficiency Using High Resolution Array-Based Comparative Genome Hybridization in Ovarian Cancer" International Journal of Molecular Sciences 24, no. 24: 17467. https://doi.org/10.3390/ijms242417467