3.9.1. Special Case: SSMF3 and F-SAPT Partitioning Analyses for Complexes of CX⋯Gly
The SAPT0 interaction energies of 24 pairs (fragment⋯Gly), resulting from the SSMF3 fragmentation of the CX, are presented in
Table 4 for
al,
pc, and
wc conformers of CX, while the corresponding F-SAPT partitionings are presented in
Figure 6. One immediately sees that the resulting interaction energies are completely different for these three cases. Let us analyze these differences in more detail, starting from the
pc conformer. In this case, the SSMF3-SAPT0 fragmentation energies with the largest absolute values correspond to fragments 7, 8, and 9 (see
Figure 7). All these fragments contain the same hydroxy group OH-4, which donates its oxygen atom to a strong H-bond with the hydroxy group of Gly (the 1.78 Å distance between the H-ending of the glycine hydroxy group and the oxygen atom from the OH-4). Removal or addition of the methyl group does not have any influence on this energy, which is equal to
mH. However, it would be incorrect to attribute this energy to the isolated H-bond since the removal of the phenyl ring with the attached OH-3 group reduces the attraction by as much as 5 mH (see fragment 10). The weaker attraction cannot be simply explained as a lack of the attraction between Gly and Ph-3 or OH-3, because the fragment 11, which possesses the same OH-4 group bound to Gly but additionally has the phenyl ring with the OH-5 group on another side, reduces the attraction with Gly by another 5 mH. Therefore, the relative position of the second unit is crucial. One can presume that for fragments 7 to 9 the interaction of OH-4 with OH-3 plays a role since according to the I-SAPT partitioning (see
Table 3) there is a strong intramolecular H-bond between these two groups. From geometrical considerations and from a more detailed analysis of induction components, one can see that the OH-4 group donates the H atom while the OH-3 group is the H acceptor, which results in shifting more negative charge to oxygen in OH-4 and making it a better H acceptor for the COOH-Gly. An opposite situation arises for the fragment 11, where the OH-4 group accepts the hydrogen atom from the OH-5 group, what makes the O-4 atom less negative, leading to a weakening of the attraction between the OH-4 and the OH-Gly group. The SSMF3 fragmentation is not detailed enough to directly separate contributions from phenyl ring, methylene, or hydroxy groups, but the F-SAPT partitioning of the complex reveals that indeed
(i) the attraction of the carboxy group of Gly with the OH-4 is the strongest one (
mH),
(ii) there is an additional attraction of COOH-Gly to the OH-3 and Ph-3 groups (
mH), which explains why fragment 10, deprived of those groups, shows a weaker attraction, and
(iii) there is a relatively strong repulsion between COOH and OH-5 (+4 mH), which explains a reduction in attraction for fragment 11. It should be noted that the attraction between the carboxy group of Gly and the OH-4 group is a net result of a balance between several components of similar importance, such as electrostatics, induction, dispersion and exchange. A significant exchange term indicates that electron clouds of these two groups overlap as in the case of covalent bonds, but the magnitude, which is smaller than for typical covalent bonds, classifies this bond as noncovalent. Therefore, the nonzero exchange and other SAPT components signify that the H-bond should exist between some atoms of COOH-Gly and OH-4 (note that according to the IUPAC criteria atoms participating in H-bonds should be close to each other so that the distance between them was smaller than the sum of atomic Van der Waals radii [
121]). If the SSMF3 contributions of a range of about
mH are analyzed in the same manner (not shown in the figure), a secondary binding is revealed, which can be attributed to the interaction between NH
2-Gly and the OH-1 groups with the amino group being the H donor. As it could be guessed from a relatively large distance between the hydrogen of NH
2 and oxygen of the OH-1 (2.23 Å) and from the value of the H⋯OH-1 angle (143
), which is quite different from the full angle, this interaction should be quite weak, but nonetheless, it is still composed of nonzero polarization and exchange contribution, which sum up to
mH according to F-SAPT, therefore we can still classify it as an H-bond.
For the
al-CX⋯Gly complex, there exists much more important terms since the Gly molecule resides in the
al cavity, and in
Figure 8, we present only those fragments, which are the most relevant to the discussion below. The important fragments can be segregated into those corresponding to the binding to the NH
2-Gly and COOH-Gly sites and–contrary to the
pc case–these two sets are of similar importance. The most attractive contributions for the first and second sites come from fragments 1 and 13 and amount to
and
mH, respectively. Fragments 1 to 3 have an H-bond between the OH-2 and NH
2 groups, while fragments 13 to 15–between OH-5 and COOH. Similarly, as in the
pc-CX case above, the removal of the phenyl ring with the hydroxy group, serving as a hydrogen donor in the intramolecular H-bond, reduces the interaction energy by about 7 mH (fragments 2 and 14). However, contrary to the
pc-CX case, the addition of the
th calixarene unit (number 3 for the fragment 3 and number 6 for the fragment 15) does not lead to a further reduction in attraction. Quite the opposite, a small rise of the attraction (by 3-4 mH) is observed in comparison to fragments 2 and 14. The geometry analysis shows that these additional phenyl and hydroxy groups are more twisted in comparison to the
pc case, so that the creation of the H-bond between OH-2 and OH-3 or between OH-5 and OH-6 is prevented, and such bonds would weaken the negative character of the O-2 and O-5 atoms. What remains to be explained is the increase in the attraction for fragment 3 in comparison to fragment 2 (and for fragment 15 in comparison to 14). Since these fragments differ by other phenyl plus hydroxy groups, one (or both) of these groups should be responsible for this phenomenon. Because of the limitations of the SSMF3 partitioning, the explanation of this fact should be postponed till the F-SAPT analysis is made. Now let us move to a complementary view of the F-SAPT partitioning. As expected, there is a strong (
mH) binding between the OH-2 and NH
2 groups. Surprisingly, the interaction between OH-5 and COOH amounts to
mH only, but the carboxy group is strongly attracted to the Ph-5 group (
mH). All F-SAPT components, including the exchange one, are significant for the COOH⋯Ph-5 interaction; therefore, the F-SAPT analysis reveals the existence of the untypical H
H-bond. Such interactions were reported, e.g., in molecules containing aromatic rings with those with the S-H bond [
122]. Another candidate for the H
H-bond would be the H atom from the NH
2 group interacting with the Ph-3 group. However, in this case, the total interaction energy is close to zero, which is a result of a perfect cancellation of several contributions of similar magnitude (the electrostatics of
mH and dispersion of
mH are counterbalanced by the large exchange component). The question remains why fragments 2 and 3 differ in attraction by as much as 4 mH. A perusal of the F-SAPT partitioning table reveals that this difference is due to the electrostatic attraction (
mH) between the Ph-3 and COOH groups. Finally, a similar difference for fragments 14 and 15 can be explained by the net attraction to the OH-6 group (
mH).
The most important contributions from the SSMF3 partitioning for the
wc-CX⋯Gly complex provide interaction energies of about
to
mH (see two representative fragments 3 and 7 in
Figure 9) and have in common the H-bond between the OH-3 and NH
2 with the latter group as the H acceptor (the H⋯N bond of 1.67 Å). As in the previous cases, this energy cannot be attributed to this one H-bond only, since, e.g., fragment 6 containing this H-bond attracts the Gly molecule weaker by 8 mH. The geometry considerations allow us to point to a possible additional interaction with the OH-2 group for fragment 3 since the O⋯H distance in this case is equal to 2.10 Å, that is, the amino group of Gly donates its hydrogen to create a weak H-bond. The mechanism of an increased attraction in case of the fragment 7 is different: here the intramolecular H-bond between the OH-3 and OH-4 groups makes the O-3 oxygen less negative, thus allowing the H-3 to interact more strongly with the nitrogen from the amino group. Not shown in the figure are fragments 15 to 17, for which two interactions: between the amino group and OH-5 (the NH
2 as H donor) and between the carboxy group and OH-6 (COOH as the H acceptor) can be guessed from the geometry considerations. The F-SAPT partitioning confirms these predictions. First, a strong interaction between the OH-3 and NH
2 groups has been obtained with this method (
mH). There are also three weak H-bonds (again recognized by the significance of all SAPT components, including exchange): between OH-6 and COOH (
mH), OH-5 and NH
2 (
mH), and OH-2 and NH
2 (
mH). The latter interaction is too weak to explain a difference between the attraction from fragments 3 and 6, but the F-SAPT provides the additional strong attraction of a purely electrostatic type between the OH-2 and carboxy group (
mH) to fill this gap.
Summarizing, all three cases of the interaction with the simplest Gly amino acid provide different mechanisms for secondary stabilizing interactions, which would be difficult to elucidate just from the analysis of the total energies.
3.9.2. Special Case: SSMF3 and F-SAPT Partitioning Analyses for Complexes of BCX⋯Gly
The SSMF3 model for hexa-
p-
tert-butylcalix[6]arene produces as many as 150 fragments; therefore, the tables analogous to
Table 4 were shifted to the
Supplementary Information. The SAPT0 interaction energies for complexes
al-BCX⋯Gly and
wc-BCX⋯Gly are close to those for the CX counterpart (
mH and
mH for
al,
mH and
mH for
wc, for CX and BCX, respectively), but the for
pc-BCX the interaction energy is 11 mH lower than for the unsubstituted case. Naively, one could assume that it is the attraction with the
tert-butyl groups, which makes the total interaction energy more negative, but the sum of interaction energies between fragments made from
tert-butyl with Gly gives a negligible contribution. Therefore, the influence of the
tert-butyl substituents is more subtle–their presence leads to geometry modifications, which, in turn, allow for a better arrangement of Gly on top of the
pc-BCX. Similarly, as in the unsubstituted case, there is a set of interaction energies of about
mH each, which contain the same type of an H-bond between the carboxyl group of Gly and the OH-1 group (note that for the pristine calixarene OH-1 and OH-4 groups etc. are equivalent, see
Figure 2). Again, the presence of the calixarene unit 2 causes an enhancement of the electronegative character of the O-1 atom through the intramolecular H-bond with the OH-2 group, which explains its weaker attraction to Gly (to
mH for fragments with the unit 2 removed). The first significant difference appears for numerical values of interaction energies for fragments with the unit added on the other side (the unit 6) of the unit 1. Although the intramolecular H-bond between OH-1 and OH-6 is created, as in the CX case, it weakens the attraction to Gly by 2 mH only, while for the CX case this change amounts to 5 mH. Still, this difference alone does not explain the 11 mH gap between the total interaction energies for the CX and BCX for this conformer, and other differing factors should be looked for. It turns out that the missing difference can be found from the examination of fragments, which contain units 4 and 3 of BCX and have interaction energies of
to
mH. In these fragments, one H-bond between the H atom from the OH-4 group and the NH
2 group of Gly can be found, while the neighboring OH-3 group forms the intramolecular H-bond acting as the H donor, i.e., it enhances the negative character of the O-4 atom. The latter explanation is confirmed by comparison with fragments without the OH-3 group, for which the interaction energy changes to
mH. Summarizing, in comparison to the CX case, the interaction with the NH
2 group is much stronger and it is this interaction which according to the SSMF3 partitioning is responsible for the stronger attractive force for the
pc-BCX in comparison to the unsubstituted calixarene. It is also worth noting that the direction of the primary H-bond for the NH
2 group changes, i.e., this group becomes the H acceptor for the BCX case. The F-SAPT partitioning confirms these findings: there is still a strong bond of
mH between the COOH and OH-1 groups, but the attraction of the OH-4 to the amino group is stronger (
mH) than for the CX case. This new intermolecular H-bond causes a distortion of one from the intramolecular H-bonds in the BCX, what can be also observed from the pattern of I-SAPT interaction energies between the hydroxy groups, where instead of one strong attraction of
mH between OH-1 and OH-6 only a weak one of
mH remains.
The al-BCX⋯Gly complex is of the inclusion type, and the Gly molecule resides in the cavity created by units 3, 4, and 5 of the BCX, as for the al-CX counterpart. The fragments which give the largest attractive SSMF3 contributions ( mH) contain the phenyl and hydroxy groups from units 1 and 2, and–similarly as for the al-CX case – one of them donates its H atom to the NH2-Gly group forming an H-bond, and another one enhances this interaction through a creation of the intramolecular H-bond with the O-2 atom. This effect can be estimated as about 6 mH, based on the energy of the fragment without the unit 1 ( mH). There is another group of fragments with no analogs for the CX case, which gives rise to contributions of about mH. It turns out that they all contain the H-bond between NH2 (the H donor) and the OH-6 group (H⋯O distance of 2.005 Å) and additionally possess the phenyl and hydroxy groups from the neighboring unit 5. The lack of these two groups leads to a strong decrease in attraction (to mH), but for the al-BCX geometry, the intramolecular H-bond between the OH-5 and OH-6 groups cannot exist; therefore, the reason for a large attraction in the former case is the direct interaction of the phenyl and hydroxy groups of unit 5 with Gly. The latter conclusion is confirmed by the interaction energy of about mH for fragments, which contain the Ph-5 and OH-5 groups, but no other groups of these types. It should be noted that a relatively large distance between Gly and such fragments suggests that the interaction should be of an electrostatic type since electrostatic contributions are known to be long-ranged. There are also fragments containing the phenyl and hydroxy groups of the unit 4, which attract Gly with the strength of mH. It is evident from the geometry analysis that the OH-4 group cannot effectively participate in any H-bond with Gly, but since the carboxy group is positioned quite close to the Ph-4 (the closest distance between the hydrogen atom of COOH and the carbon atom is about 2.5 Å), one can predict a formation of an unusual H-bond between this hydrogen and the cloud of the phenyl ring. Note that this distribution of interaction strengths is different from the al-CX case, where contributions as large as mH are present, so–surprisingly–such close total interaction energies result from the summation of contributions of a partially different origin. The F-SAPT partitioning confirms the existence of strong bonding between the OH-2 and NH2 groups ( mH), as in the CX case. However, contrary to the CX case, the carboxy group is attracted mostly not to the Ph-5 group, but to a closer Ph-4 group with a remarkable strength of mH. Nevertheless, in both cases, the existence of the H- bond can be postulated based on the position of the hydrogen atom and on the analysis of SAPT components. Another feature of the binding pattern is the existence of a secondary H-bond, in which the NH2 group donates a hydrogen atom. This H-bond can be identified based on the analysis of the interaction between the NH2 and OH-6 ( mH, with about +10 mH from the exchange component). The strong attraction from unit 5 found in the SSMF3 partitioning is also reproduced here as a strong electrostatic-dominated interaction between the COOH and Ph-5 ( mH). It should be emphasized that the F-SAPT and SSMF3 partitionings for the al-CX and al-BCX complexes with Gly reveal that the Gly molecule is attracted by the cavity from several sites with similar strength. The geometry analysis shows that this relatively small molecule seems to fit well into the small cavity of the al conformer; therefore, these complexes represent examples of the enhancement of the interaction due to a confinement effect.
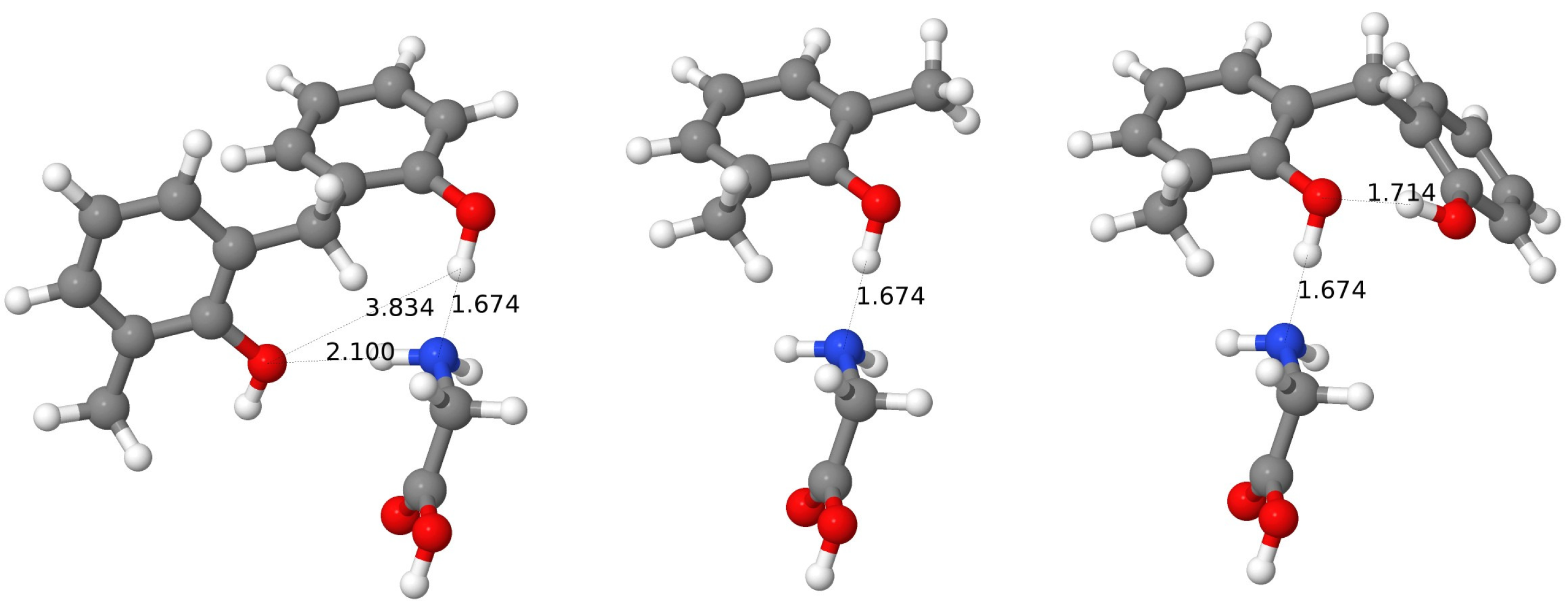
The main features of the complex of Gly with the wc-BCX are similar to the wc-CX case. The first set of interaction energies of about mH corresponds to fragments containing the phenyl groups plus hydroxy groups from units 5 and 6. The OH-5 group serves as an H donor, and the OH-6–as an H acceptor for two H-bonds with the NH2-Gly. Since the fragment without the OH-5 group, but with the OH-6 group remaining, has the interaction energy reduced to mH, the strength of the second H bond can be estimated as about 7 mH. The addition of the phenyl and OH groups from the unit 1 leads to the increased attraction ( mH), which can be explained by the intramolecular H-bond creation between the OH-6 and OH-1 groups, which increases the electronegative character of the O-6 atom (this H-bond is also seen from the I-SAPT analysis). The next sets of interaction energies correspond to the fragments containing either both phenyl and hydroxy groups from units 2 and 3 (energies of mH), or having groups from unit 3 only, which reduces the interaction to mH. Since the OH-3 group donates its H atom to the oxygen from the carbonyl group of Gly (the H⋯O distance of 1.99 Å) and the OH-2 group is a hydrogen acceptor for the NH2 group; therefore, two H-bonds are present here, and the H-bond between the OH-2 and amino groups can be estimated as about 5.5 mH from a difference analysis. The F-SAPT partitioning results are in line with these findings. Firstly, the most important interaction of mH exists between the amino and OH-6 groups. The carboxy group forms a weaker bond with the OH-3 group with the strength of mH. The analysis of SAPT components confirms that these two bonds are H-bonds. The amino group is also connected with the groups OH-1 and OH-2, but from these two pairs, the first one is dominated by electrostatics, while the second again represents an H-bond. It is interesting to note that the distance between the corresponding hydrogen atoms of the amino group and oxygen atoms of the hydroxy groups is only 0.15 Å longer (2.22 versus 2.04 Å) for the electrostatic driven interaction and differences in the total interaction energies are also not very large ( mH versus mH). Nevertheless, in the second case, one has a significant first-order exchange component of 7 mH, which is counterbalanced by other components, from which the electrostatics gives the most negative contribution ( mH). It should be noted that for the CX case, both these interactions were of the H-bond type.
3.9.3. Special Cases: Analysis of pc- CX Inclusion Complexes
Let us use the F-SAPT partitioning to examine in detail the only four cases, for which the
pc conformer of CX prefers to form an inclusion complex with an amino acid (Ile, Lys, Val, and Pro). Since the dispersion energy can be only negative and its magnitude is correlated with the number of electrons, it can be used to locate electron-rich environments. Indeed, for all four cases, one can identify pairs of functional groups (one from CX and one from an amino acid), which are close to each other and have a large number of electrons. In particular, a perusal of the dispersion graphs (see the
Supplementary Information) allows us to instantly locate the guest molecule into one from two parts of the
pc-conformer cavity. (Note that if the dispersion component is significant, then usually other SAPT contributions are nonzero and we deal with a potential candidate for a noncovalent bonding, such as the H-bond.) The next components to examine are the electrostatic and induction energies. One can see that indeed, depending on a relative orientation, some pairs attract each other more strongly than it would be possible with dispersion only. However, the analysis of these three components only is not sufficient to guess the total interaction strength, since many of these components are quenched by short-range first-order exchange contribution. One should emphasize again that the presence of nonzero exchange components together with the polarization (induction and dispersion) components show that we have to deal with a noncovalent bond.
Let us first consider the pc-CX⋯Ile case. The F-SAPT reveals here a strong attraction between the OH-2 group of the calixarene and the COOH group of Ile, which can be explained by the creation of a weak H-bond (the O⋯H distance of 2.0 Å). The F-SAPT estimation of this bond strength can be given by the interaction energy of the OH-2 and COOH groups and it amounts to mH, which should be compared with mH for the intramolecular attraction of two adjacent hydroxy groups in calixarene. This interaction is not the only one in this complex. Moreover, the Ph-6 ring is attracted to the carboxy group, but it is also strongly repelled by the neighboring CH group. It is also interesting that the chain of six intra-calixarene H-bonds is not destroyed in spite of a contribution of OH-2 to another H-bond, which can be seen in the examination of the interaction between the hydroxy groups of calixarene with help of I-SAPT (there are still six interaction energies between these groups of value below mH).
For the complex of pc-CX with Pro the F-SAPT partitioning of dispersion energies reveals that the ring of Pro is strongly attracted by the calixarene phenyl rings on one part of the macrocycle (with Ph-4, Ph-3, and Ph-5), which agrees with the fact that the Pro amino acid occupies just one side of the calixarene cavity. For the interaction with Ph-4 and Ph-5, there is some attractive electrostatic component, too. However, the major part of the interaction comes from the attraction of the COOH group of Pro with Ph-3, which has a predominantly electrostatic origin. It turns out that a strong attractive dispersion interaction of the Pro-ring with three phenyl rings is to a large extent counterweighted by a repulsive exchange interaction, leaving a weak net attraction for these three pairs. It is also interesting to note that practically no interaction exists between Pro and the hydroxy groups of calixarene, in spite of the fact that both Pro fragments are polar and close to at least the OH-3, OH-4, and OH-5 groups. Apparently, the sextet of H-bonds makes the hydroxy group less polar, so they are not involved even in a long-range electrostatic interaction (it should be also noted that the H-bonds’ distances range between 1.63 to 1.69 Å, i.e., they do not elongate upon the complexation with Pro).
In the complex with the Lys molecule, there are three Ph groups, which are mostly responsible for the net attraction. From the Lys side, all major polar groups contribute to the interaction and it is impossible to select one or two major interacting pairs. One can note that one from two NH2 groups of Lys is involved in a secondary bond with the nearest Ph-2 ring. This interaction of mH is composed of significant attractive electrostatic and dispersion components and is quenched by the exchange. Interestingly, the interaction of another amino group with the same ring is of a very similar value ( mH), but is predominantly of the electrostatic character. The hydroxy groups of calixarene do not contribute to these interactions in a significant way (what can be also seen by practically fixed lengths of H-bonds similar to the empty calixarene). It turns out that the COOH and NH2 groups of Lys connect through an intramolecular H-bond, which apparently depolarizes this molecule to some extent and makes it a weaker target for calixarene.
Finally, for the complex with Val again the net attraction comes from the same three Ph groups, but in this case, one can pinpoint the COOH group as the main partner on the Val side. The interaction between the COOH group and Ph-1, Ph-2, and Ph-6 is of mostly electrostatic character. There are several cases where the exchange contribution is relatively large, but, e.g., for the largest exchange contribution amounting to mH (the Ph-1 with the amino group) the electrostatic one is close to zero and after adding the dispersion component a small net interaction energy below 1 mH remains. The alkyl skeleton gives together a weak attraction of mH, but the closest CH group is oriented in a way that produces repulsive electrostatics. Therefore, all potential candidates for a formation of weak noncovalent bonds are excluded in this way and altogether the Val@pc-CX complex can be described as the electrostatically bound complex.
3.9.4. Types of Noncovalent Bonding in Calixarene-Amino Acid Complexes
As shown in
Section 3.9.1,
Section 3.9.2 and
Section 3.9.3, the F-SAPT analysis is very useful as a tool for the identification of various types of noncovalent bonds. A comprehensive study of all 120 complexes reveals many more interesting bindings, among which the various sorts of H-bonds form an important group. The IUPAC classification of H-bonds [
51,
54,
121] provides the following H-bond criteria, which may be useful to browse through the present data:
(i) their energy (63–167
for strong, 17–36
for medium, and less than 17
for weak hydrogen bonds);
(ii) a large contribution to the electrostatic interaction (with a nonnegligible dispersion), and geometry constraints; such as
(iii) a preferable alignment of a hydrogen donor, a hydrogen atom, and a hydrogen acceptor and;
(iv) distances between interacting atoms, which should be significantly smaller than the sum of the Van der Waals radii. The SAPT approach has been applied many times for studying H-bonds, such as, e.g., in the comprehensive study of the behavior of various SAPT components for the water dimer [
123], where the near-linear preference of the H-O⋯H atoms has been explained by a different angle dependence of first- and second-order SAPT energy components, with the prevailing importance of the second-order induction and dispersion. Therefore, from the SAPT point of view, one should examine not only the pure electrostatic contribution as a fingerprint of the H-bond but also the exchange component, which indicates how large the overlap of the electron clouds of the hydrogen is, the hydrogen donor and the hydrogen acceptor (which is indirectly implied in the H-bond classification by the requirement of a small enough distance between atoms).
The interaction types for all pairs of groups (i.e., 24 groups for a calixarene and a differing number of groups for amino acids) can be first classified according to their total interaction energy . According to the IUPAC criteria (64–24 mH–strong, 24–6.5 mH–medium, below 6.5 mH–weak H-bonds), almost all bondings present in calixarenes belong to medium or weak types (note, however, that this criterion is of a limited value here, as the F-SAPT energies are calculated between functional groups containing more atoms than just three involved in an H-bond). The strength of the pair interaction does not imply automatically the H-bond character, although most of the strongest pairs do contain typical H-bonds, i.e., these interactions occur between hydroxy groups of calixarenes and, e.g., hydroxy groups (usually from the carboxy group) or amino groups of amino acids.
The interaction type can be established through the analysis of weights of SAPT components with respect to the interaction energy, i.e., by examining the ratios:
,
,
, and
, where the “eff" subscript denotes effective components as described in
Section 2.5. In particular, a large contribution of the exchange energy signifies that both functional groups are close enough for the electron exchange and are a necessary prerequisite of a true bonding (of course, it could also signify a repulsion). A large exchange contribution usually implies that the remaining ratios are nonnegligible. If the only significant contribution is the electrostatic energy, then the functional groups do not share the electron cloud and their attraction or repulsion can be described classically
via the Coulomb law. The selection of a cutoff value for such ratios is to a large extent arbitrary, and we assumed that if a given component accounts for at least 30% of the interaction energy, the contribution from this component is significant.
The F-SAPT partitioning for selected pairs is presented in
Table 5, while tables for all pairs are shifted to the
Supplementary Information. A representative of a typical moderately strong H-bond can be characterized by high ratios for electrostatic, exchange, and induction–usually above 1.0 for both first-order components and above 0.5 for the induction and by a smaller contribution from the dispersion (below 0.3). The contribution from the dispersion rises for special cases, such as Pro or His, where a ring with a heteroatom (nitrogen) with the attached hydrogen serves as a hydrogen donor, but additionally, the ring itself interacts with the calixarene hydroxy group, which occurs to a large extent through the dispersion interaction. It should be noted that in such cases, the first three ratios remain high, similar to other H-bonds discussed so far.
Apart from such cases, there are also several moderately strong bondings, which are characterized by a different ratio pattern. Usually, they arise from the interaction of a phenyl ring of calixarene with a polar group belonging to an amino acid. They are characterized by a lower ratio for the exchange (much lower than 1, but not less than 0.3), the still high ratio for electrostatics, and usually by smaller ratios for induction and dispersion (about 0.2–0.3).
An examination of the relative position of the calixarene phenyl ring in question and the hydrogen atom from the polar group of the amino acid reveals that such cases correspond to a partial donation of the hydrogen atom to the ring, i.e., here, the untypical H-bond is detected (H⋯ bond), where the phenyl ring serves as a hydrogen acceptor. One should add that usually an elongation of the H–X bond in comparison to the isolated amino acid is found in such cases, similarly to typical H-bonds.
The next characteristic pattern occurs for the interaction between a calixarene phenyl ring and a ring of an amino acid (e.g., for His). For such interactions there is still a high electrostatics ratio, the exchange ratio is higher than for the H⋯ case, but lower than for a typical H-bond (usually about 0.5), and finally, a large dispersion contribution amounts to at least 0.5. Summarizing, in comparison to all other cases, these pairs are to a large extent dispersion-bound, especially if one takes into account that the electrostatics and first-order exchange contributions partially cancel each other.
One should add that even among quite strong interactions one could find electrostatic-driven ones, especially for the interaction of a calixarene ring with a polar group of the amino acid; therefore, the ratio check of components is usually necessary in order to determine the interaction type. In the case of calixarenes, the macrocyclic structure represents an obstacle in relaxing the geometry after the complexation. In some cases, this means that apart from the dominant attractive pairs, some other pairs exist, for which the interaction energy becomes zero or is even positive (repulsive). In the interaction table, the largest such contributions amount to 7–8 mH. Among them, there are pairs that repel each other electrostatically, but also those that are close enough to have a large exchange contribution.
The sorting of F-SAPT total energies for pair interactions reveals in many cases two or more pairs with interaction energies of a similar value (which one can set arbitrarily for, e.g., no more than 50% difference). Among such cases, both inclusion and outer complexes can be found. For the inclusion complexes, this analysis helps to identify the confinement (encapsulation) effects, i.e., those cases where an amino acid occupies the calixarene cavity and utilizes its active groups to bind with several groups of the host. One such example has already been presented in
Section 3.9.1 and
Section 3.9.2, where the
al conformers of both CX and BCX use both polar groups of Gly to encapsulate it effectively in the cavity. More such complexes can be found, e.g., for the
pc-CX case for complexes with Val, Pro, and Ile and for the
pc-BCX one–with Pro, GluH, Ala, and AspH. In all these cases, the guest molecule fits into one of two cavities created by three calixarene units and the dihydrogen bridge. Other complexes with the confinement effect are: complexes with Ile, Val, Tyr, Thr, and Ser for the
al-CX, and complexes with Val, Trp, Thr, Ser, Pro, Met, HisD, Gln, AspH, and Asn for the
al-BCX. It should be noted that in the case of partial confinement, which we have in the case of calixarenes, the cage (or better saying: half-cage) has more flexibility to adapt itself for a particular guest than an inherently rigid full cage, such as in fullerenes. In all these cases, the guest molecules are attached in the cavity by at least two comparable interactions, which are usually (but not always) the proper H-bonds.
The same analysis for the outer complexes reveals that a general classification into: either cases for which one molecule has a dominating group interacting with at least two groups of another molecule, or cases for which each molecule has at least two such groups. The first scheme corresponds to the pattern , and the second–to , where and are functional groups of molecules A and B. The first case can be named, similarly as in the ligand theory, as di- (or in general: poly-) dentate complex, while the second case–as di- (or in general: poly-) site complex. The di-dentate complexes are the most common. Among several examples, one can name the complex of the pc-CX with Gln, where the Gln molecule uses its carboxy group to make two H-bonds with the OH-1 and OH-6 groups (of similar strength of about mH), or with Tyr, for which the carboxy group binds electrostatically with the Ph-3 and Ph-2 groups. Yet another example of this type is the pc-BCX complex with Asn, where the carboxy group forms two H-bonds with two hydroxy groups of calixarene, with the interaction energies of and mH.
The poly-site complexes are, e.g., complexes of the pc-CX with Ala, where two H-bonds are created: between the OH-4 and NH2 groups and between the OH-1 and COOH groups with the interaction energies of mH and mH, respectively, or with AspH, where again two opposite hydroxy groups (OH-3 and OH-6) form H-bonds with two AspH carboxy groups. For the wc-CX case, the complex with AspH is particularly interesting, since the examination of the SAPT energy decomposition allows us to identify two H-bonds as for the pc case, but additionally both COOH groups interact electrostatically with other groups of calixarene with total energies of the same magnitude. Other wc-CX examples of poly-site binding are: (i) Gln, where two large groups (COOH and CONH) each form two H-bonds with hydroxy groups of calixarene (the strongest two interactions amount to and mH); (ii) HisE, where one typical H-bond is formed between the OH-3 and NH2 groups, but additionally a strong dispersion-dominated interaction between two rings (the Ph-2 group with the HisE ring) occurs (the first interaction amounts to mH, and the second–to mH); (iii) Leu or Lys, where two H-bonds are formed: one with help of the COOH, and the second–with the NH2 group.
There is also a number of poly-site complexes for the hexa-
p-
tert-butylcalix[6]arene case, such as, e.g., already discussed
pc-BCX⋯Gly case, see
Section 3.9.2. For the
wc-BCX complex with HisE an analogous situation appears as for the CX case with very similar interaction energies of
and
mH for the H-bond and the dispersion-dominated bond, respectively. The same pair of interactions exists for the complex with Trp, where apart from a typical H-bond between the OH-6 and NH
2 groups there exists a dispersion-dominated interaction between the Ph-5 and Trp rings (of energies of
and
mH, respectively). Other interesting cases are:
(i) the complex with Thr, where apart from a typical H-bond between the OH-3 and NH
2, the Ph-6 and carboxy groups attract each other electrostatically (both bonds of strength
mH) and;
(ii) a similar pair of H-bond and electrostatic interactions in the complex with Pro, where the nitrogen from the Pro ring forms the H-bond, while the COOH group is attracted by the phenyl ring (with the interaction energies of
and
mH, respectively), or;
(iii) the interaction with Met, where again pairs OH-6 with NH
2 and Ph-3 with COOH form the H-bond and the electrostatic-dominated interaction of the same strength of
mH. There are also cases of typical H-bonds of similar strength, such as in the complex with Ser (the OH-6 with NH
2 and the OH-3 with OH-Ser groups with interaction energies of
and
mH, respectively), with Lys (interaction energies of about
mH) or with GluH (
mH).
Many more similar examples can be found after the analysis of the F-SAPT interaction energy partitioning, listed in the
Supplementary Information.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}