Dear Colleagues,
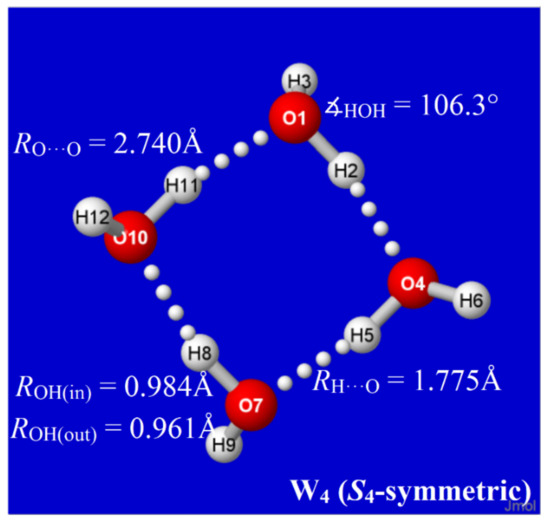
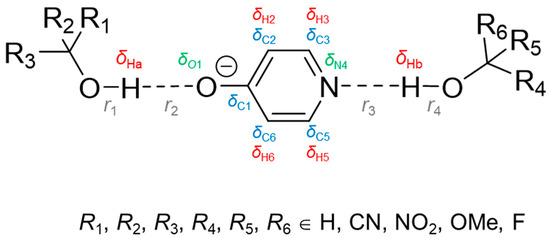
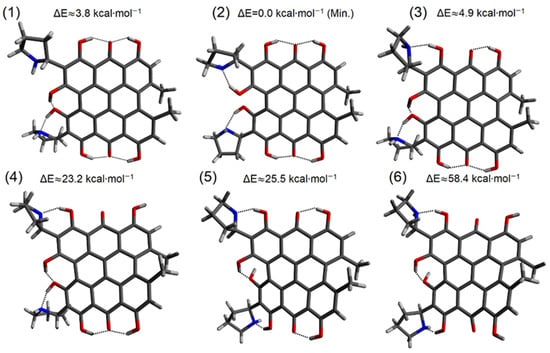
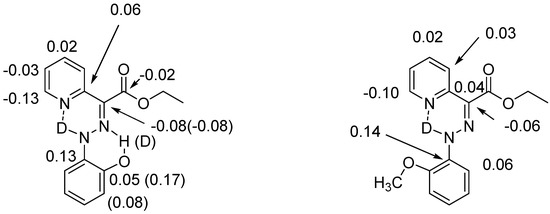
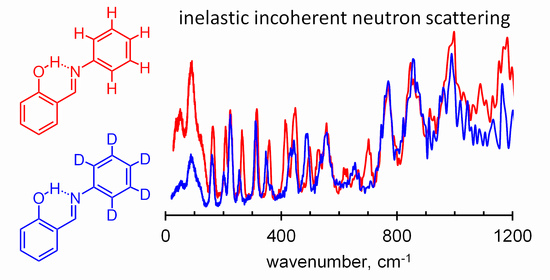
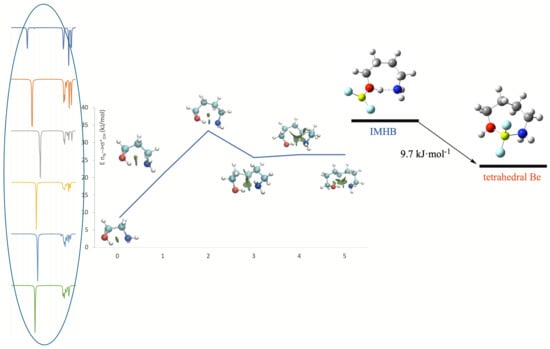
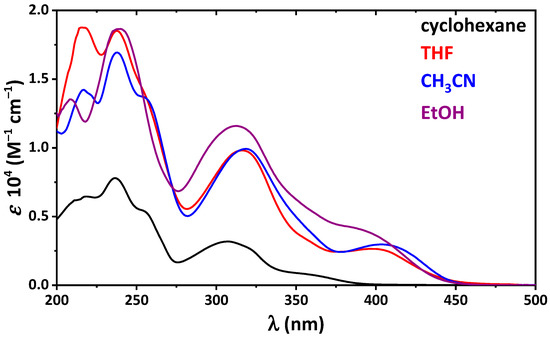
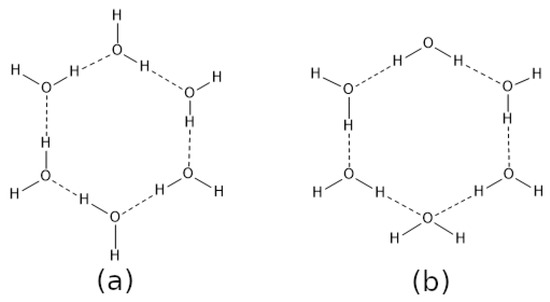
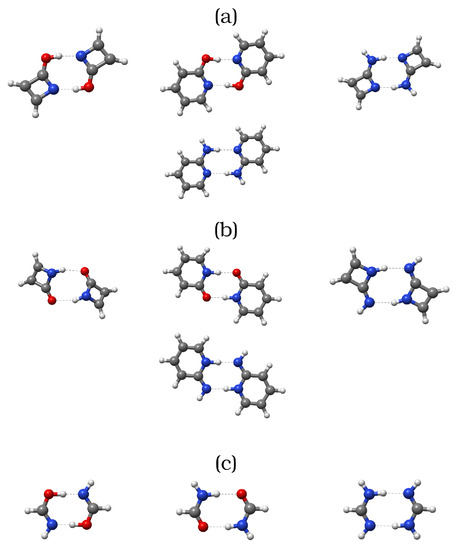
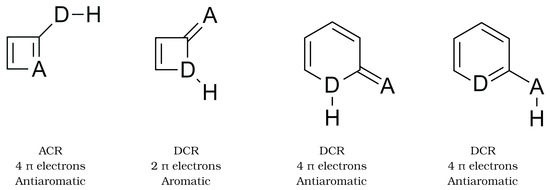
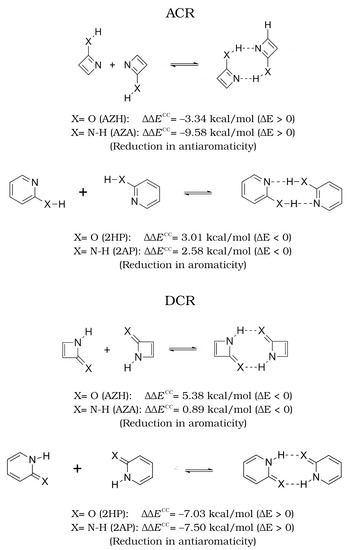
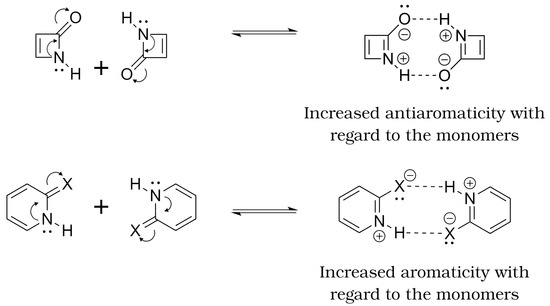
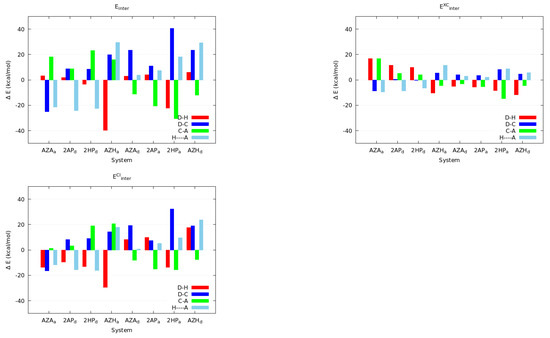
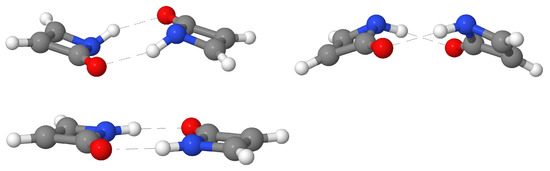
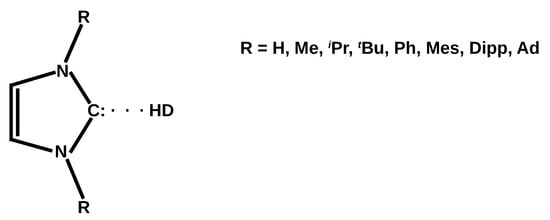
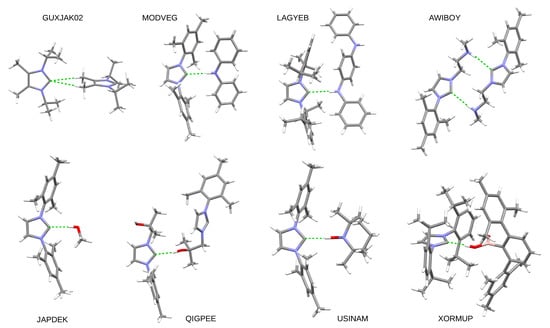
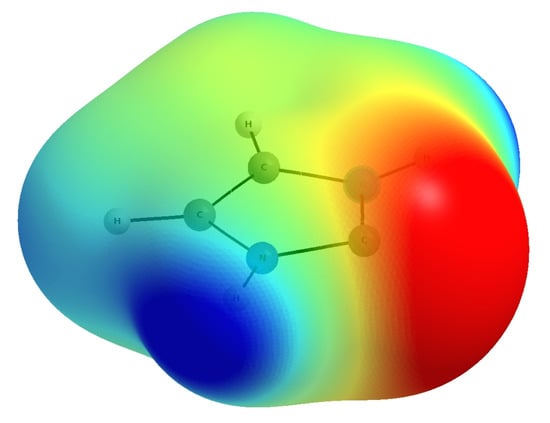
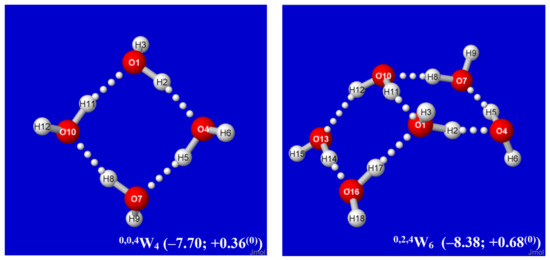
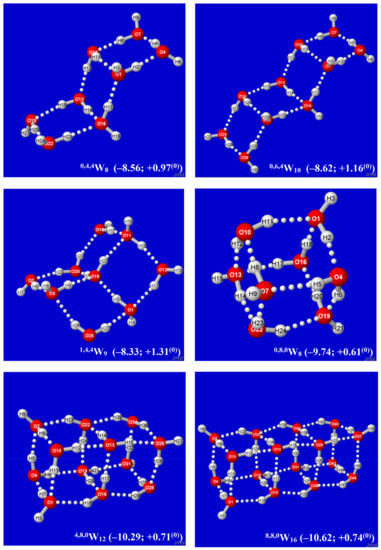
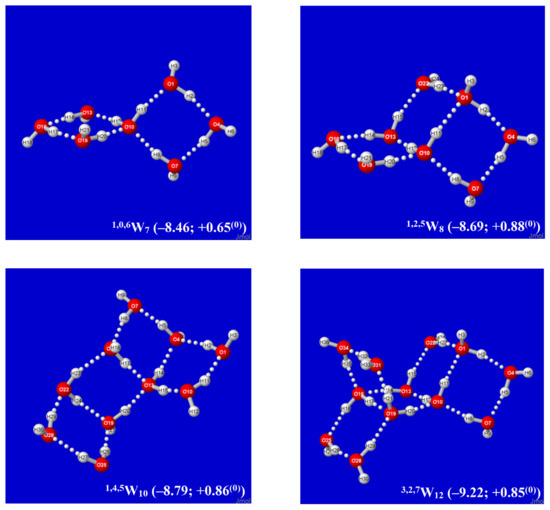
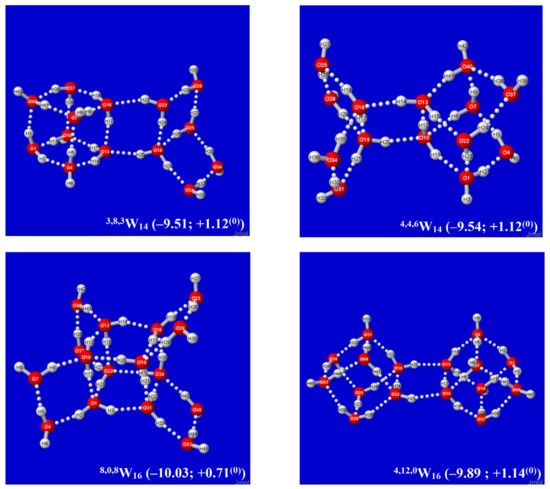
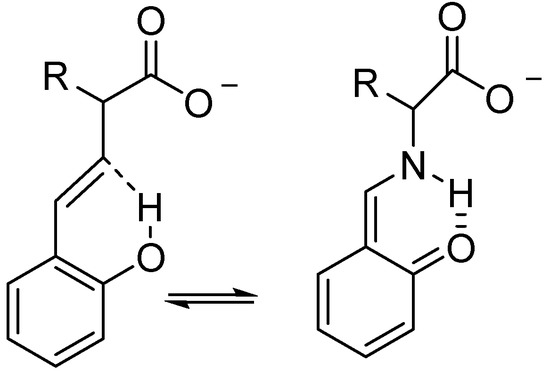
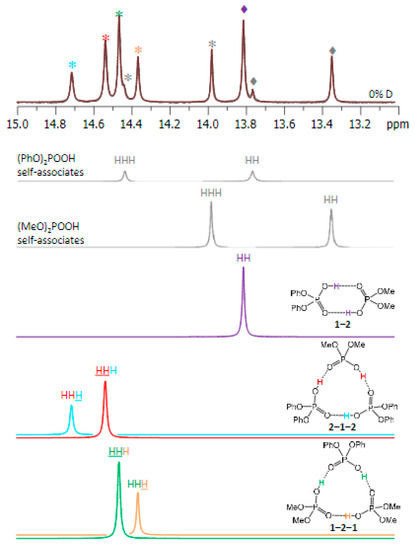
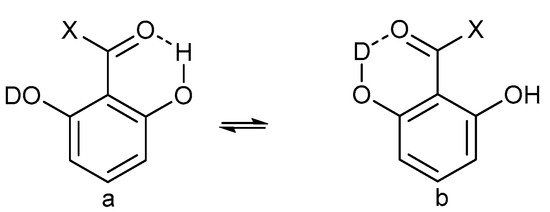
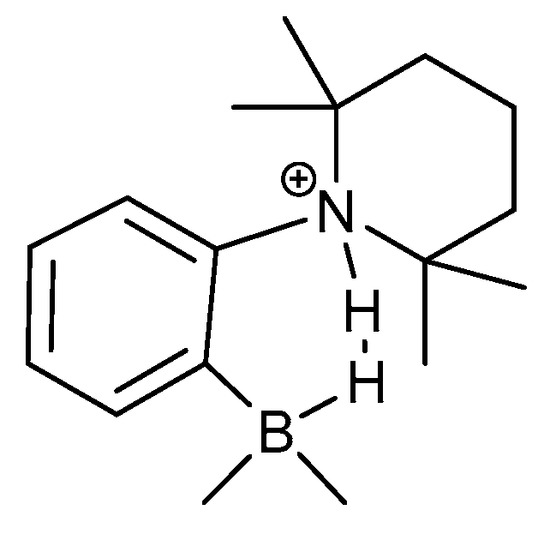
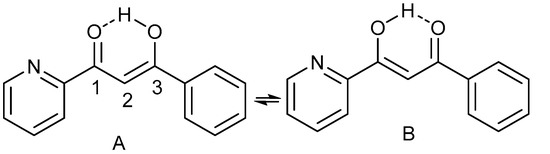
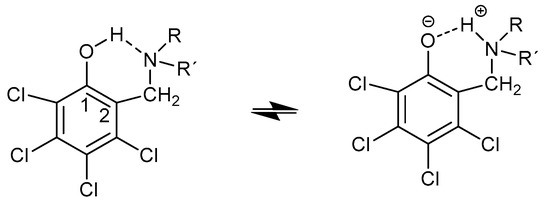
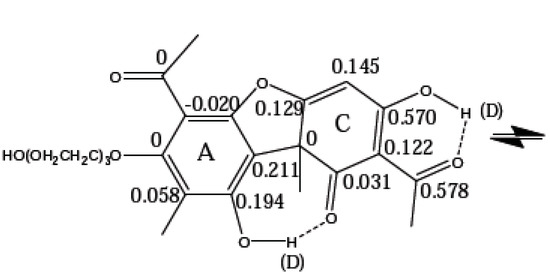
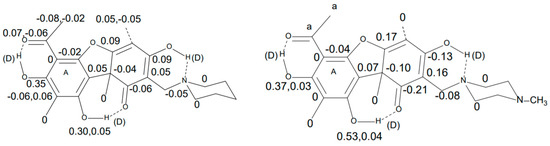
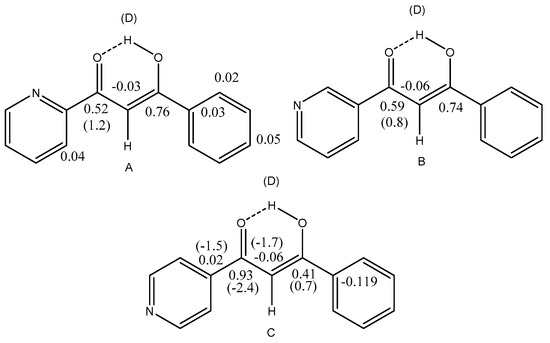
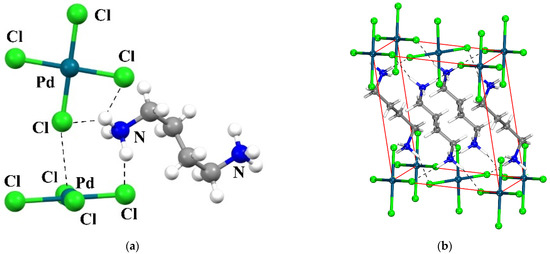
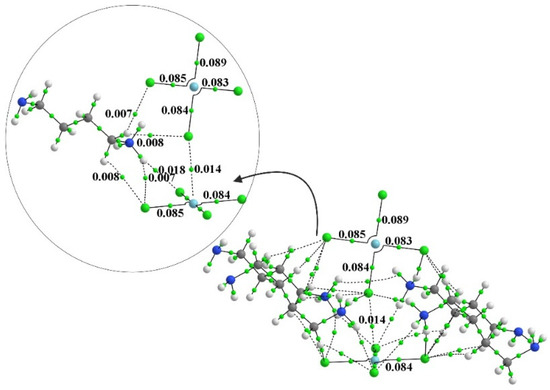
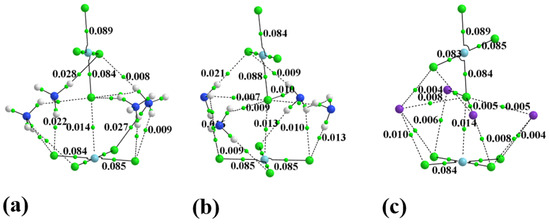
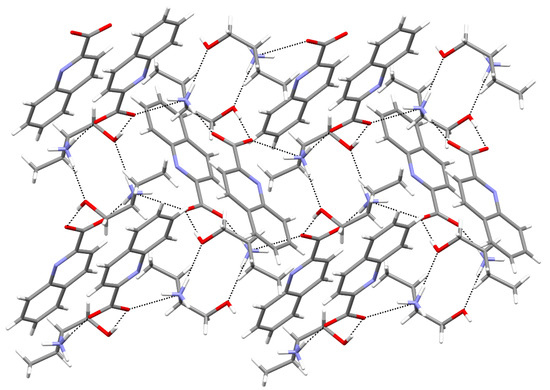
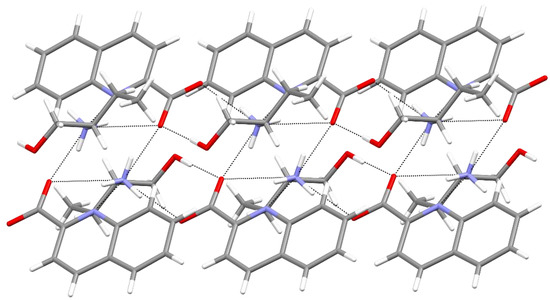
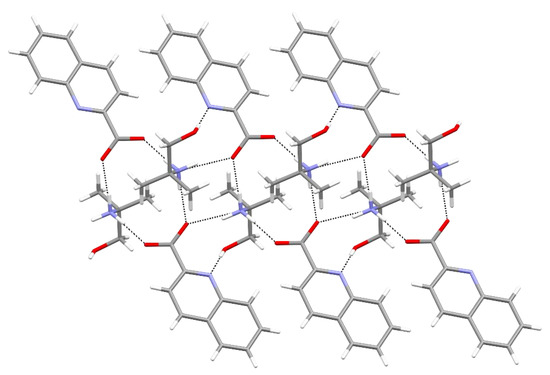
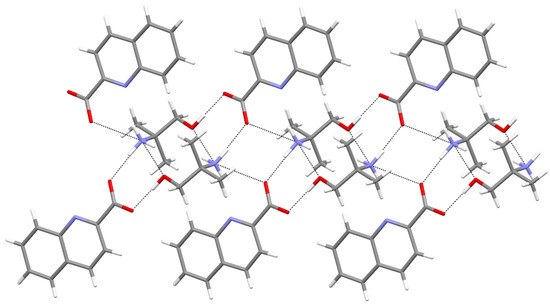
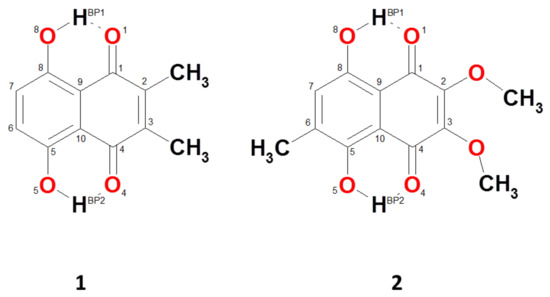
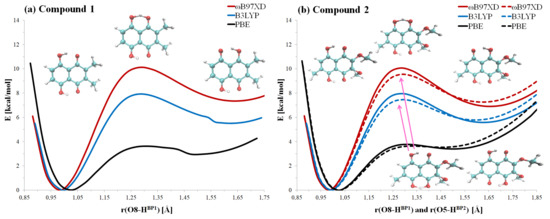
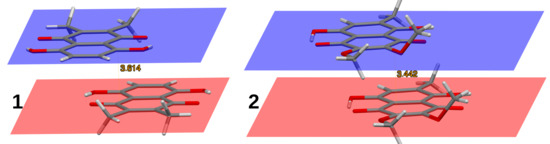
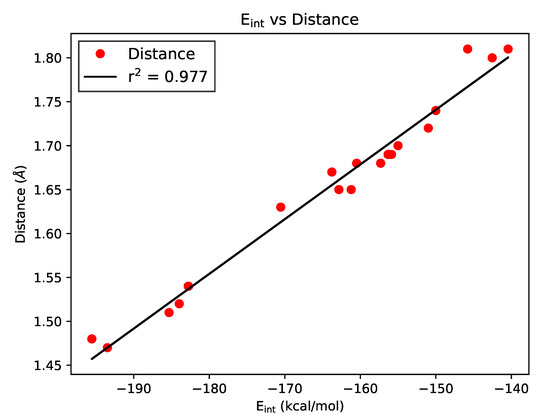
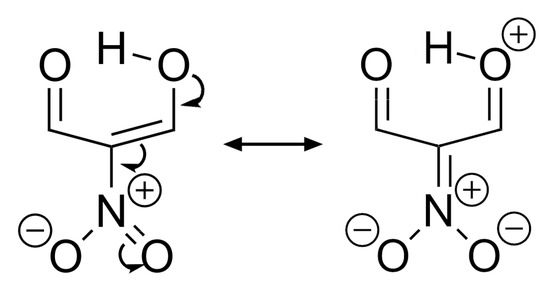
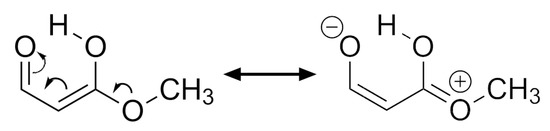
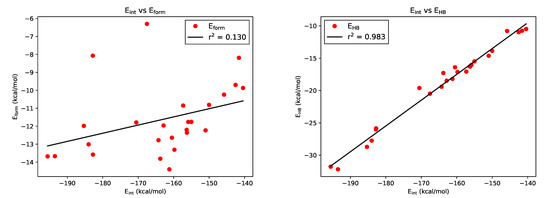
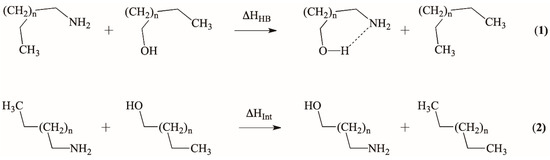
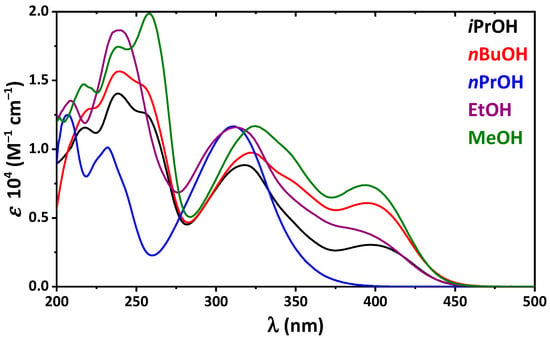
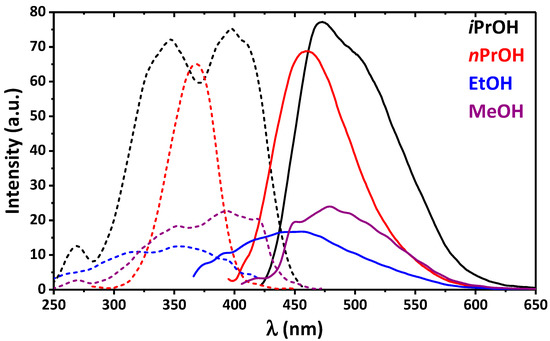
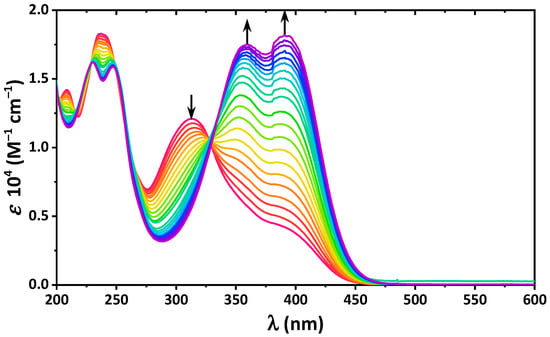
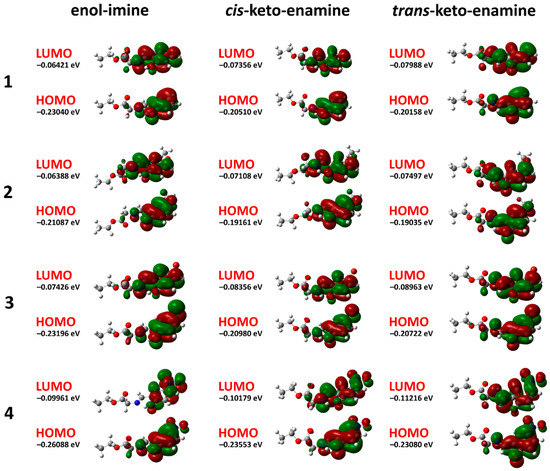
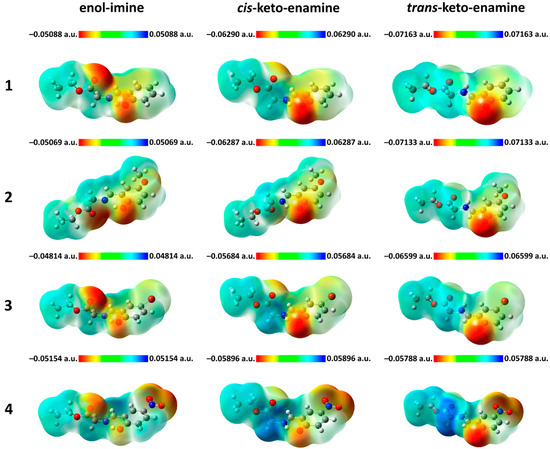
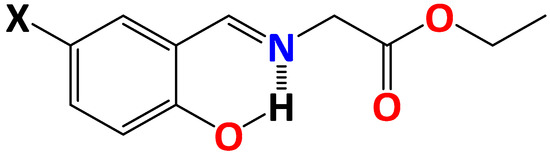
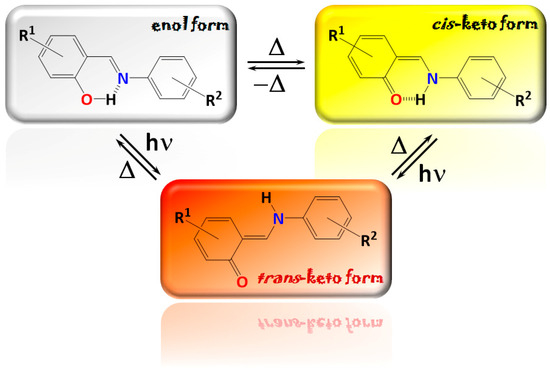
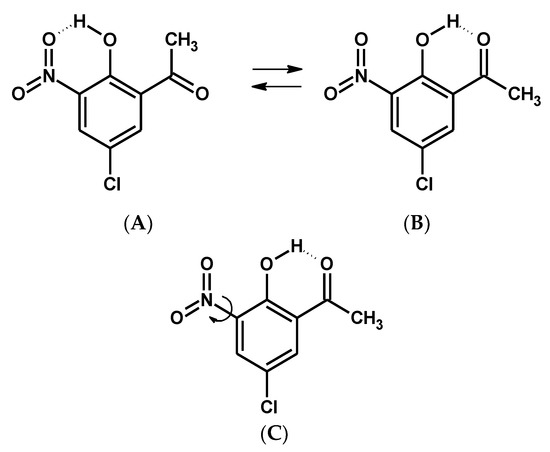
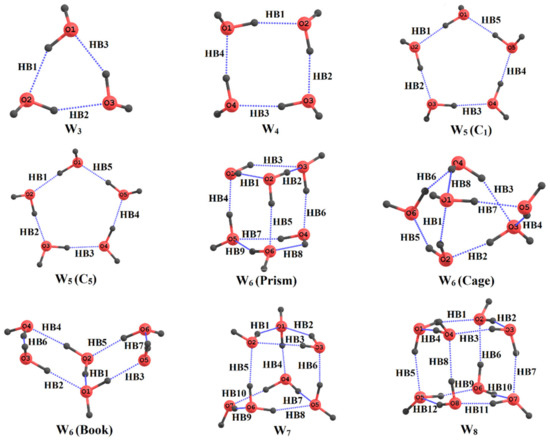
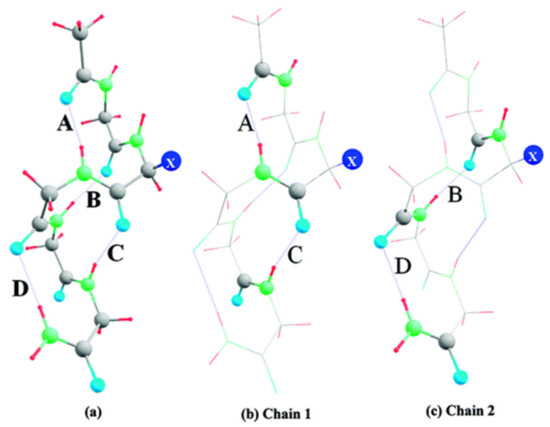
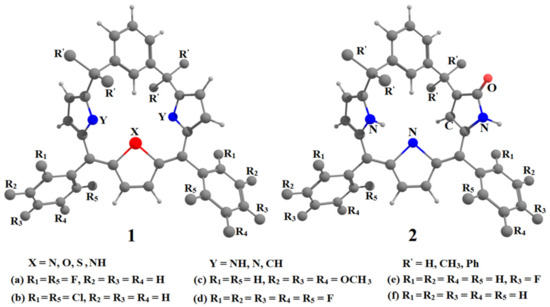
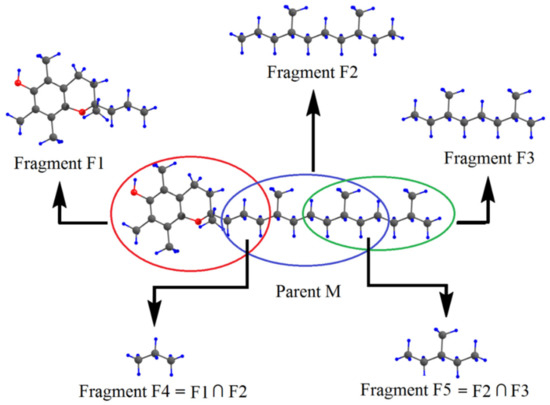
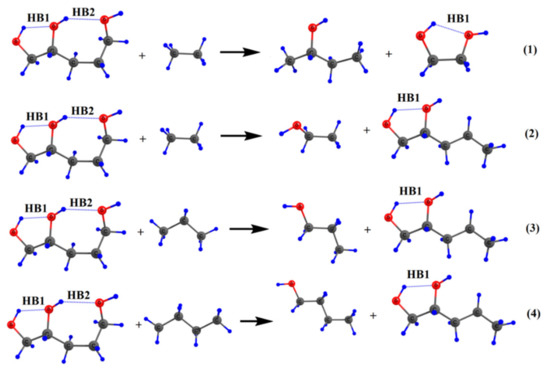
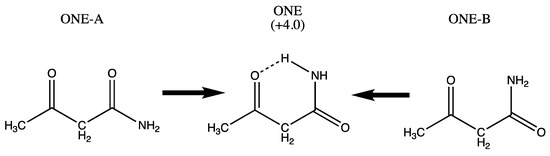
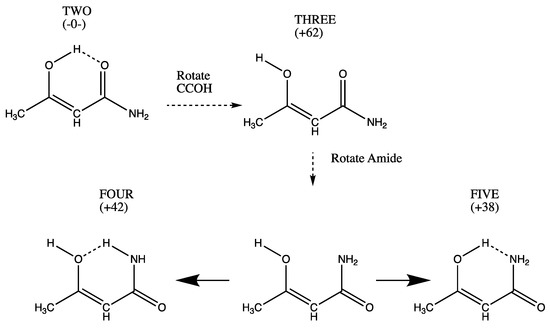
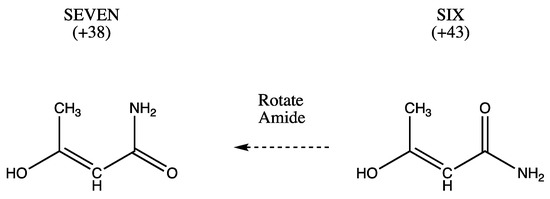
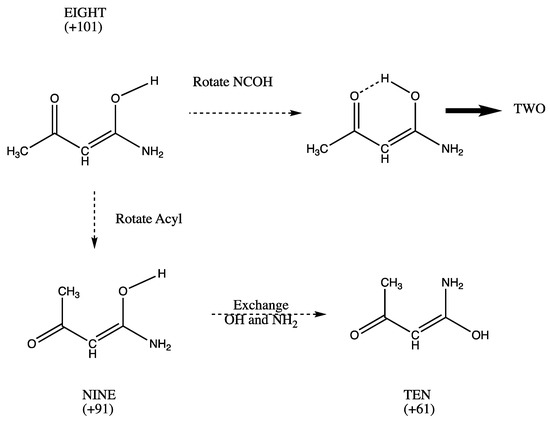
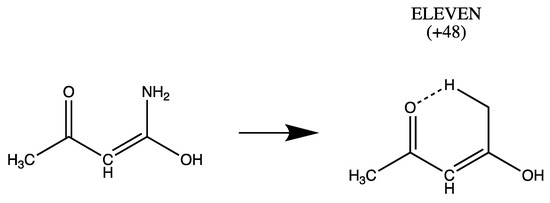
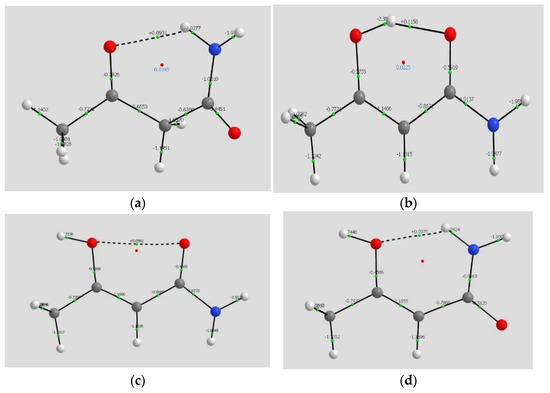
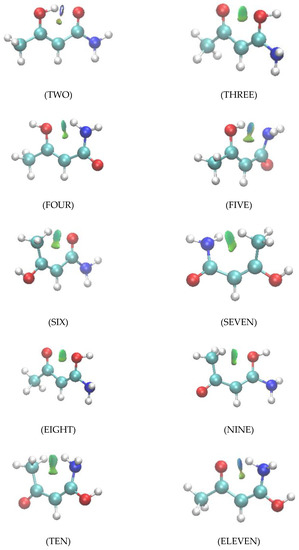
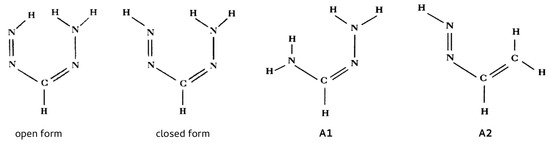
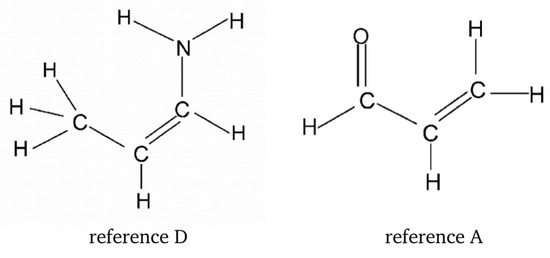
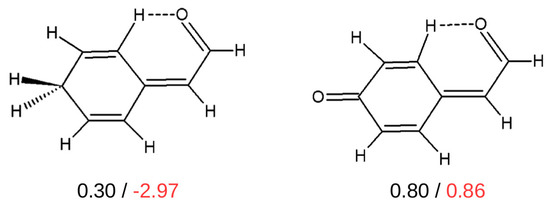
Hydrogen bonds are, quite rightly, considered the most important type of non-covalent interactions. For example, they have a fundamental impact on the properties of water—the compound that has the most important influence on life processes. Namely, when proclaiming the manifestations of the presence of hydrogen bonds, most often one refers to the exceptionally high boiling point of liquid water compared to its slightly heavier counterpart H2S. Another manifestation of the presence of intermolecular hydrogen bonds is that the density of ice is lesser than the density of liquid water. Consequently, the ice floats on the surface of water, enabling the fish to survive the cold winters. Good solubility of polar substances in water also results from the formation of hydrogen bonds between water molecules and the solute. However, hydrogen bonds are not all about water. Their great importance is also manifested in the case of much larger molecules, such as, e.g., carbohydrates and proteins. It is well known that the helical structure of DNA is due to the presence of intermolecular hydrogen bonds between complementary nitrogen bases (adenine–thymine and cytosine–guanine). Intermolecular hydrogen bonds also affect the so-called protein folding and maintenance of cellulose or polymer chains. As in the case of intermolecular hydrogen bonds, the role of intramolecular hydrogen bonds cannot be overestimated. For example, they can significantly affect the conformational equilibrium (a good example is the keto-enol equilibrium in the gas and liquid phases). Such an equilibrium, in turn, often affects the crystallographic structure of the compound in the solid. A very important effect related to the concept of hydrogen bonding is the effect of proton transfer in the hydrogen bridge X-H···Y with the formation of X··H··Y or X···H-Y. As it most often occurs in the excited state of a system, its study requires different techniques.
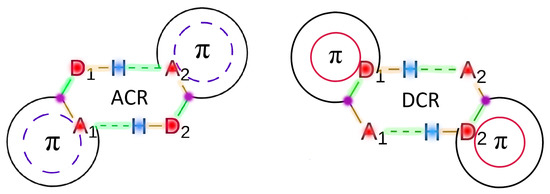
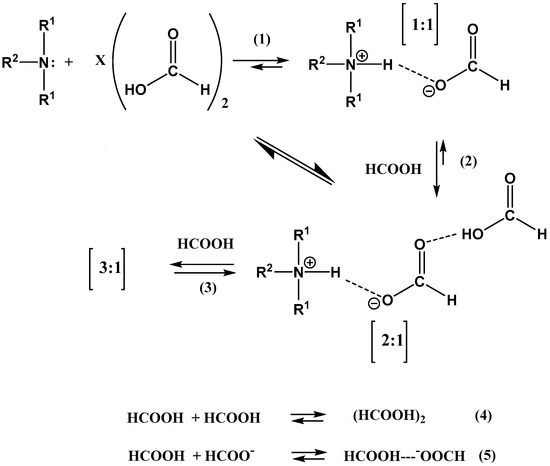
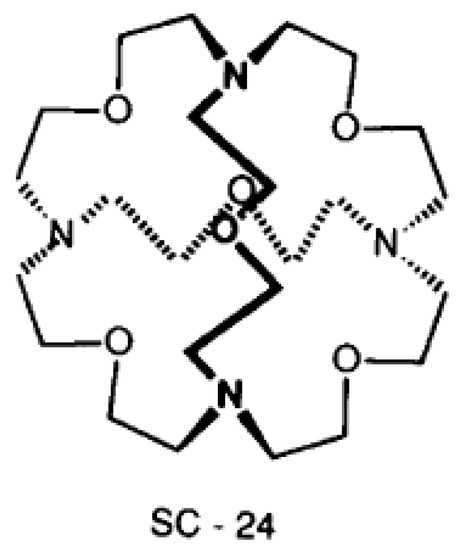
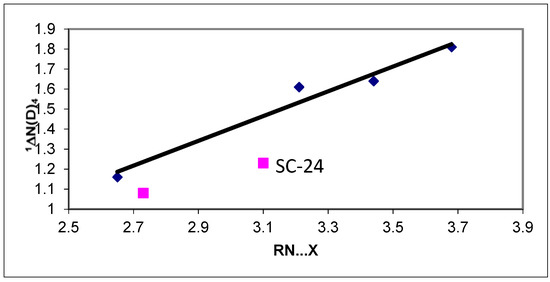
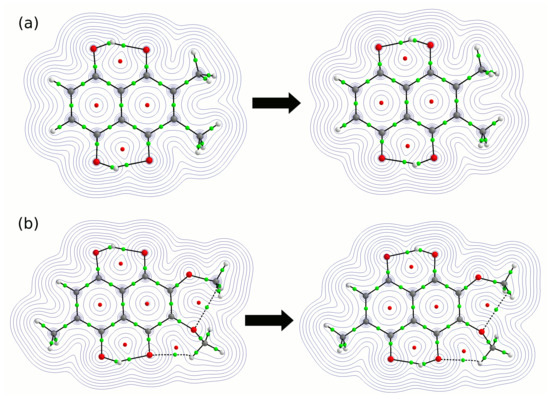
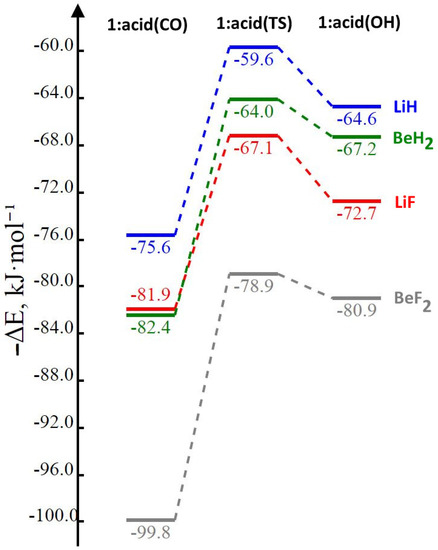
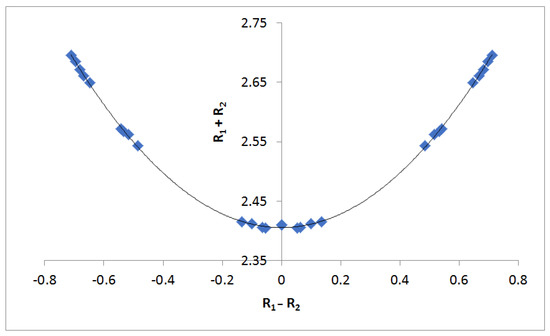
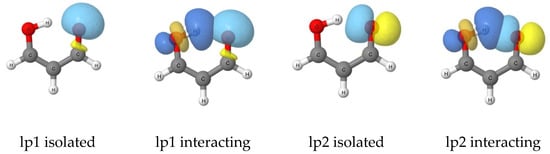
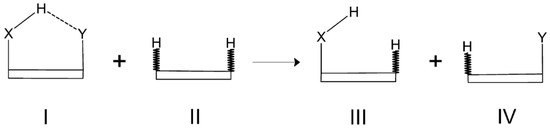
The reason for the extraordinary importance of hydrogen bonds in the rich world of various bonds and interactions is their unique location on the strength scale. Namely, on this scale, hydrogen bonds are in the middle, between the weak van der Waals interactions and the stronger covalent bonds. This intermediate strength of hydrogen bonds means that they are created relatively easily and generally in large amounts. Like a glue, they bind individual molecules into dimers, trimers, molecular clusters, or even larger agglomerates. However, the network of hydrogen bonds is dynamic, old bonds are broken and new ones are formed in their place. Therefore, the evolution of such a system is generally described by Molecular Dynamics (MD). A specific property of hydrogen bonds is that, as the smallest among all the elements in the Periodic Table, hydrogen atom allows a relatively close contact of the X and Y atoms in the X-H···Y bridge, most often strongly electronegative and therefore having the same charge sign, acting as a buffer between them.
The aim of this Topical Collection is to bring together scientific articles on various aspects of hydrogen bonding, both inter- and intramolecular. Articles can be based either on experimental research or theoretical calculations or combine both ways of cognition. I hope that the articles collected here will also be a great overview of the multitude of research techniques currently occurring as well as the multiple topics related to the nature of hydrogen bonding and will contribute to an even greater emphasis on its role not only in chemistry, but also in various related sciences. I hope that this Topical Collection will also be a successful continuation of the recent Special Issue on intramolecular hydrogen bonding (see Book and Editorial Molecules 2021, 26(20), 6319), that is extended to intermolecular hydrogen bonds as well.
Dr. Mirosław Jabłoński
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Molecules is an international peer-reviewed open access semimonthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
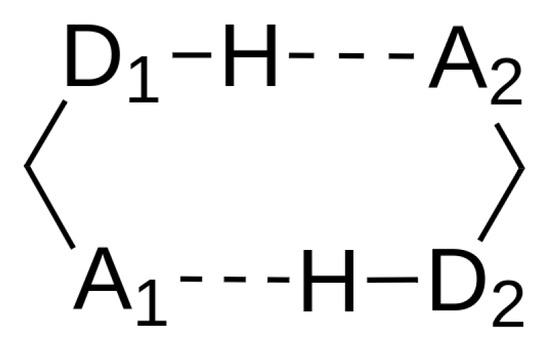{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}