
Viral Diversity in Samples of Freshwater Gastropods Benedictia baicalensis (Caenogastropoda: Benedictiidae) Revealed by Total RNA-Sequencing
 , ,
, ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Taxonomic Viral Composition Based on Direct Analysis of Reads
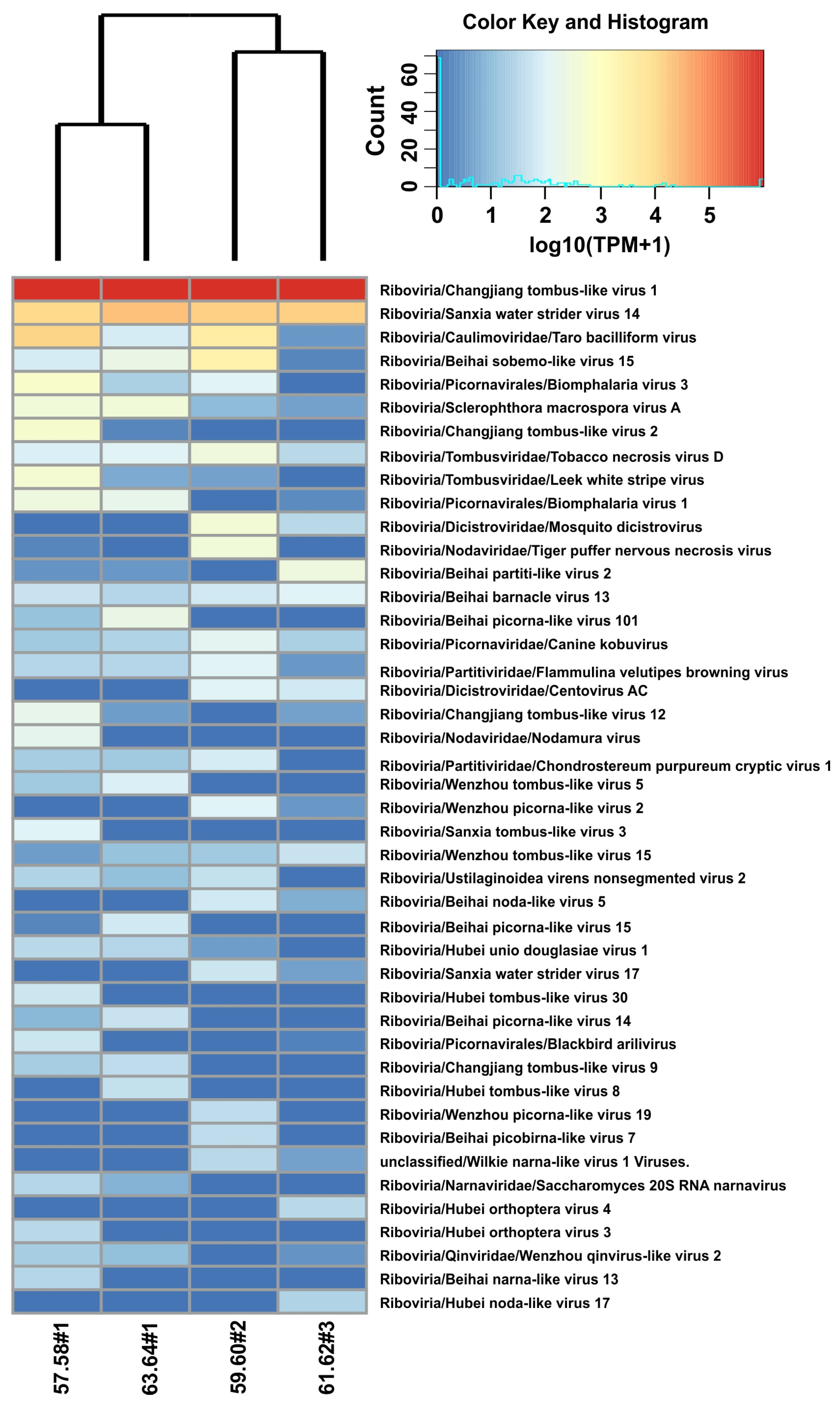
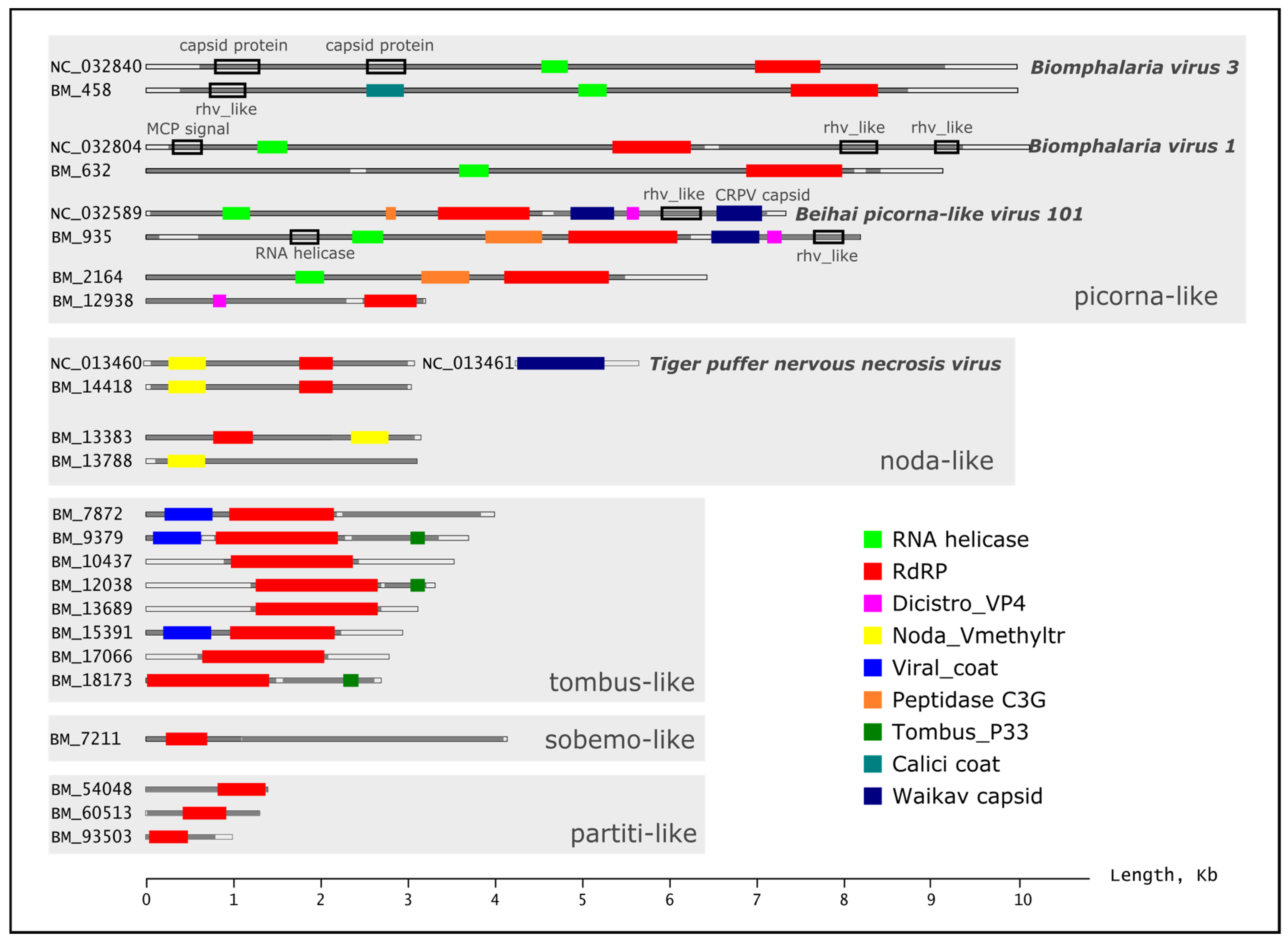
2.2. Analysis of Assembled Scaffolds in Samples of Benedictia baicalensis
2.3. Phylogenetic Analysis of RNA-Dependent RNA Polymerase Genes
3. Discussion
4. Materials and Methods
4.1. Sampling Sites and Total RNA Sequencing
4.2. Primary Processing of Reads
4.3. Taxonomic Analysis of Original Genome Reads
4.4. Assembly of Metatranscriptomic Reads
4.5. Viral Scaffolds Detection
4.6. Filtering out False Positive Viral Scaffolds
4.7. Taxonomic Assignment of Viral Scaffolds
4.8. Functional Assignment of Viral Communities
4.9. Phylogenetic Analysis of RNA-Dependent RNA Polymerase Genes
4.10. Statistical Analysis of Taxonomic Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouchet, P. The Exploration of Marine Biodiversity Scientific and Technological Challenges; Duarte, C.M., Ed.; Fundación BBVA: Granada, Spain, 2015. [Google Scholar]
- Dang, V.T.; Benkendorff, K.; Green, T.; Speck, P. Marine Snails and Slugs: A Great Place to Look for Antiviral Drugs. J. Virol. 2015, 89, 8114–8118. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Raftos, D.; Speck, P.; Montagnani, C. Antiviral immunity in marine molluscs. J. Gen. Virol. 2015, 96, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Philipp, E.E.R.; Abele, D. Masters of longevity: Lessons from long-lived bivalves—A mini-review. Gerontology 2010, 56, 55–65. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Matteson, A.R. Freshwater and marine virioplankton: A brief overview of commonalities and differences. Freshw. Biol. 2008, 53, 1076–1089. [Google Scholar] [CrossRef]
- Le Guyader, F.S.; Atmar, R.L.; Le Pendu, J. Transmission of viruses through shellfish: When specific ligands come into play. Curr. Opin. Virol. 2012, 2, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Shuping, L.S.; Human, I.S.; Lues, J.F.R.; Paulse, A.N. The Prevalence of Viruses Related to the Production of Mussels and Oysters in Saldanha Bay: A Systematic Review. Aquac. J. 2023, 3, 90–106. [Google Scholar] [CrossRef]
- Farley, C.A. Viruses and viruslike lesions in marine mollusks. Mar. Fish. Rev. 1978, 40, 18–20. [Google Scholar]
- Johnson, P.T. Viral diseases of marine invertebrates. Helgoländer Meeresunters 1984, 37, 65–98. [Google Scholar] [CrossRef]
- Renault, T.; Novoa, B. Viruses infecting bivalve molluscs. Aquat. Living Resour. 2004, 17, 397–409. [Google Scholar] [CrossRef]
- Galinier, R.; Tetreau, G.; Portet, A.; Pinaud, S.; Duval, D.; Gourbal, B. First characterization of viruses from freshwater snails of the genus Biomphalaria, the intermediate host of the parasite Schistosoma mansoni. Acta Trop. 2017, 167, 196–203. [Google Scholar] [CrossRef]
- Goldberg, T.L.; Blevins, E.; Leis, E.M.; Standish, I.F.; Richard, J.C.; Lueder, M.R.; Cer, R.Z.; Bishop-Lilly, K.A. Plasticity, Paralogy, and Pseudogenization: Rhabdoviruses of Freshwater Mussels Elucidate Mechanisms of Viral Genome Diversification and the Evolution of the Finfish-Infecting Rhabdoviral Genera. J. Virol. 2023, 97, e00196-23. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Galatowitsch, M.L.; Argüello-Astorga, G.R.; van Bysterveldt, K.; Kraberger, S.; Stainton, D.; Harding, J.S.; Roumagnac, P.; Martin, D.P.; Lefeuvre, P.; et al. Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect. Genet. Evol. 2016, 39, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.L.; Dunn, C.D.; Leis, E.; Waller, D.L. A Novel Picorna-Like Virus in a Wabash Pigtoe (Fusconaia flava) from the Upper Mississippi River, USA. Freshw. Mollusk Biol. Conserv. 2019, 22, 81–84. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, S.M.; Buddenborg, S.K.; Loker, E.S.; Bonning, B.C. Virus-derived sequences from the transcriptomes of two snail vectors of schistosomiasis, Biomphalaria pfeifferi and Bulinus globosus from Kenya. PeerJ 2021, 9, e12290. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Liu, G.F.; Liu, C.; Yang, L.L.; Liu, M.; Xie, K.M.; Shi, S.K.; Shi, M.; Jiang, J.Z. Novel RNA viruses in oysters revealed by virome. iMeta 2022, 1, e65. [Google Scholar] [CrossRef]
- Jiang, J.Z.; Fang, Y.F.; Wei, H.Y.; Zhu, P.; Liu, M.; Yuan, W.G.; Yang, L.L.; Guo, Y.X.; Jin, T.; Shi, M.; et al. A remarkably diverse and well-organized virus community in a filter-feeding oyster. Microbiome 2023, 11, 2. [Google Scholar]
- Meyers, T. Marine bivalve mollusks as reservoirs of viral finfish pathogens: Significance to marine and anadromous finfish aquaculture. Mar. Fish. Rev. 1984, 46, 14–17. [Google Scholar]
- Renault, T. Viruses Infecting Marine Mollusks. Stud. Viral Ecol. Second Ed. 2011, 2, 277–303. [Google Scholar]
- Arzul, I.; Corbeil, S.; Morga, B.; Renault, T. Viruses infecting marine molluscs. J. Invertebr. Pathol. 2017, 147, 118–135. [Google Scholar] [CrossRef]
- Zannella, C.; Mosca, F.; Mariani, F.; Franci, G.; Folliero, V.; Galdiero, M.; Tiscar, P.G.; Galdiero, M. Microbial diseases of bivalve mollusks: Infections, immunology and antimicrobial defense. Mar. Drugs 2017, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Agius, J.; Ackerly, D.; Beddoe, T.; Helbig, K. The Role of Anti-Viral Effector Molecules in Mollusc Hemolymph. Biomolecules 2022, 12, 345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Chen, Y.; Wei, X.; Cui, J. Viromes in marine ecosystems reveal remarkable invertebrate RNA virus diversity. Sci. China Life Sci. 2022, 65, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.C.; Leis, E.; Dunn, C.D.; Agbalog, R.; Waller, D.; Knowles, S.; Putnam, J.; Goldberg, T.L. Mass mortality in freshwater mussels (Actinonaias pectorosa) in the Clinch River, USA, linked to a novel densovirus. Sci. Rep. 2020, 10, 14498. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.C.; Leis, E.M.; Dunn, C.D.; Harris, C.; Agbalog, R.E.; Campbell, L.J.; Knowles, S.; Waller, D.L.; Putnam, J.G.; Goldberg, T.L. Freshwater Mussels Show Elevated Viral Richness and Intensity during a Mortality Event. Viruses 2022, 14, 2603. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.C.; Blevins, E.; Dunn, C.D.; Leis, E.M.; Goldberg, T.L. Viruses of Freshwater Mussels during Mass Mortality Events in Oregon and Washington, USA. Viruses 2023, 15, 1719. [Google Scholar] [CrossRef]
- Porter, A.F.; Shi, M.; Eden, J.S.; Zhang, Y.Z.; Holmes, E.C. Diversity and evolution of novel invertebrate DNA viruses revealed by meta-transcriptomics. Viruses 2019, 11, 1092. [Google Scholar] [CrossRef]
- Sitnikova, T.Y.; Roepstorf, P. Mollusks that Live Exclusively in Lake Baikal. First Hand 2004, 2, 84–99. [Google Scholar]
- Maximova, N.; Koroleva, A.; Sitnikova, T.; Khanaev, I.; Bukin, Y.; Kirilchik, S. Age dynamics of telomere length of Baikal gastropods is sex-specific and multidirectional. Folia Biol. 2017, 65, 187–197. [Google Scholar] [CrossRef]
- Ropstorf, P.; Sitnikova, T.Y.; Timoshkin, O.A.; Pomazkina, G.V. Observation on Stomach Contents, Food Uptake and Feeding Strategies of Endemic Baikalian Gastropods. Berl. Palaobiologische Abh. 2003, 4, 151–156. [Google Scholar]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Waldron, F.M.; Stone, G.N.; Obbard, D.J. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2018, 14, e1007533. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Clements, J.; Eddy, S.R.; Hubley, R.; Jones, T.A.; Jurka, J.; Smit, A.F.A.; Finn, R.D. Dfam: A database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013, 41, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. MBio 2018, 9, 02329-18. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef]
- Culley, A. New insight into the RNA aquatic virosphere via viromics. Virus Res. 2018, 244, 84–89. [Google Scholar] [CrossRef]
- Kolundžija, S.; Cheng, D.-Q.; Lauro, F.M. RNA Viruses in Aquatic Ecosystems through the Lens of Ecological Genomics and Transcriptomics. Viruses 2022, 14, 702. [Google Scholar] [CrossRef]
- Paraskevopoulou, S.; Käfer, S.; Zirkel, F.; Donath, A.; Petersen, M.; Liu, S.; Zhou, X.; Drosten, C.; Misof, B.; Junglen, S. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus Evol. 2021, 7, veab030. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Y.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.; Suzuki, N. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Galaziy, G. (Ed.) Atlas of Lake Baikal; Roskartografiya: Moscow, Russia, 1993. [Google Scholar]
- Inoue, Y.; Takeda, H. Teratorn and its relatives—A cross-point of distinct mobile elements, transposons and viruses. Front. Vet. Sci. 2023, 10, 1158023. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Yutin, N. Evolution of double-stranded DNA viruses of eukaryotes: From bacteriophages to transposons to giant viruses. Ann. N. Y. Acad. Sci. 2015, 1341, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Saga, T.; Aikawa, T.; Kumagai, M.; Shimada, A.; Kawaguchi, Y.; Naruse, K.; Morishita, S.; Koga, A.; Takeda, H. Complete fusion of a transposon and herpesvirus created the Teratorn mobile element in medaka fish. Nat. Commun. 2017, 8, 551. [Google Scholar] [CrossRef]
- Dayaram, A.; Galatowitsch, M.; Harding, J.S.; Argüello-Astorga, G.R.; Varsani, A.; Goldstien, S.; Zawar-Reza, P.; Gomez, C.; Harding, J.S.; Varsani, A. Novel ssDNA virus recovered from estuarine mollusc (Amphibola crenata) whose replication associated protein (Rep) shares similarities with Rep-like sequences of bacterial origin. J. Gen. Virol. 2013, 22, 1104–1110. [Google Scholar] [CrossRef]
- Rosani, U.; Gerdol, M. A bioinformatics approach reveals seven nearly-complete RNA-virus genomes in bivalve RNA-seq data. Virus Res. 2017, 239, 33–42. [Google Scholar] [CrossRef]
- Scapolatiello, A.; Rosani, U.; Manfrin, C.; Puljas, S.; Pallavicini, A.; Gerdol, M. Identification of five picorna-like viruses associated with the endangered cavedwelling bivalve Congeria kusceri (Bole, 1962). ISJ 2022, 19, 28–36. [Google Scholar]
- Ip, H.S.; Desser, S.S. A picornavirus-like pathogen of Cotylogaster occidentalis (Trematoda: Aspidogastrea), an intestinal parasite of freshwater mollusks. J. Invertebr. Pathol. 1984, 43, 197–206. [Google Scholar] [CrossRef]
- Rasmussen, L.P.D.; Ip, H.S.; Desser, S.S. Virus-associated granulocytomas in the marine mussel, Mytilus edulis, from three sites in Denmark. J. Invertebr. Pathol. 1986, 48, 117–123. [Google Scholar] [CrossRef]
- Comps, M.; Herbaut, C.; Fougerouse, A. Virus-like particles in pearl oyster Pinctada margaritifera. Bull. Eur. Assoc. Fish. Pathol. 1999, 19, 85–88. [Google Scholar]
- Novoa, B.; Figueras, A. Virus-like particles associated with mortalities of the carpet-shell clam Ruditapes decussatus. Dis. Aquat. Organ. 2000, 39, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Carballal, M.J.; Villalba, A.; Iglesias, D.; Hine, P.M. Virus-like particles associated with large foci of heavy hemocytic infiltration in cockles Cerastoderma edule from Galicia (NW Spain). J. Invertebr. Pathol. 2003, 84, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Bandín, I.; Souto, S. Betanodavirus and VER Disease: A 30-year. Pathogens 2020, 9, 106. [Google Scholar] [CrossRef]
- Volpe, E.; Errani, F.; Mandrioli, L.; Ciulli, S. Advances in Viral Aquatic Animal Disease Knowledge: The Molecular Methods’ Contribution. Biology 2023, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, C.; Pérez, M.; Arizcun, M.; García-Ruiz, C.; Chaves-Pozo, E. Reservoirs of Red-Spotted Grouper Nervous Necrosis Virus (RGNNV) in Squid and Shrimp Species of Northern Alboran Sea. Viruses 2022, 14, 328. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Liu, S.; Li, J.; Xu, T.T.; Wang, X.H.; Fu, G.M.; Li, X.P.; Sang, S.W.; Bian, X.D.; Hao, J.W. Evidence for cross-species transmission of covert mortality nodavirus to new host of Mugilogobius abei. Front. Microbiol. 2018, 9, 1447. [Google Scholar] [CrossRef]
- Wang, C.; Yao, L.; Wang, W.; Sang, S.; Hao, J.; Li, C.; Zhang, Q. First report on natural infection of nodavirus in an echinodermata, sea cucumber (Apostichopus japonicas). Viruses 2021, 13, 636. [Google Scholar] [CrossRef]
- Warrilow, D.; Huang, B.; Newton, N.D.; Harrison, J.J.; Johnson, K.N.; Chow, W.K.; Hall, R.A.; Hobson-Peters, J. The taxonomy of an Australian nodavirus isolated from mosquitoes. PLoS ONE 2018, 13, e0210029. [Google Scholar] [CrossRef]
- Karmanova, A.N.; Zimin, A.A. Experimental model for study bacteriophage bioaccumulation by a bivalve Unio pictorium (L.1758). J. Phys. Conf. Ser. 2020, 1701, 012012. [Google Scholar] [CrossRef]
- Errani, F.; Ponti, M.; Volpe, E.; Ciulli, S. Spatial and seasonal variability of human and fish viruses in mussels inside and offshore of Ravenna’s harbour (Adriatic Sea, Italy). J. Appl. Microbiol. 2021, 130, 994–1008. [Google Scholar] [CrossRef]
- Grachev, M.A.; Kumarev, V.P.; Mamaev, L.V.; Zorin, V.L.; Baranova, L.V.; Denikina, N.N.; Belikov, S.I.; Petrov, E.A.; Kolesnik, V.S.; Kolesnik, R.S.; et al. Distemper virus in Baikal seals. Nature 1989, 338, 209. [Google Scholar] [CrossRef]
- Butina, T.V.; Denikina, N.N.; Belikov, S.I. Canine distemper virus diversity in Lake Baikal seal (Phoca sibirica) population. Vet. Microbiol. 2010, 144, 192–197. [Google Scholar] [CrossRef]
- Kondratov, I.G.; Denikina, N.N.; Belikov, S.I.; Durymanova, A.A.; Ustinova, E.N.; Shestopalov, A.M. Mollusks as a natural reservoir of morbilliviruses. Dokl. Biol. Sci. 2003, 389, 154–156. [Google Scholar] [CrossRef]
- Petrova, N.M.; Kondratov, I.G.; Dzyuba, E.V.; Denikina, N.N.; Belikov, S.I. Study of the mechanisms of circulation of the canine distemper virus in the lake ecosystem. Baikal. Bull. East Sib. Sci. Cent. SB RAMS 2003, 7, 135–136. [Google Scholar]
- Butina, T.V.; Khanaev, I.V.; Petrushin, I.S.; Bondaryuk, A.N.; Maikova, O.O.; Bukin, Y.S. The RNA Viruses in Samples of Endemic Lake Baikal Sponges. Diversity 2023, 15, 835. [Google Scholar] [CrossRef]
- Kozhova, O.M.; Izmesteva, L.R. Lake Baikal: Evolution and Biodiversity; Backhuys Publisher: Leiden, The Netherlands, 1998. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Butina, T.V.; Petrushin, I.S.; Khanaev, I.V.; Bukin, Y.S. Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera. Microorganisms 2022, 10, 480. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. Rna 2020, 26, 903–909. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Jari Oksanen, F.; Blanchet, G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Sólymos, M.P.; Stevens, H.H.; Wagner, H. Package ‘vegan’. Community ecology package, version 2.0-10. J. Stat. Softw. 2013, 48, 103–132. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. R Package, Package “gplots”: Various R Programming Tools for Plotting Data, Version 2.17.0; ScienceOpen: Berlin, Germany, 2015.
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; López-Pérez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New viral biogeochemical roles revealed through metagenomic analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef]
- Butina, T.V.; Bukin, Y.S.; Petrushin, I.S.; Tupikin, A.E.; Kabilov, M.R.; Belikov, S.I. Extended evaluation of viral diversity in lake baikal through metagenomics. Microorganisms 2021, 9, 760. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample_ID | Sampling Site | Date | Depth, m | Latitude and Longitude |
|---|---|---|---|---|
| 57.58#1 | #1—Listvennichny Bay (Listvyanka) | 31 October 2022 | 3–15 | 51°51′51.77″ N 104°50′37.80″ E |
| 59.60#2 | #2—Ushkany Islands | 6 June 2021 | 2–9 | 53°51′05.76″ N 108°42′28.46″ E |
| 61.62#3 | #3—Bolshie Koty | 31 May 2022 | 4–8 | 51°54′08.66″ N 105°06′13.04″ E |
| 63.64#1 | #1—Listvennichny Bay | 10 June 2022 | 3–15 | 51°51′51.77″ N 104°50′37.80″ E |
| Scaffold ID_Length (nt) | Number of ORFs | Similarity of ORFs with RefSeq Proteins | BLAST Hit_RefSeq (Virotype) | Virotype Taxa (Known Order or Family) | Host/Source Lineage | TPM per Sample | |||
|---|---|---|---|---|---|---|---|---|---|
| 57.58 | 59.60 | 61.62 | 63.64 | ||||||
| BM_458/8746 | 1 | 24.8 | Biomphalaria virus 3 | Picornavirales | Mollusca; Gastropoda | 550 | 0 | 0 | 3 |
| BM_632/8432 | 3 | 29.6–32.2 | Biomphalaria virus 1 | Picornavirales | Mollusca; Gastropoda | 250 | 0 | 1 | 183 |
| BM_935/8225 | 3 | 25.5–29 | Beihai picorna-like virus 101 | unclassified | /Arthropoda; Decapoda | 12 | 0 | 0 | 203 |
| BM_2164/5832 | 2 | 33.2 | Centovirus AC | Dicistroviridae | Insecta; Kerteszia | 0 | 116 | 0 | 0 |
| BM_7211/4124 | 2 | 28.1–39.3 | Beihai sobemo-like virus 15 | unclassified | /Arthropoda; Decapoda | 71 | 2376 | 1 | 192 |
| BM_7872/3857 | 4 | 30.6–49.2 | Sanxia water strider virus 14 | unclassified | /Insecta; Hemiptera | 10,958 | 14,615 | 13,680 | 23,072 |
| BM_9379/3384 | 3 | 38.3–45.3 | Tobacco necrosis virus D | Tombusviridae | Viridiplantae; Magnoliopsida | 91 | 323 | 31 | 112 |
| BM_12038/3224 | 4 | 32.4–67.3 | Changjiang tombus-like virus 1 | unclassified | /Arthropoda; Decapoda | 966,801 | 969,290 | 978,336 | 970,063 |
| BM_12938/3213 | 2 | 42–49.8 | Beihai picorna-like virus 15 | unclassified | /Mollusca; Octopoda | 1 | 0 | 0 | 64 |
| BM_10437/3187 | 3 | 31.4–43.0 | Changjiang tombus-like virus 12 | unclassified | /Arthropoda; Decapoda | 168 | 0 | 3 | 2 |
| BM_13788/3115 | 1 | 33.9 | Sclerophthora macrospora virus A | unclassified | Oomycota; Sclerophthora | 299 | 9 | 0 | 334 |
| BM_13383/3065 | 2 | 45.2–52.8 | Nodamura virus | Nodaviridae | Insecta; Diptera | 156 | 0 | 0 | 0 |
| BM_14418/3051 | 1 | 99.3 | Tiger puffer nervous necrosis virus | Nodaviridae | Vertebrata; Tetraodontiformes | 1 | 329 | 0 | 0 |
| BM_13689/2980 | 3 | 33.9–40.6 | Sanxia water strider virus 14 | unclassified | /Insecta; Hemiptera | 1 | 9 | 386 | 1 |
| BM_17066/2761 | 3 | 39.3–76.8 | Changjiang tombus-like virus 2 | unclassified | /Arthropoda; Decapoda | 387 | 0 | 0 | 1 |
| BM_15391/2722 | 3 | 39.3–46.8 | Sanxia water strider virus 14 | unclassified | /Insecta; Hemiptera | 0 | 16 | 417 | 3 |
| BM_18173/2610 | 2 | 47.3 | Leek white stripe virus | Tombusviridae | Viridiplantae; Liliopsida | 460 | 4 | 0 | 5 |
| BM_23235/2360 | 1 | 23.7 | Hubei tombus-like virus 30 | unclassified | /Arthropoda; Arachnida | 58 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butina, T.V.; Zemskaya, T.I.; Bondaryuk, A.N.; Petrushin, I.S.; Khanaev, I.V.; Nebesnykh, I.A.; Bukin, Y.S. Viral Diversity in Samples of Freshwater Gastropods Benedictia baicalensis (Caenogastropoda: Benedictiidae) Revealed by Total RNA-Sequencing. Int. J. Mol. Sci. 2023, 24, 17022. https://doi.org/10.3390/ijms242317022
Butina TV, Zemskaya TI, Bondaryuk AN, Petrushin IS, Khanaev IV, Nebesnykh IA, Bukin YS. Viral Diversity in Samples of Freshwater Gastropods Benedictia baicalensis (Caenogastropoda: Benedictiidae) Revealed by Total RNA-Sequencing. International Journal of Molecular Sciences. 2023; 24(23):17022. https://doi.org/10.3390/ijms242317022
Chicago/Turabian StyleButina, Tatyana V., Tamara I. Zemskaya, Artem N. Bondaryuk, Ivan S. Petrushin, Igor V. Khanaev, Ivan A. Nebesnykh, and Yurij S. Bukin. 2023. "Viral Diversity in Samples of Freshwater Gastropods Benedictia baicalensis (Caenogastropoda: Benedictiidae) Revealed by Total RNA-Sequencing" International Journal of Molecular Sciences 24, no. 23: 17022. https://doi.org/10.3390/ijms242317022