
Quantifying Parameter Interdependence in Stochastic Discrete Models of Biochemical Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Background
2.1.1. Chemical Master Equation
Gillespie’s Algorithm
- Initialize the time and the state of the system, .
- While
- Calculate each propensity for and the sum
- Sample two uniform random variables over , to obtain , .
- Evaluate the time and the index j of the next occurring reaction, according to
- (a)
- (b)
- the smallest integer fulfilling
- Update the state and the time .
- End while.
2.1.2. Chemical Langevin Equation
2.1.3. Reaction Rate Equation
2.2. Parametric Correlations
2.2.1. Parametric Sensitivity for the Chemical Master Equation
2.2.2. Common Random Number
2.2.3. Common Reaction Path
2.2.4. Coupled Finite-Difference
2.2.5. Parametric Sensitivity for the Chemical Langevin Equations
2.2.6. Parametric Sensitivity for the Reaction Rate Equations
2.3. Practical Identifiability Analysis
2.3.1. Sensitivity-Based Identifiability Analysis
2.3.2. Parameter Collinearity
2.3.3. Method for Selecting Subsets of Identifiable Parameters
| Algorithm 1 Computing the Normalized Sensitivity Matrix |
| Algorithm 2 Selecting a Subset of Identifiable Parameters |
|
3. Results
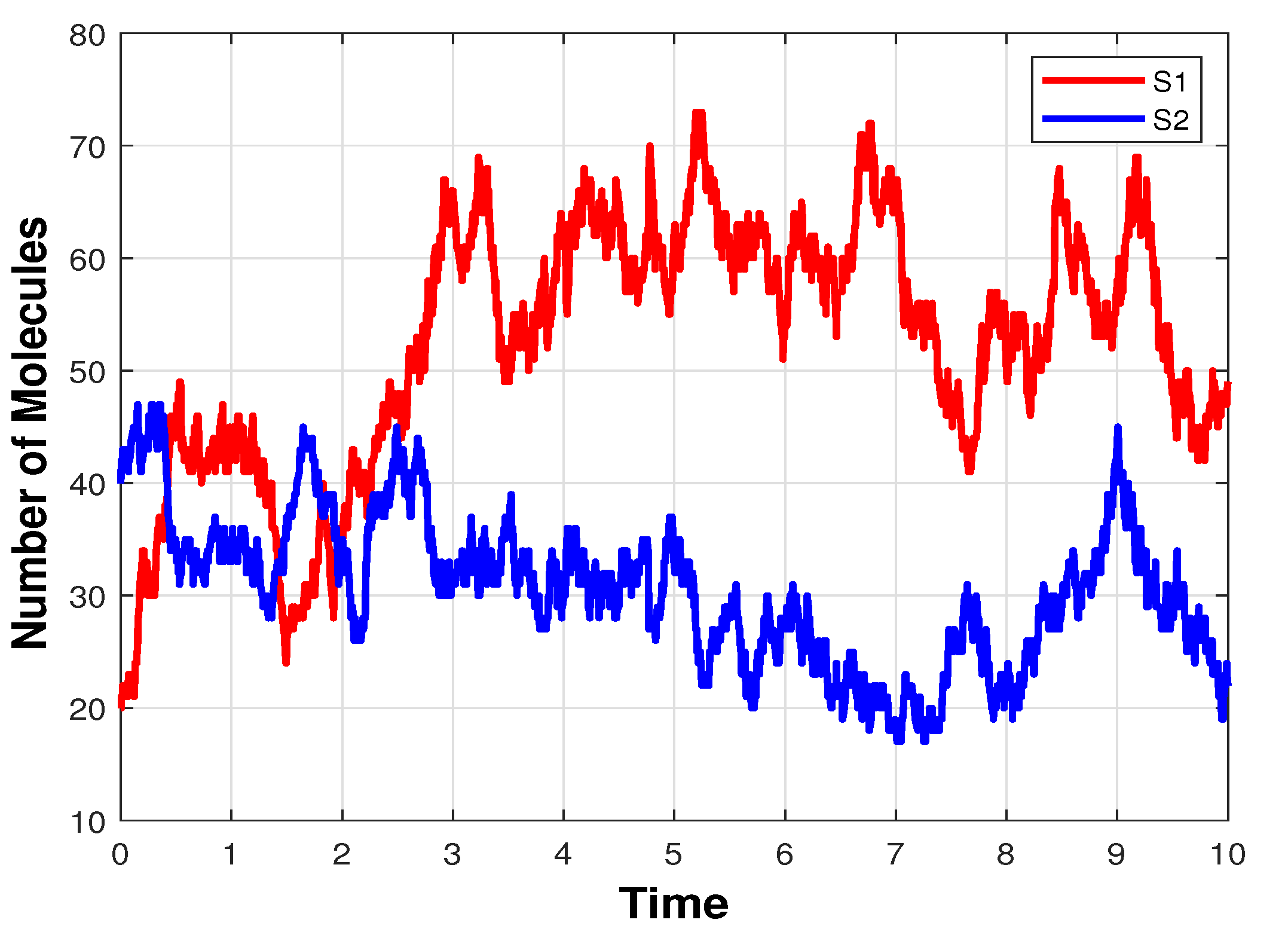
3.1. Infectious Disease Model
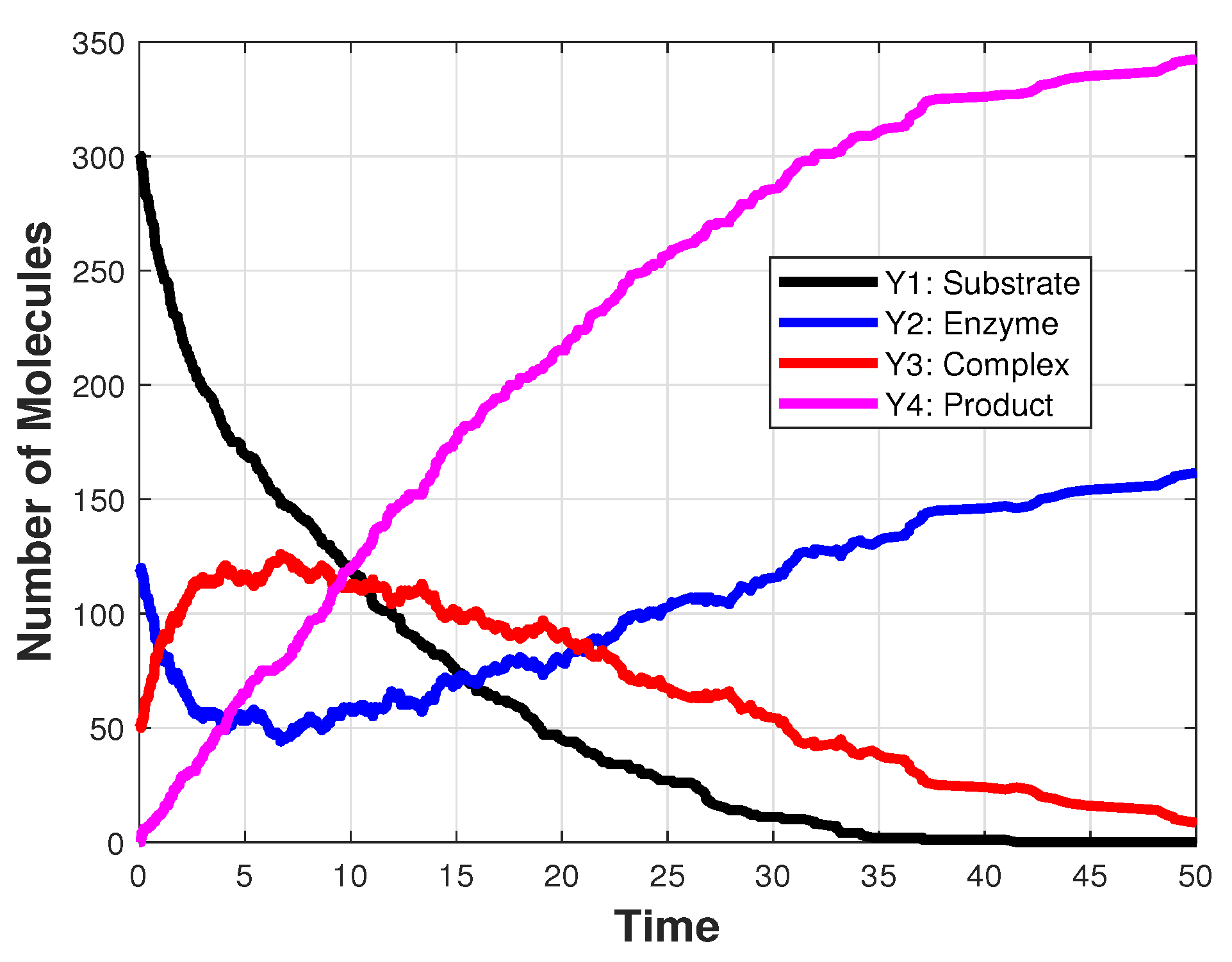
3.2. Michaelis–Menten Model
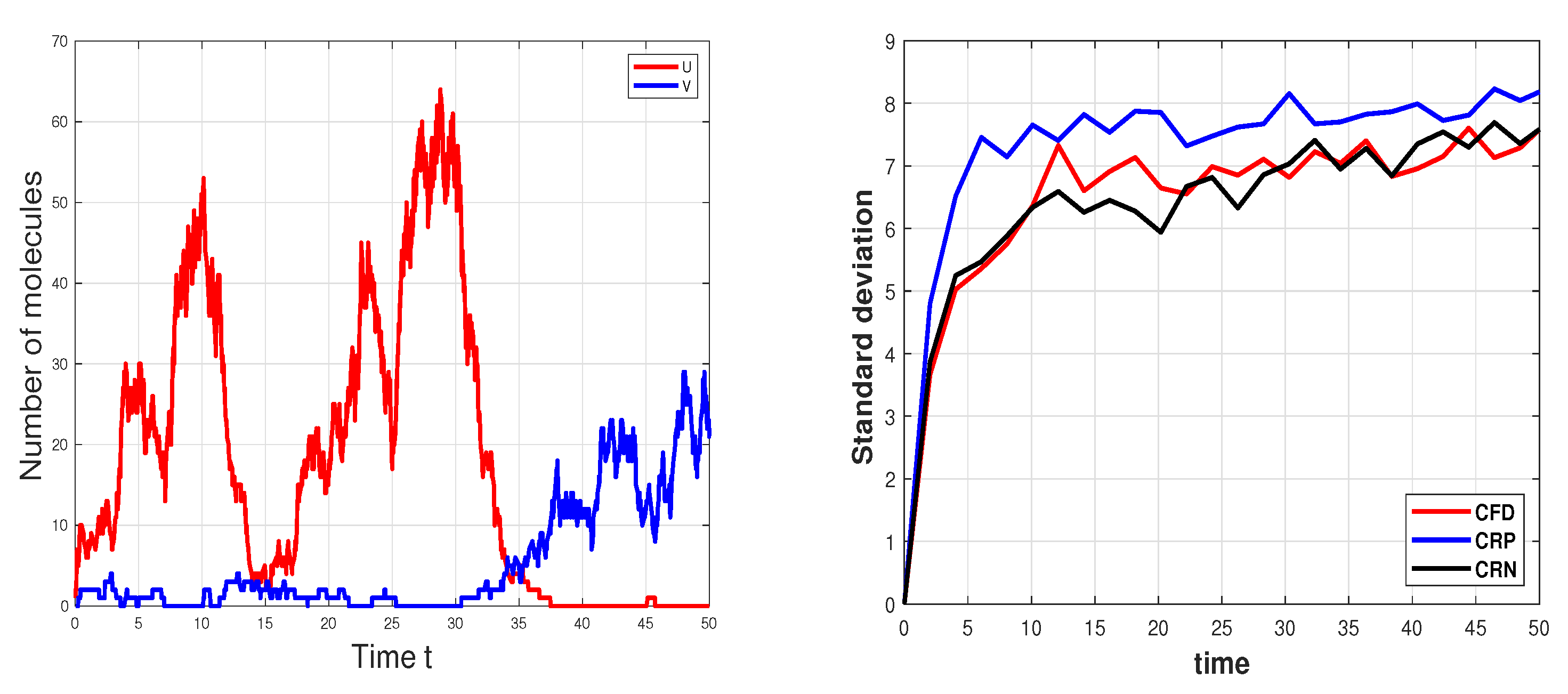
3.3. Genetic Toggle Switch Model
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Parameter | of CFD Sensitivity | of CRP Sensitivity | of CRN Sensitivity | of RRE Sensitivity |
|---|---|---|---|---|
| 1.03 | 1.04 | 1.04 | 1.07 | |
| 0.002 | 0.005 | 0.003 | 0.002 | |
| 1.22 | 1.23 | 1.23 | 1.29 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 3.43 | 2.18 | 2.59 | 4.85 | |
| 2.21 | 2.13 | 2.13 | 2.17 | |
| 1.67 | 1.48 | 1.49 | 1.87 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 4.08 | 2.78 | 3.4 | 5.3 |
References
- Kitano, H. Computational systems biology. Nature 2002, 420, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Maheshri, N.; O’Shea, E.K. Living with noisy genes: How cells function reliably with inherent variability in gene expression. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 413–434. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozbudak, E.M.; Thattai, M.; Kurtser, I.; Grossman, A.D.; van Oudenaarden, A. Regulation of noise in the expression of a single gene. Nat. Genet. 2002, 31, 69–73. [Google Scholar] [CrossRef]
- Raser, J.M.; O’Shea, E.K. Noise in gene expression: Origins, consequences, and control. Science 2005, 309, 2010–2013. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, D.T. A rigorous derivation of the chemical master equation. Stat. Mech. Its Appl. 1992, 188, 404–425. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Stochastic approach to chemical kinetics. J. Appl. Probab. 1967, 4, 413. [Google Scholar] [CrossRef]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Ethier, S.N.; Kurtz, T.G. Markov Processes: Characterization and Convergence; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Rathinam, M.; Sheppard, P.W.; Khammash, M. Efficient computation of parameter sensitivities of discrete stochastic chemical reaction networks. J. Chem. Phys. 2010, 132, 034103–034116. [Google Scholar] [CrossRef]
- El Samad, H.; Khammash, M.; Petzold, L.; Gillespie, D.T. Stochastic modelling of gene regulatory networks. Int. J. Robust Nonlinear Control 2005, 15, 691–711. [Google Scholar] [CrossRef]
- Strehl, R.; Ilie, S. Hybrid stochastic simulation of reaction-diffusion systems with slow and fast dynamics. J. Chem. Phys. 2015, 143, 234108. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.J. Stochastic Modelling for Systems Biology; Taylor & Francis: Boca Raton, FL, USA, 2019. [Google Scholar]
- Thanh, V.H.; Zunino, R.; Priami, C. On the rejection-based algorithm for simulation and analysis of large-scale reaction networks. J. Chem. Phys. 2015, 142, 244106. [Google Scholar] [CrossRef] [Green Version]
- Barrows, D.; Ilie, S. Parameter estimation for the reaction-diffusion master equation. AIP Adv. 2023, 13, 065318. [Google Scholar] [CrossRef]
- Petre, I.; Mizera, A.; Hyder, C.L.; Meinander, A.; Mikhailov, A.; Morimoto, R.I.; Sistonen, L.; Eriksson, J.E.; Back, R.J. A simple mass-action model for the eukaryotic heat shock response and its mathematical validation. Nat. Comput. 2011, 10, 595–612. [Google Scholar] [CrossRef]
- Vajda, S.; Rabitz, H.; Walter, E.; Lecourtier, Y. Qualitative and quantitative identifiability analysis of nonlinear chemical kinetic models. Chem. Eng. Commun. 1989, 83, 191–219. [Google Scholar] [CrossRef]
- Brun, R.; Reichert, P.; Künsch, H.R. Practical identifiability analysis of large environmental simulation models. Water Resour. Res. 2001, 37, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Brun, R.; Kühni, M.; Siegrist, H.; Gujer, W.; Reichert, P. Practical identifiability of ASM2d parameters—Systematic selection and tuning of parameter subsets. Water Res. 2002, 36, 4113–4127. [Google Scholar] [CrossRef] [PubMed]
- Chis, O.T.; Banga, J.R.; Balsa-Canto, E. Structural identifiability of systems biology models: A critical comparison of methods. PLoS ONE 2011, 6, e27755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmberg, A. On the practical identifiability of microbial growth models incorporating Michaelis-Menten type nonlinearities. Math. Biosci. 1982, 62, 23–43. [Google Scholar] [CrossRef]
- Jacquez, J.; Greif, P. Numerical parameter identifiability and estimability: Integrating identifiability, estimability, and optimal sampling design. Math. Biosci. 1985, 77, 201–227. [Google Scholar] [CrossRef] [Green Version]
- Komorowski, M.; Costa, M.J.; Rand, D.A.; Stumpf, M.P.H. Sensitivity, robustness, and identifiability in stochastic chemical kinetics models. Proc. Natl. Acad. Sci. USA 2011, 108, 8645–8650. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fernandez, M.; Egea, J.A.; Banga, J.R. Novel metaheuristic for parameter estimation in nonlinear dynamic biological systems. BMC Bioinform. 2006, 7, 483. [Google Scholar] [CrossRef] [Green Version]
- Villaverde, A.F. Observability and structural identifiability of nonlinear biological systems. Complexity 2019, 2019, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.F. An efficient finite-difference method for parameter sensitivities of continuous time Markov chains. SIAM J. Num. Anal. 2012, 50, 2237–2258. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Anderson, D.F.; Rawlings, J.B. Comparison of finite difference based methods to obtain sensitivities of stochastic chemical kinetic models. J. Chem. Phys. 2013, 138, 074110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morshed, M. Efficient Finite-Difference Methods for Sensitivity Analysis of Stiff Stochastic Discrete Models of Biochemical Systems. Ph.D. Thesis, University of Waterloo, Waterloo, ON, Canada, 2017. [Google Scholar]
- Gábor, A.; Villaverde, A.F.; Banga, J.R. Parameter identifiability analysis and visualization in large-scale kinetic models of biosystems. BMC Syst. Biol. 2017, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.E.; Schnell, S.; Burrage, K. Stochastic approaches for modelling in vivo reactions. Comput. Biol. Chem. 2004, 28, 165–178. [Google Scholar] [CrossRef]
- Gillespie, D.T. The chemical Langevin equations. J. Phys. Chem. 2000, 113, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Ilie, S.; Gholami, S. Simplifying stochastic mathematical models of biochemical systems. Appl. Math. 2013, 4, 248–256. [Google Scholar] [CrossRef]
- Glasserman, P. Monte Carlo Methods in Financial Engineering; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Ilie, S. Variable time-stepping in the pathwise numerical solution of the chemical Langevin equation. J. Phys. Chem. 2012, 137, 234110. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulos, V.; Kaznessis, Y.N. An adaptive time step scheme for a system of stochastic differential equations with multiple multiplicative noise: Chemical Langevin equation, a proof of concept. J. Chem. Phys. 2008, 128, 014103. [Google Scholar] [CrossRef] [PubMed]
- Corless, R.M.; Fillion, N. An Introduction to Numerical Methods from the Point of View of Backward Error Analysis; Springer: New York, NY, USA, 2013. [Google Scholar]
- Golub, G.; Van Loan, C. Matrix Computations, 3rd ed.; The Johns Hopkins University Press: London, UK, 1996. [Google Scholar]
- Weijers, S.R.; Vanrolleghem, P.A. A procedure for selecting best identifiable parameters in calibrating activated sludge model no. 1 to full-scale plant data. Water Sci. Technol. 1997, 36, 69–79. [Google Scholar] [CrossRef]
- Jahnke, T. On reduced models for chemical master equation. Multiscale Model. Simul. 2011, 9, 1646–1676. [Google Scholar] [CrossRef] [Green Version]



| Reaction Channel | Rate Parameter Value | |
|---|---|---|
| : | ||
| : | ||
| : | ||
| : | ||
| : |
| Parameter | of CFD Sensitivity | of CRP Sensitivity | of CRN Sensitivity | of RRE Sensitivity | Path-Wise Sensitivity |
|---|---|---|---|---|---|
| 0.97 | 0.96 | 0.94 | 0.97 | 0.98 | |
| 0.02 | 0.02 | 0.1 | 0.02 | 0.02 | |
| 0.26 | 0.29 | 0.26 | 0.26 | 0.26 | |
| 0.55 | 0.66 | 0.54 | 0.55 | 0.55 | |
| 0.68 | 0.69 | 0.67 | 0.71 | 0.71 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity | Collinearity Index of Path-Wise Sensitivity |
|---|---|---|---|---|---|
| 1.18 | 1.13 | 1.18 | 1.2 | 1.19 | |
| 1.93 | 1.17 | 1.94 | 1.92 | 1.95 | |
| 1.339 | 1.25 | 1.32 | 1.32 | 1.31 | |
| 1.103 | 1.15 | 1.13 | 1.18 | 1.17 | |
| 4.69 | 2.37 | 1.16 | 9.77 | 9.96 | |
| 1.43 | 1.27 | 1.02 | 1.34 | 1.33 | |
| 1.86 | 1.89 | 1.9 | 1.85 | 1.86 | |
| 1.35 | 1.28 | 1.33 | 1.34 | 1.34 | |
| 10.816 | 3.04 | 7.2 | 11.34 | 11.22 | |
| 1.466 | 1.31 | 1.00 | 1.36 | 1.35 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity | Collinearity Index of Path-Wise Sensitivity |
|---|---|---|---|---|---|
| 21.19 | 2.63 | 9.6 | 21.3 | 21.77 | |
| 5.0444 | 2.38 | 1.2 | 9.97 | 10.15 | |
| 7.7768 | 2.91 | 2.01 | 10.48 | 10.51 | |
| 4.88 | 2.38 | 1.43 | 9.83 | 10.01 | |
| 9.92 | 3.65 | 9.4 | 10.83 | 10.98 | |
| 11.07 | 3.12 | 7.2 | 11.68 | 11.73 | |
| 10.87 | 3.05 | 7.2 | 11.46 | 11.45 | |
| 7.44 | 4.8 | 2 | 7.87 | 7.95 | |
| 4.95 | 2.38 | 1.43 | 9.82 | 10.01 | |
| 11.02 | 3.06 | 7.3 | 11.45 | 11.44 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity | Collinearity Index of Path-Wise Sensitivity |
|---|---|---|---|---|---|
| 11.509 | 3.06 | 7.3 | 11.53 | 11.49 | |
| 11.092 | 4.88 | 7.3 | 13.65 | 13.53 | |
| 10.2347 | 4.94 | 9.4 | 13.82 | 14.20 | |
| 22.6313 | 3.87 | 10.54 | 22.19 | 22.49 | |
| 21.4369 | 2.91 | 9.6 | 25.71 | 27.77 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity | Collinearity Index of Path-Wise Sensitivity |
|---|---|---|---|---|---|
| 22.65 | 5.01 | 10.54 | 26.17 | 28.09 | |
| singular values | 16.31, 9.48, | 16.27, 10.35, | 15.86, 9.28, | 36.73, 21.86, | 37.03, 21.76, |
| 1.06, 0.21, 0.06 | 2.98, 1.79, 0.14 | 1.31, 1.1, 0.52 | 2.21, 0.48, 0.09 | 2.19, 0.48, 0.09 |
| Reaction Channel | Rate Parameter Value | |
|---|---|---|
| : | ||
| : | ||
| : |
| Parameter | of CFD Sensitivity | of CRP Sensitivity | of CRN Sensitivity | of RRE Sensitivity |
|---|---|---|---|---|
| 1.11 | 1.1 | 1.07 | 1.07 | |
| 0.002 | 0.01 | 0.003 | 0.002 | |
| 1.31 | 1.30 | 1.29 | 1.29 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 2.9 | 1.35 | 1.47 | 4.85 | |
| 2.21 | 2.17 | 2.17 | 2.17 | |
| 1.56 | 1.21 | 1.2 | 1.87 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 3.92 | 2.25 | 2.43 | 5.3 |
| Reaction Channel | Propensity Function | |
|---|---|---|
| : | ||
| : | ||
| : | ||
| : |
| Parameter | of CFD Sensitivity | of RRE Sensitivity |
|---|---|---|
| 2.22 | 0.89 | |
| 0.6762 | 0 | |
| 4.21 | 0.31 | |
| 4.3 | 0 |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 1 | 1.01 | 1.72 | 2.22 | |
| 1.32 | 1.08 | 1.12 | * | |
| 1.27 | 1.17 | 1.07 | * | |
| 1.01 | 1.1 | 1.56 | * | |
| 1.00 | 1.35 | 2.13 | * | |
| 1.19 | 1.25 | 1.1 | * |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 1.38 | 1.37 | 2.46 | * | |
| 1.42 | 1.25 | 1.80 | * | |
| 1.01 | 1.38 | 2.18 | * | |
| 1.52 | 1.19 | 1.73 | * |
| Parameter Subset | Collinearity Index of CFD Sensitivity | Collinearity Index of CRP Sensitivity | Collinearity Index of CRN Sensitivity | Collinearity Index of RRE Sensitivity |
|---|---|---|---|---|
| 1.64 | 1.39 | 2.45 | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gholami, S.; Ilie, S. Quantifying Parameter Interdependence in Stochastic Discrete Models of Biochemical Systems. Entropy 2023, 25, 1168. https://doi.org/10.3390/e25081168
Gholami S, Ilie S. Quantifying Parameter Interdependence in Stochastic Discrete Models of Biochemical Systems. Entropy. 2023; 25(8):1168. https://doi.org/10.3390/e25081168
Chicago/Turabian StyleGholami, Samaneh, and Silvana Ilie. 2023. "Quantifying Parameter Interdependence in Stochastic Discrete Models of Biochemical Systems" Entropy 25, no. 8: 1168. https://doi.org/10.3390/e25081168