
Inhibition of CDK1 by RO-3306 Exhibits Anti-Tumorigenic Effects in Ovarian Cancer Cells and a Transgenic Mouse Model of Ovarian Cancer
 , ,
, ,
Abstract
:1. Introduction
2. Results
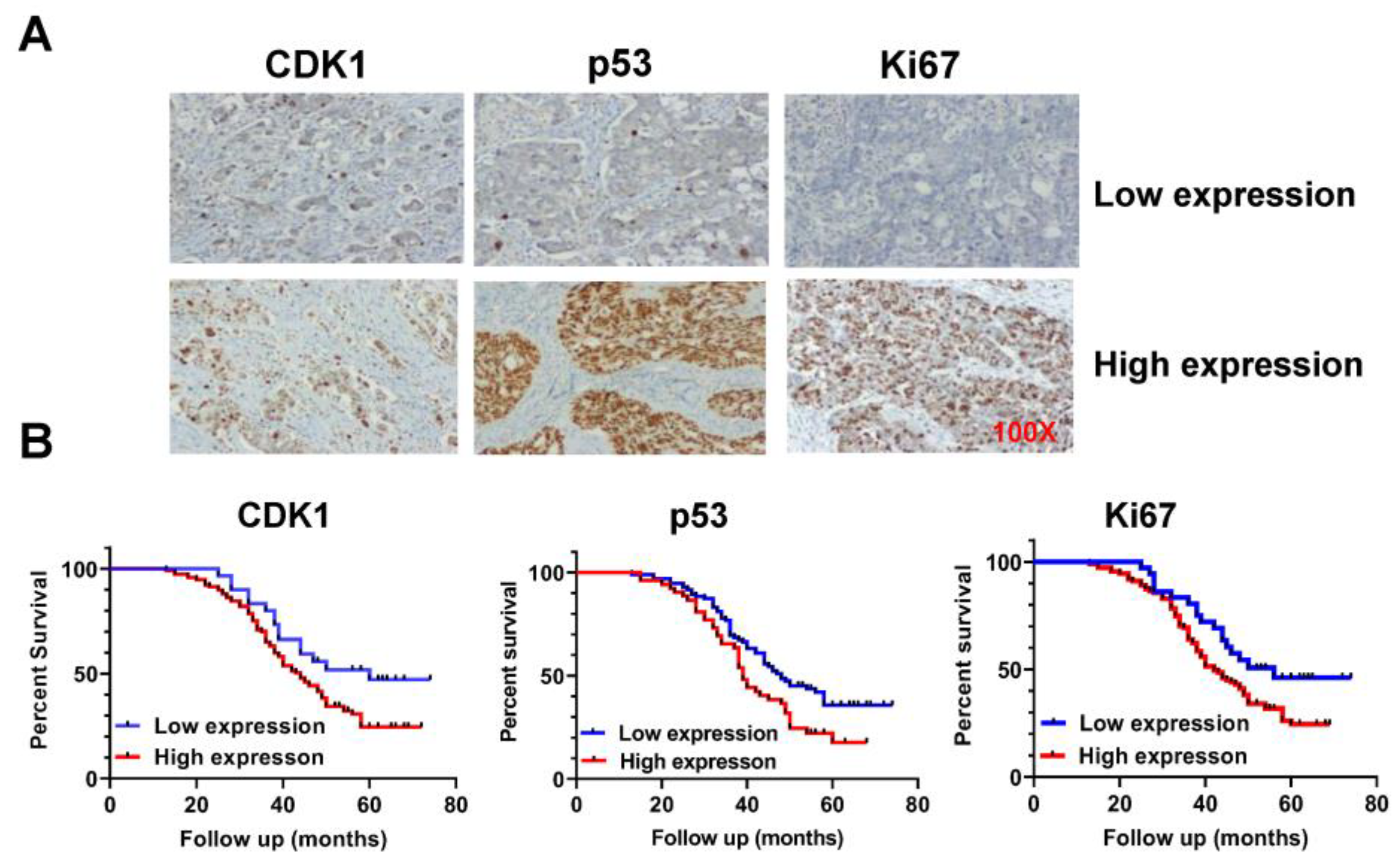
2.1. Effect of CDK1 on Prognosis in Patients with OC
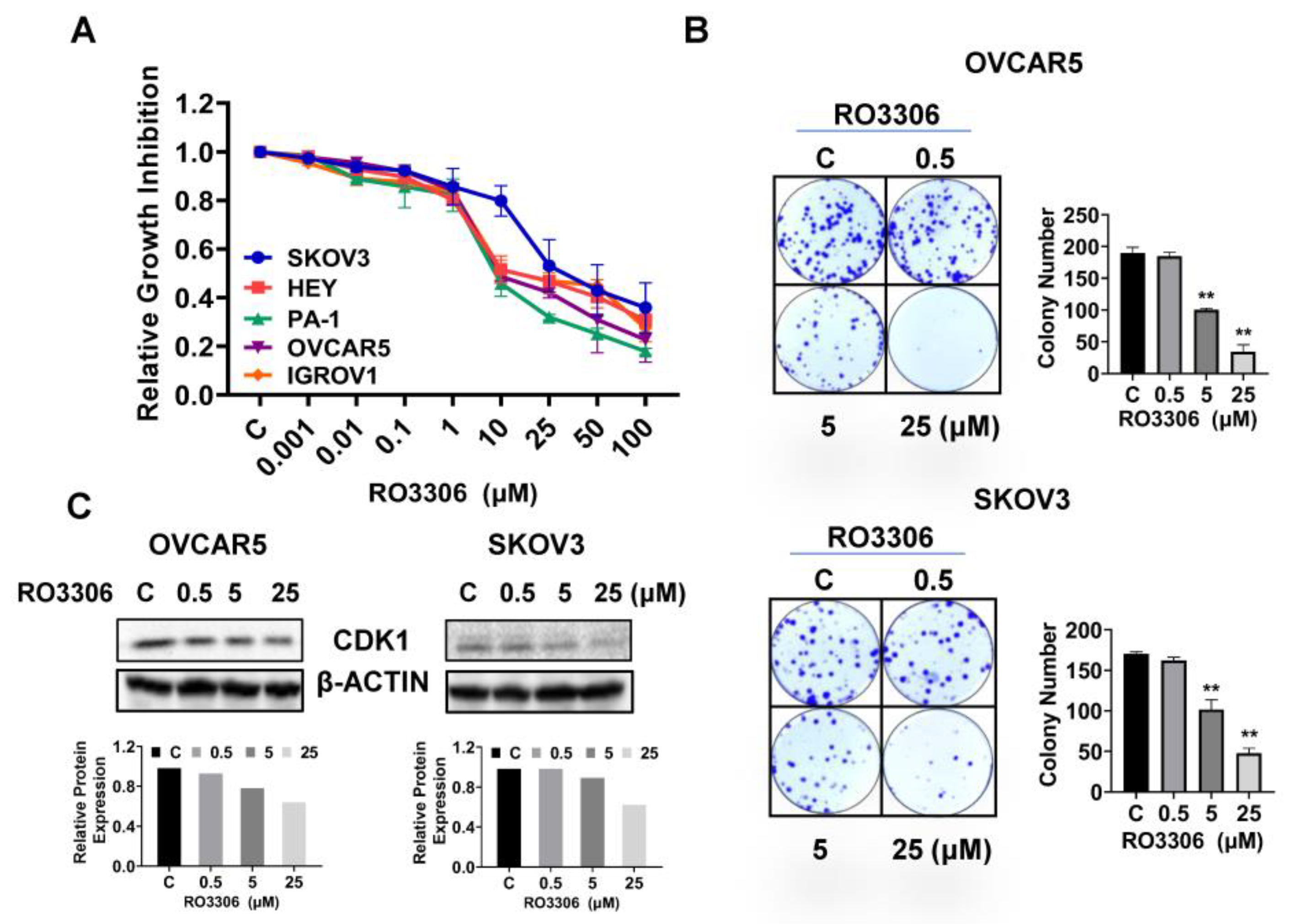
2.2. Effect of RO-3306 on Cell Proliferation in OC Cells
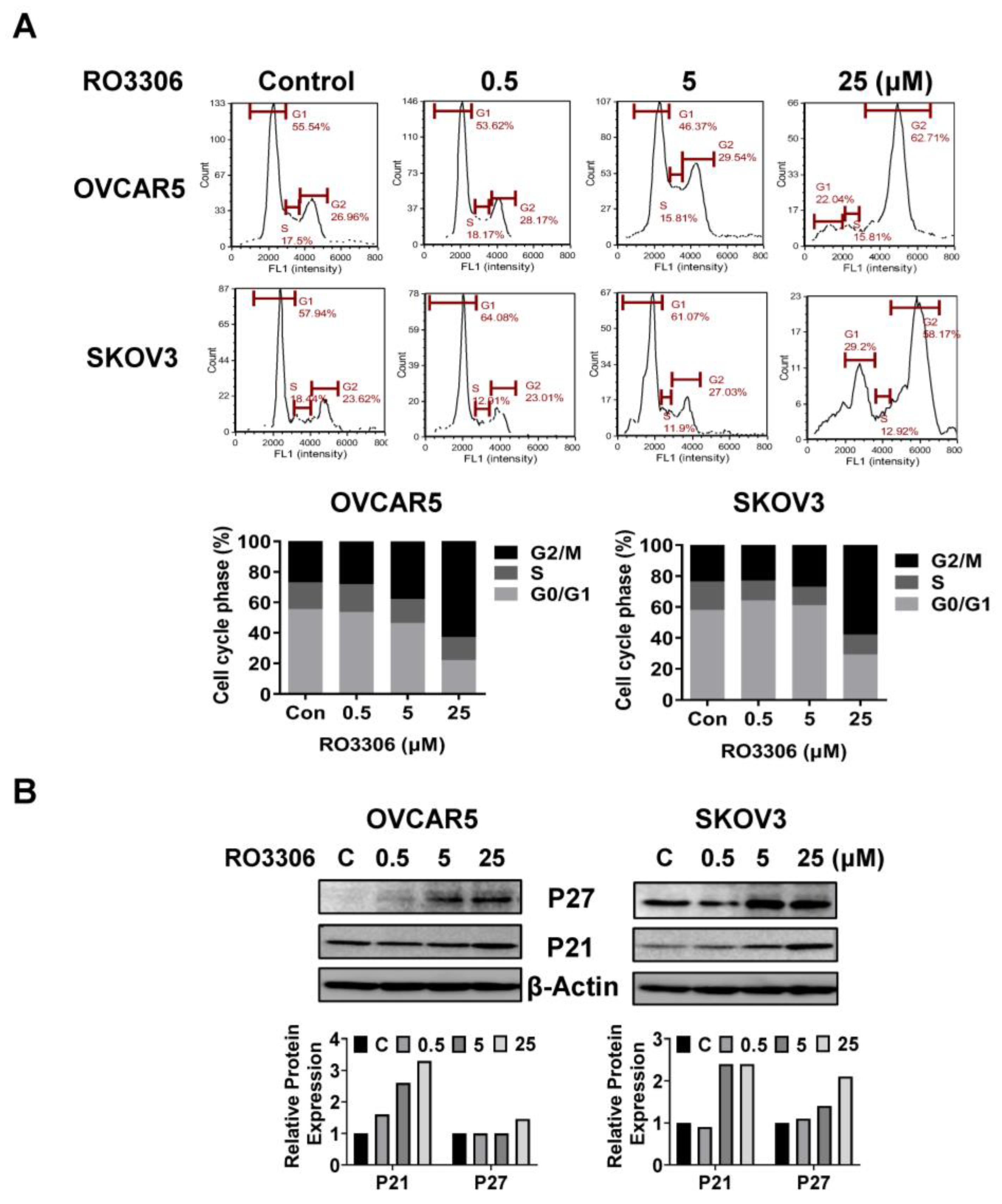
2.3. Effect of RO-3306 on Cell Cycle Progression in OC Cells
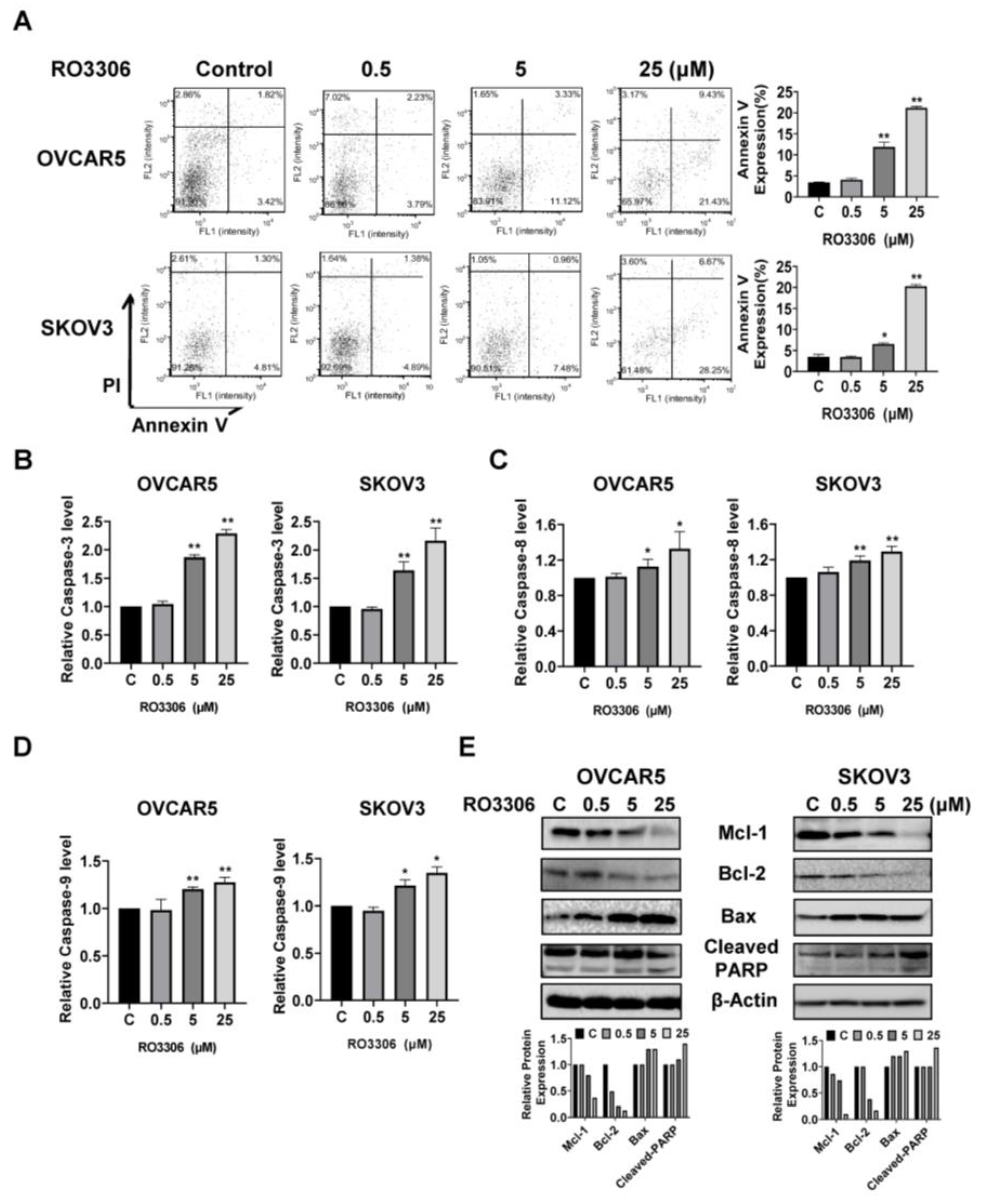
2.4. Effect of RO-3306 on Apoptosis in OC Cells
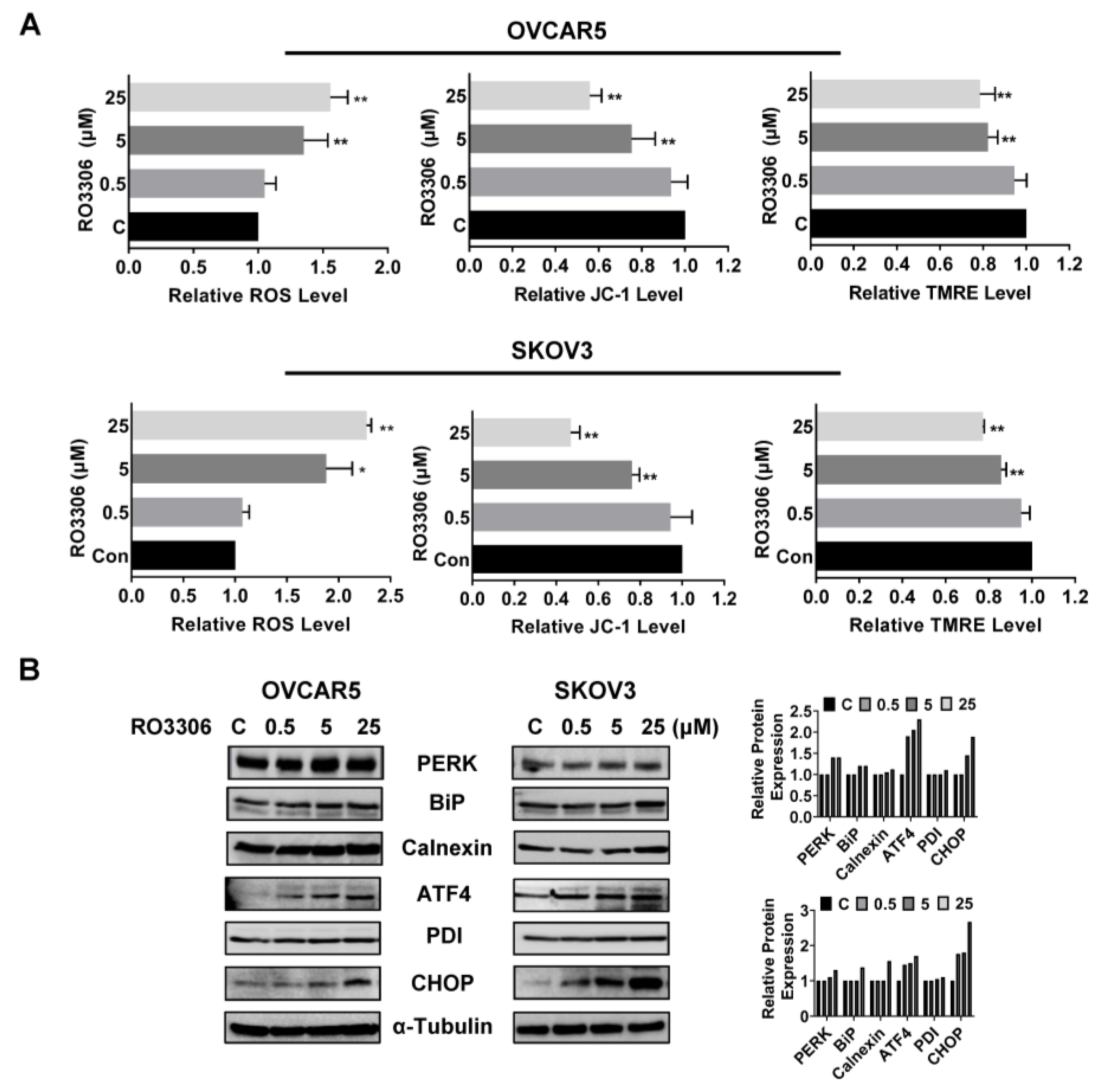
2.5. Effect of RO-3306 on Cellular Stress in OC Cells
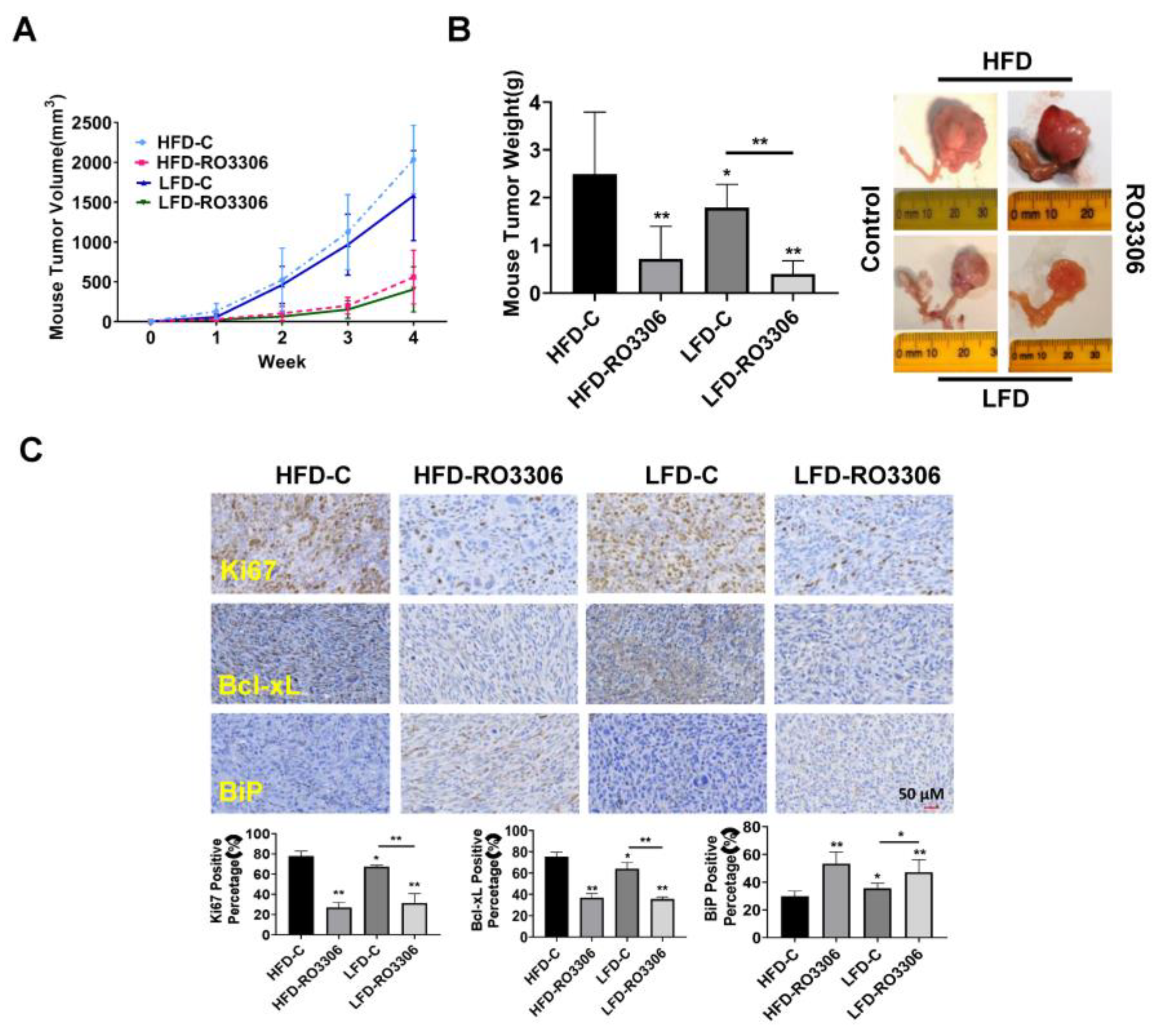
2.6. Effect of RO-3306 on Tumor Growth in the KpB Mouse Model of OC
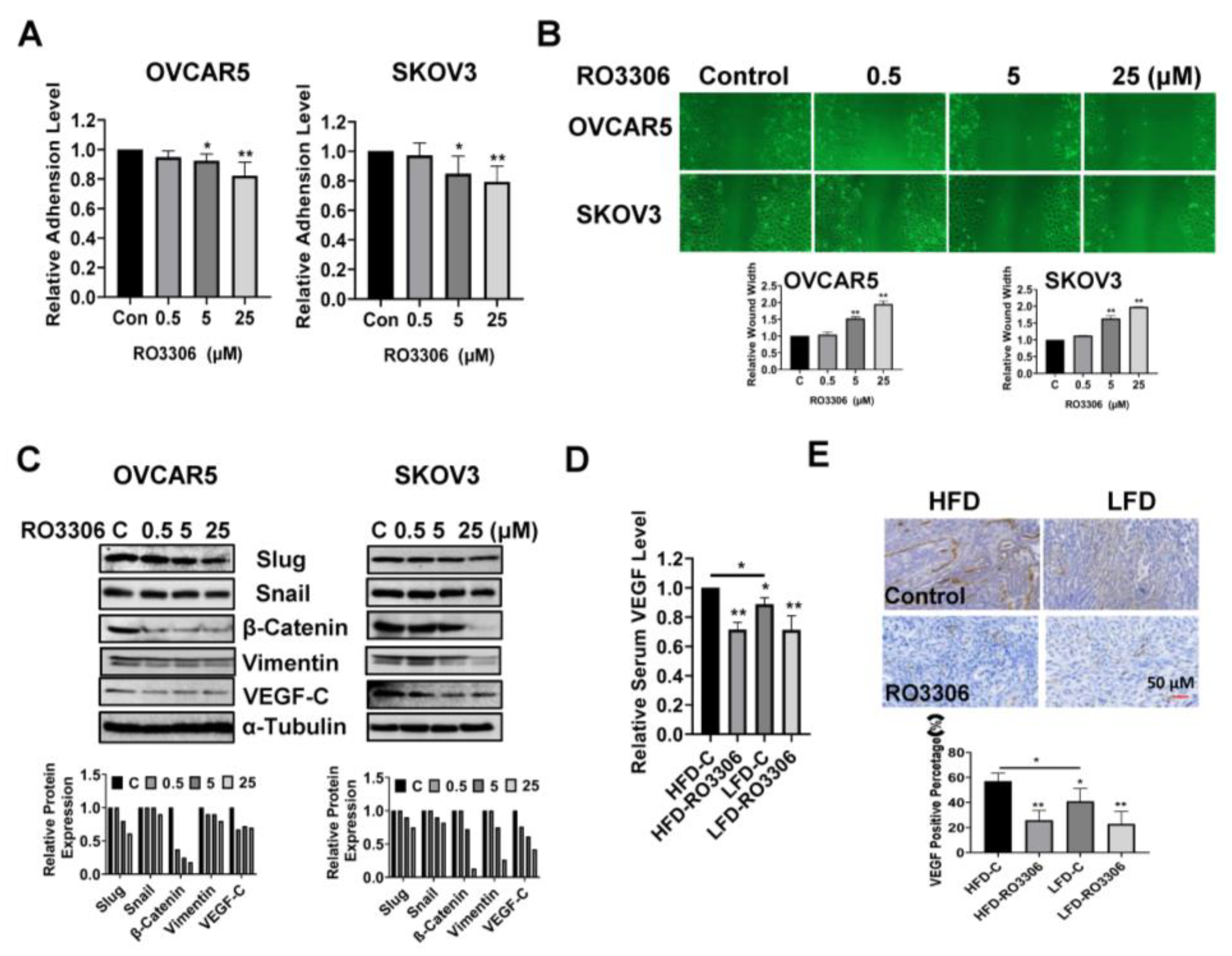
2.7. Effect of RO-3306 on Adhesion and Invasion In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Patients and OC Samples
4.2. Cell Culture and Reagents
4.3. Cell Proliferation Assay
4.4. Colony Assays
4.5. Cell Cycle Assay
4.6. Annexin V Assay
4.7. Cleaved Caspase 3, 8, and 9 ELISA Assays
4.8. Reactive Oxygen Species (ROS) Assay
4.9. Mitochondrial Membrane Potential Assays
4.10. Adhesion Assay
4.11. Wound Healing Assay
4.12. Western Immunoblotting
4.13. KpB Transgenic Mouse Model of OC
4.14. Serum VEGF Assay
4.15. IHC for Ovarian Tumors of KpB Mice
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Penny, S.M. Ovarian Cancer: An Overview. Radiol. Technol. 2020, 91, 561–575. [Google Scholar] [PubMed]
- Rauh-Hain, J.A.; Krivak, T.C.; Del Carmen, M.G.; Olawaiye, A.B. Ovarian cancer screening and early detection in the general population. Rev. Obs. Gynecol. 2011, 4, 15–21. [Google Scholar]
- Sambasivan, S. Epithelial ovarian cancer: Review article. Cancer Treat. Res. Commun. 2022, 33, 100629. [Google Scholar] [CrossRef]
- Oronsky, B.; Ray, C.M.; Spira, A.I.; Trepel, J.B.; Carter, C.A.; Cottrill, H.M. A brief review of the management of platinum-resistant-platinum-refractory ovarian cancer. Med. Oncol. 2017, 34, 103–110. [Google Scholar] [CrossRef]
- Mogos, R.A.; Popovici, R.; Tanase, A.E.; Calistru, T.; Popovici, P.; Grigore, M.; Carauleanu, A. New approaches in ovarian cancer based on genetics and carcinogenesis hypotheses (Review). Exp. Ther. Med. 2022, 23, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, A.S.; Dumitrescu, E.A.; Komporaly, I.A.; Mihăilă, R.I.; Lungulescu, C.V.; Stănculeanu, D.L. New Targeted Therapies and Combinations of Treatments for Cervical, Endometrial, and Ovarian Cancers: A Year in Review. Curr. Oncol. 2022, 29, 2835–2847. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Chymkowitch, P. Cell Cycle-Dependent Transcription: The Cyclin Dependent Kinase Cdk1 Is a Direct Regulator of Basal Transcription Machineries. Int. J. Mol. Sci. 2022, 23, 1293. [Google Scholar] [CrossRef]
- Chohan, T.A.; Qayyum, A.; Rehman, K.; Tariq, M.; Akash, M.S.H. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed. Pharmacother. 2018, 107, 1326–1341. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sofi, S.; Mehraj, U.; Qayoom, H.; Aisha, S.; Almilaibary, A.; Alkhanani, M.; Mir, M.A. Targeting cyclin-dependent kinase 1 (CDK1) in cancer: Molecular docking and dynamic simulations of potential CDK1 inhibitors. Med. Oncol. 2022, 39, 133. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Guo, X.; Yuan, Q.; Nian, S.; Kang, P.; Xu, Z.; Li, L.; Ye, Y. Systematic Pan-Cancer Analysis Identifies CDK1 as an Immunological and Prognostic Biomarker. J. Oncol. 2022, 2022, 8115474. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Yuan, Q.; Liu, Y.; Zeng, Y.; Ke, D.; Dai, X.; Shuai, Y.; Hu, J.; Shi, H. Identification of key molecular markers in epithelial ovarian cancer by integrated bioinformatics analysis. Taiwan. J. Obs. Gynecol. 2021, 60, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Tian, X.; He, F.; Zhang, J.; Cui, X.; He, Q.; Si, P.; Shen, Y. Integrative analysis of key candidate genes and signaling pathways in ovarian cancer by bioinformatics. J. Ovarian Res. 2021, 14, 92. [Google Scholar] [CrossRef]
- Cai, J.; Li, B.; Zhu, Y.; Fang, X.; Zhu, M.; Wang, M.; Liu, S.; Jiang, X.; Zheng, J.; Zhang, X.; et al. Prognostic Biomarker Identification Through Integrating the Gene Signatures of Hepatocellular Carcinoma Properties. EBioMedicine 2017, 19, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, M.; Sato, Y.; Takahashi, K.; Sasaki, M.; Harada, K. The cyclin-dependent kinase pathway involving CDK1 is a potential therapeutic target for cholangiocarcinoma. Oncol. Rep. 2020, 43, 306–317. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, B.; Ma, Y.; Kuang, J.; Liang, J.; Yuan, Y. NUCKS1 Promotes Proliferation, Invasion and Migration of Non-Small Cell Lung Cancer by Upregulating CDK1 Expression. Cancer Manag. Res. 2020, 12, 13311–13323. [Google Scholar] [CrossRef]
- Lu, X.; Pang, Y.; Cao, H.; Liu, X.; Tu, L.; Shen, Y.; Jia, X.; Lee, J.C.; Wang, Y. Integrated Screens Identify CDK1 as a Therapeutic Target in Advanced Gastrointestinal Stromal Tumors. Cancer Res. 2021, 81, 2481–2494. [Google Scholar] [CrossRef]
- Yang, Y.; Dai, Y.; Yang, X.; Wu, S.; Wang, Y. DNMT3A Mutation-Induced CDK1 Overexpression Promotes Leukemogenesis by Modulating the Interaction between EZH2 and DNMT3A. Biomolecules 2021, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yang, W.; Fu, Z.; Xie, H.; Guo, Z.; Liu, D.; Ge, J.; Zhong, S.; Liu, L.; Liu, J.; et al. Selected using bioinformatics and molecular docking analyses, PHA-793887 is effective against osteosarcoma. Aging 2021, 13, 16425–16444. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, H.; Ren, F.; Zhang, M.; Ji, P.; Wang, W.; Liu, C. The aberrant upstream pathway regulations of CDK1 protein were implicated in the proliferation and apoptosis of ovarian cancer cells. J. Ovarian Res. 2017, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.G.; Zahurak, M.; Shah, M.; Weekes, C.D.; Hansen, A.; Siu, L.L.; Spreafico, A.; LoConte, N.; Anders, N.M.; Miles, T.; et al. A Phase I Study of Dinaciclib in Combination With MK-2206 in Patients With Advanced Pancreatic Cancer. Clin. Transl. Sci. 2020, 13, 1178–1188. [Google Scholar] [CrossRef]
- Mita, M.M.; Mita, A.C.; Moseley, J.L.; Poon, J.; Small, K.A.; Jou, Y.M.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Statkevich, P.; et al. Phase 1 safety, pharmacokinetic and pharmacodynamic study of the cyclin-dependent kinase inhibitor dinaciclib administered every three weeks in patients with advanced malignancies. Br. J. Cancer 2017, 117, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Aspeslagh, S.; Shailubhai, K.; Bahleda, R.; Gazzah, A.; Varga, A.; Hollebecque, A.; Massard, C.; Spreafico, A.; Reni, M.; Soria, J.C. Phase I dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Che, X.; Wang, J.; Zou, G.; Yu, Q.; Zhang, X. CDK1 serves as a novel therapeutic target for endometrioid endometrial cancer. J. Cancer 2021, 12, 2206–2215. [Google Scholar] [CrossRef]
- Voce, D.J.; Bernal, G.M.; Cahill, K.E.; Wu, L.; Mansour, N.; Crawley, C.D.; Campbell, P.S.; Arina, A.; Weichselbaum, R.R.; Yamini, B. CDK1 is up-regulated by temozolomide in an NF-κB dependent manner in glioblastoma. Sci. Rep. 2021, 11, 5665–5675. [Google Scholar] [CrossRef] [PubMed]
- Swadi, R.R.; Sampat, K.; Herrmann, A.; Losty, P.D.; See, V.; Moss, D.J. CDK inhibitors reduce cell proliferation and reverse hypoxia-induced metastasis of neuroblastoma tumours in a chick embryo model. Sci. Rep. 2019, 9, 9136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Wang, J.; Fang, Z.; Pierce, S.R.; West, L.; Staley, A.; Tucker, K.; Yin, Y.; Sun, W.; Kong, W.; et al. Anti-Tumor and Anti-Invasive Effects of ONC201 on Ovarian Cancer Cells and a Transgenic Mouse Model of Serous Ovarian Cancer. Front. Oncol. 2022, 12, 789450. [Google Scholar] [CrossRef]
- Xu, G.; Kong, W.; Fang, Z.; Fan, Y.; Yin, Y.; Sullivan, S.A.; Tran, A.Q.; Clark, L.H.; Sun, W.; Hao, T.; et al. Asparagus officinalis Exhibits Anti-Tumorigenic and Anti-Metastatic Effects in Ovarian Cancer. Front. Oncol. 2021, 11, 688461. [Google Scholar] [CrossRef]
- Han, J.; Wysham, W.Z.; Zhong, Y.; Guo, H.; Zhang, L.; Malloy, K.M.; Dickens, H.K.; Huh, G.; Lee, D.; Makowski, L.; et al. Increased efficacy of metformin corresponds to differential metabolic effects in the ovarian tumors from obese versus lean mice. Oncotarget 2017, 8, 110965–110982. [Google Scholar] [CrossRef]
- Zhang, X.; Li, M.J.; Xia, L.; Zhang, H. The biological function of m6A methyltransferase KIAA1429 and its role in human disease. PeerJ 2022, 10, e14334. [Google Scholar] [CrossRef]
- Xie, R.; Li, B.; Jia, L.; Li, Y. Identification of Core Genes and Pathways in Melanoma Metastasis via Bioinformatics Analysis. Int. J. Mol. Sci. 2022, 23, 794. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, H.; Li, D.; Jiang, C.; Zhao, H.; Teng, Y. Identification of novel biomarkers in breast cancer via integrated bioinformatics analysis and experimental validation. Bioengineered 2021, 12, 12431–12446. [Google Scholar] [CrossRef]
- Yang, W.; Cho, H.; Shin, H.Y.; Chung, J.Y.; Kang, E.S.; Lee, E.J.; Kim, J.H. Accumulation of cytoplasmic Cdk1 is associated with cancer growth and survival rate in epithelial ovarian cancer. Oncotarget 2016, 7, 49481–49497. [Google Scholar] [CrossRef] [Green Version]
- Xi, Q.; Huang, M.; Wang, Y.; Zhong, J.; Liu, R.; Xu, G.; Jiang, L.; Wang, J.; Fang, Z.; Yang, S. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumour Biol. 2015, 36, 4939–4948. [Google Scholar] [CrossRef]
- Huis In’t Veld, P.J.; Wohlgemuth, S.; Koerner, C.; Müller, F.; Janning, P.; Musacchio, A. Reconstitution and use of highly active human CDK1:Cyclin-B:CKS1 complexes. Protein Sci. 2022, 31, 528–537. [Google Scholar] [CrossRef]
- Kojima, K.; Shimanuki, M.; Shikami, M.; Andreeff, M.; Nakakuma, H. Cyclin-dependent kinase 1 inhibitor RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis in AML. Cancer Sci. 2009, 100, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle 2002, 1, 391–393. [Google Scholar] [CrossRef] [Green Version]
- Mitrea, D.M.; Yoon, M.K.; Ou, L.; Kriwacki, R.W. Disorder-function relationships for the cell cycle regulatory proteins p21 and p27. Biol. Chem. 2012, 393, 259–274. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Chang, J.G.; Tien, N.; Chang, Y.C.; Lin, M.L.; Chen, S.S. Oxidative Stress-Induced Unscheduled CDK1-Cyclin B1 Activity Impairs ER-Mitochondria-Mediated Bioenergetic Metabolism. Cells 2021, 10, 1280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.K.; Grant, S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol. Cancer Ther. 2014, 13, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Hu, S.; Wang, W.; Li, Y.; Zhi, W.; Lu, S.; Shi, Q.; Wang, Y.; Yang, Y. Matrine inhibits prostate cancer via activation of the unfolded protein response/endoplasmic reticulum stress signaling and reversal of epithelial to mesenchymal transition. Mol. Med. Rep. 2018, 18, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Du, T.; Lu, Y.; Lv, Y.; Du, Y.; Wu, J.; Ma, R.; Xu, C.; Feng, J. VLX1570 regulates the proliferation and apoptosis of human lung cancer cells through modulating ER stress and the AKT pathway. J. Cell. Mol. Med. 2022, 26, 108–122. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Yue, C.; Du, K.Y.; Salem, M.; O’Brien, J.; Peng, C. The Role of microRNAs in Epithelial Ovarian Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 7093. [Google Scholar] [CrossRef]
- Dhaliwal, D.; Shepherd, T.G. Molecular and cellular mechanisms controlling integrin-mediated cell adhesion and tumor progression in ovarian cancer metastasis: A review. Clin. Exp. Metastasis 2022, 39, 291–301. [Google Scholar] [CrossRef]
- Ren, L.; Yang, Y.; Li, W.; Zheng, X.; Liu, J.; Li, S.; Yang, H.; Zhang, Y.; Ge, B.; Zhang, S.; et al. CDK1 serves as a therapeutic target of adrenocortical carcinoma via regulating epithelial-mesenchymal transition, G2/M phase transition, and PANoptosis. J. Transl. Med. 2022, 20, 444. [Google Scholar] [CrossRef]
- Chen, H.; Hu, K.; Xie, Y.; Qi, Y.; Li, W.; He, Y.; Fan, S.; Liu, W.; Li, C. CDK1 Promotes Epithelial-Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation. Int. J. Mol. Sci. 2022, 23, 7385. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, W.; Li, Q.H.; Xie, B.M.; Shen, F.; Du, Y.P.; Zong, Z.H.; Wang, L.L.; Wei, X.Q.; Zhao, Y. Circ-NOLC1 promotes epithelial ovarian cancer tumorigenesis and progression by binding ESRP1 and modulating CDK1 and RhoA expression. Cell Death Discov. 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef] [PubMed]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301. [Google Scholar] [CrossRef]
- Garziera, M.; Montico, M.; Bidoli, E.; Scalone, S.; Sorio, R.; Giorda, G.; Lucia, E.; Toffoli, G. Prognostic Role of Serum Antibody Immunity to p53 Oncogenic Protein in Ovarian Cancer: A Systematic Review and a Meta-Analysis. PLoS ONE 2015, 10, e0140351. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, K.; No, J.H.; Jeon, Y.T.; Jeon, H.W.; Kim, Y.B. Prognostic value of biomarkers related to drug resistance in patients with advanced epithelial ovarian cancer. Anticancer. Res. 2012, 32, 589–594. [Google Scholar] [CrossRef]
- Sylvia, M.T.; Kumar, S.; Dasari, P. The expression of immunohistochemical markers estrogen receptor, progesterone receptor, Her-2-neu, p53 and Ki-67 in epithelial ovarian tumors and its correlation with clinicopathologic variables. Indian J. Pathol. Microbiol. 2012, 55, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Zhou, C.; Zhong, Y.; Kuan, P.F.; Fan, C.; Sampey, B.P.; Difurio, M.; Bae-Jump, V.L. Obesity increases tumor aggressiveness in a genetically engineered mouse model of serous ovarian cancer. Gynecol. Oncol. 2014, 133, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benot-Dominguez, R.; Cimini, A.; Barone, D.; Giordano, A.; Pentimalli, F. The Emerging Role of Cyclin-Dependent Kinase Inhibitors in Treating Diet-Induced Obesity: New Opportunities for Breast and Ovarian Cancers? Cancers 2022, 14, 2709. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Number of Patients | CDK1 Expression | p Value | X2 | ||
|---|---|---|---|---|---|---|
| Low (≤3) | High (>3) | |||||
| Age | ≥50 | 67 | 19 | 48 | <0.05 | 3.933 |
| <50 | 80 | 11 | 69 | |||
| Pathological Type | Mucinous | 25 | 8 | 17 | 0.32 | 3.477 |
| Endometrioid | 24 | 5 | 19 | |||
| Clear Cell | 26 | 6 | 20 | |||
| Serous | 72 | 11 | 61 | |||
| FIGO Stage | I | 25 | 12 | 13 | <0.01 | 51.346 |
| II | 34 | 4 | 30 | |||
| III | 68 | 1 | 67 | |||
| IV | 20 | 13 | 7 | |||
| BMI | ≤24 | 91 | 14 | 77 | >0.05 | 2.943 |
| >24 | 56 | 16 | 40 | |||
| Metastasis | Yes | 88 | 14 | 74 | >0.05 | 2.085 |
| No | 59 | 16 | 43 | |||
| p53 Expression | Low (≤3) | 53 | 20 | 33 | <0.01 | 13.698 |
| High (>3) | 94 | 10 | 84 | |||
| Ki67 Expression | Low (≤3) | 53 | 20 | 33 | <0.01 | 26.656 |
| High (>3) | 94 | 10 | 84 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Fan, Y.; Zhao, Z.; Zhang, X.; Tucker, K.; Staley, A.; Suo, H.; Sun, W.; Shen, X.; Deng, B.; et al. Inhibition of CDK1 by RO-3306 Exhibits Anti-Tumorigenic Effects in Ovarian Cancer Cells and a Transgenic Mouse Model of Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 12375. https://doi.org/10.3390/ijms241512375
Huang Y, Fan Y, Zhao Z, Zhang X, Tucker K, Staley A, Suo H, Sun W, Shen X, Deng B, et al. Inhibition of CDK1 by RO-3306 Exhibits Anti-Tumorigenic Effects in Ovarian Cancer Cells and a Transgenic Mouse Model of Ovarian Cancer. International Journal of Molecular Sciences. 2023; 24(15):12375. https://doi.org/10.3390/ijms241512375
Chicago/Turabian StyleHuang, Yu, Yali Fan, Ziyi Zhao, Xin Zhang, Katherine Tucker, Allison Staley, Hongyan Suo, Wenchuan Sun, Xiaochang Shen, Boer Deng, and et al. 2023. "Inhibition of CDK1 by RO-3306 Exhibits Anti-Tumorigenic Effects in Ovarian Cancer Cells and a Transgenic Mouse Model of Ovarian Cancer" International Journal of Molecular Sciences 24, no. 15: 12375. https://doi.org/10.3390/ijms241512375