
Focal Point Evaluation of Energies for Tautomers and Isomers for 3-hydroxy-2-butenamide: Evaluation of Competing Internal Hydrogen Bonds of Types -OH…O=, -OH…N, -NH…O=, and CH…X (X=O and N)


Abstract
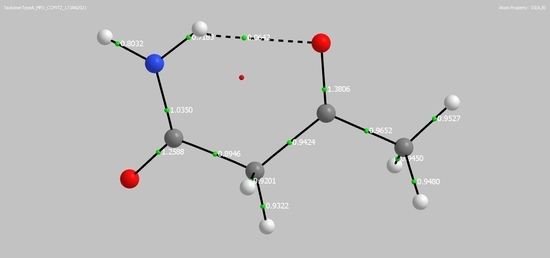
:
1. Introduction
2. Results
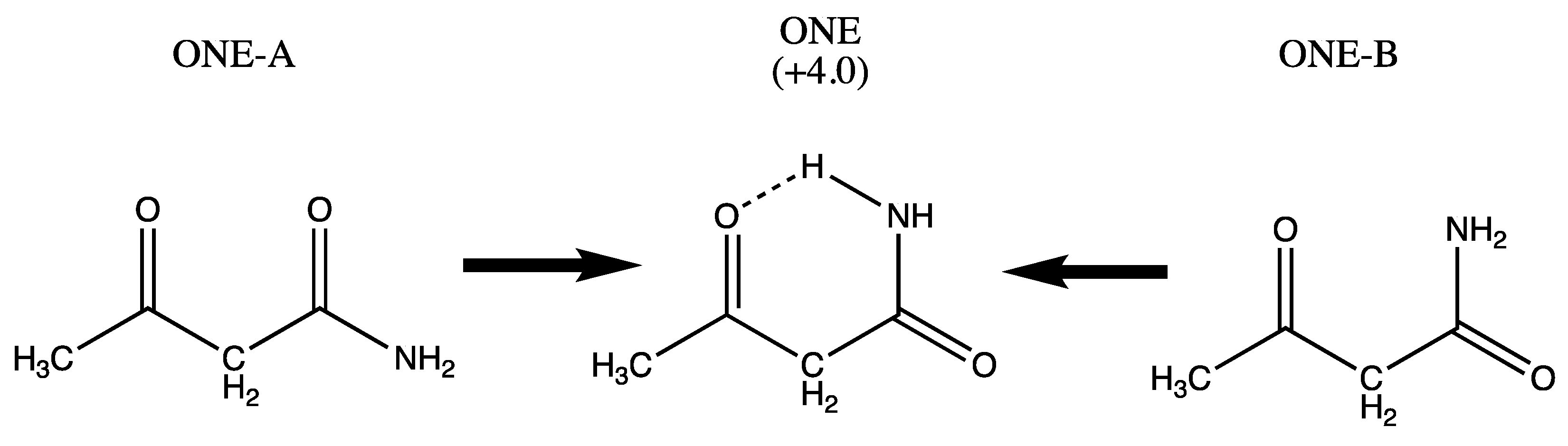
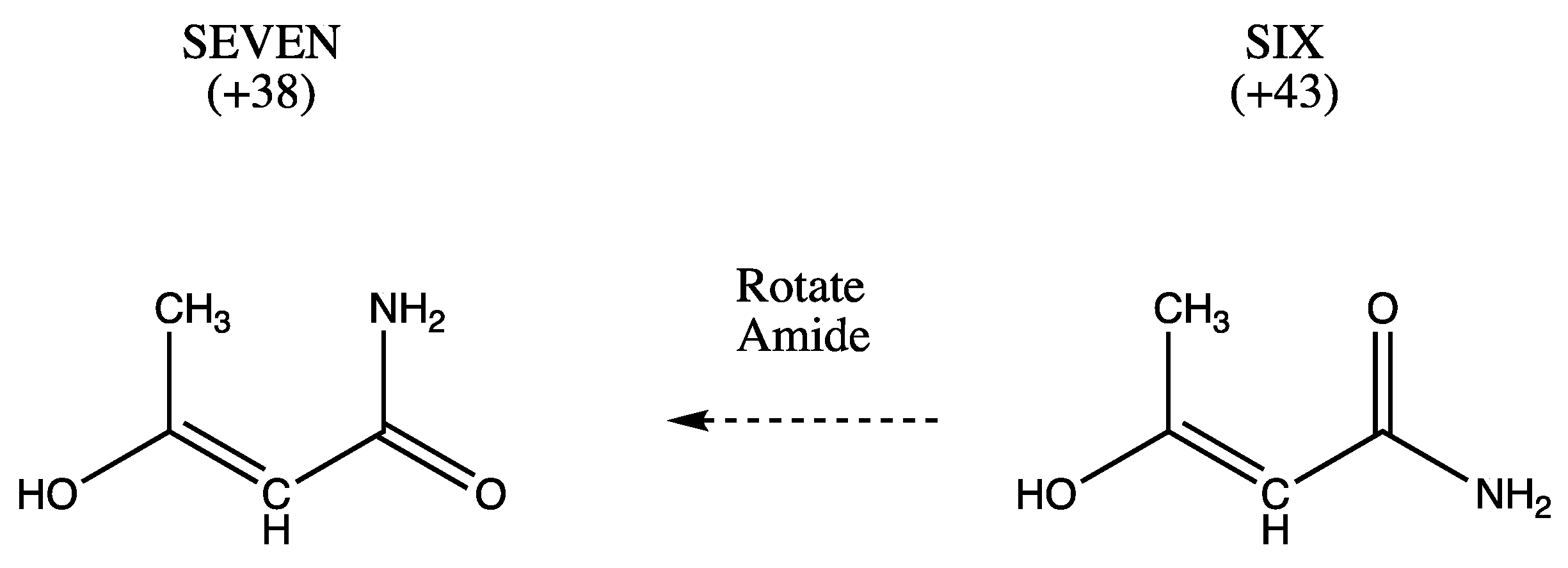
2.1. Systems under Study
2.2. Extrapolation of Accurate Relative Energies
2.3. Atoms in Molecules (AIM) Characterization of Interactions
2.4. Non-Covalent Interaction (NCI) Characterization of Interactions
2.5. Expression of Hydrogen Bonding in MP2/cc-pVTZ Computed Harmonic Vibrations
2.6. Local Mode Analysis
3. Discussion
4. Materials and Methods
4.1. Extrapolation Techniques
4.2. Atoms in Molecules
- Topology: There should appear a bond path between atoms considered to be linked by a H bond. This path should contain a BCP.
- Density: At this point the density should have a “reasonable” value, about an order of magnitude smaller than the values for covalent bonds or ca 0.01 atomic units, with a range up to 0.035 atomic units. There is a correlation between the BCP density and intermolecular attraction.
- H atom charge: q(H) involved in a hydrogen bond XH…Y should be substantially smaller than q(H) for H atoms not participating in a H bond but with the same X atom in XH. Here q(H) is the charge in the basin containing the nuclear attractor for H atom.
4.3. Non-Covalent Interactions and the Reduced Density Gradient
4.4. Canonical Vibrational Spectra and Local Mode Analysis
4.5. Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Needham, P. Hydrogen bonding: Homing in on a tricky chemical concept. Stud. Hist. Philos. Sci. 2013, 44, 51–65. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford U Press: New York, NY, USA; Oxford, UK, 1997. [Google Scholar]
- Taylor, R.; Kennard, O. Crystallographic Evidence for the Existence of CH…O, CH…N, and CH…Cl Hydrogen Bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. [Google Scholar] [CrossRef]
- Desiraju, G.R. The CH…O Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C. Strong hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Havlas, Z. Blue-Shifting Hydrogen. Bonds Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; José Elguera, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27, 163–170. [Google Scholar] [CrossRef]
- Hadži, D. (Ed.) Theoretical Treatments of Hydrogen Bonding; Wiley Research Series in Theoretical Chemistry; John Wiley and Sons, Ltd.: Chichester, UK, 1997. [Google Scholar]
- Schuster, P.; Wolschann, P. Hydrogen Bonding: From Small Clusters to Biopolymers. Mon. Chem. 1999, 130, 947–960. [Google Scholar]
- Grabowski, S.J. (Ed.) Hydrogen Bonding—New Insights; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond. Outline of a Comprehensive Hydrogen Bond Theory; Oxford U Press: Oxford, UK, 2009. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond. J. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Scheiner, S. Special Issue: Intramolecular Hydrogen Bonding 2017. Molecules 2017, 22, 1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, G. Introduction to “Intramolecular Hydrogen Bonding 2018”. Molecules 2019, 24, 2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonin, A.V.; Vashchenko, A.V. Benchmark calculations of intramolecular hydrogen bond energy based on molecular tailoring and function-based approaches: Developing hybrid approach. Int. J. Quantum. Chem. 2019, 119, 26001. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comp. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Yannacone, S.F.; Sethio, D.; Kraka, E. Quantitative assessment of intramolecular hydrogen bonds in neutral histidine. Theor. Chem. Acc. 2020, 139, 125. [Google Scholar] [CrossRef]
- Zavitsas, A.A. Quantitative relationship between bond dissociation energies, infrared stretching frequencies, and force constants in polyatomic Molecules. J. Phys. Chem. 1987, 91, 5573–5577. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Characterization of CF Bonds with Multiple-Bond Character: Bond Lengths, Stretching Force Constants, and Bond Dissociation Energies. Chem. Phys. Chem. 2009, 10, 686–698. [Google Scholar]
- Espinosa, J.E.; Molins, E.; LeComte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Jablónski, M.A. Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef]
- Jablónski, M.; Monaco, G. Different Zeroes of Interaction Energies as the Cause of Opposite Results on the Stabilizing Nature of C-H…O Intramolecular Interactions. J. Chem. Inf. Modeling 2013, 53, 1661–1675. [Google Scholar] [CrossRef]
- Jablónski, M. Bond paths between distant atoms do not necessarily indicate dominant interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Jablónski, M. On the Uselessness of Bond Paths Linking Distant Atoms and on the Violation of the Concept of Privileged Exchange Channels. Chem. Open 2019, 8, 497–507. [Google Scholar]
- Jablónski, M. Counterintuitive bond paths: An intriguing case of the C(NO2)3– ion. Chem. Phys. Lett. 2020, 759, 137946. [Google Scholar] [CrossRef]
- Bader, R.F.W. Bond Paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 891–892. [Google Scholar] [CrossRef]
- Bauer, J.S.H.; Wilcox, C.F. On malonaldehyde and acetylacetone: Are theory and experiment compatible? Chem. Phys. Lett. 1997, 279, 122–128. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.J. Quantitative decomposition of resonance-assisted hydrogen bond energy in β-diketones into resonance and hydrogen bonding (π- and σ-) components using molecular tailoring and function-based approaches. Comput. Chem. 2020, 41, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Belova, N.V.; Sliznev, V.V.; Oberhammer, H.; Girichev, G.V.J. Tautomeric and conformational properties of β-diketones. Mol. Struct. 2010, 978, 282–293. [Google Scholar] [CrossRef]
- Ochterski, J.W.; Peterson, G.A. Complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density–Does the Difference Electron Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Varandas, A.J.C. Straightening the Hierarchical Staircase for Basis Set Extrapolations: A Low-Cost Approach to High-Accuracy Computational Chemistry. Annu. Rev. Phys. Chem. 2018, 69, 177–203. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Wilson, A.K.; Mourik, T.; Dunning, J.T.H. Gaussian Basis Sets for use in Correlated Molecular Calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. (Theochem.) 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Lesiuk, M.; Jeziorski, B.J. Complete Basis Set Extrapolation of Electronic Correlation Energies Using the Riemann Zeta Function. Chem. Theory Comput. 2019, 15, 5298–5403. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Martin, J.M.L.; de Oliveira, G. Toward standard methods for benchmark quality thermochemistry—W1 and W2 theory. J. Chem. Phys. 1999, 111, 1843–1856. [Google Scholar] [CrossRef] [Green Version]
- Parthiban, J.; Martin, J.M.L. Assessment of W1 and W2 theories for the computation of electron affinities, heats of formation, and proton affinities. J. Chem. Phys. 2001, 114, 6014–6029. [Google Scholar] [CrossRef] [Green Version]
- Tajti, A.; Szalay, P.G.; Császár, A.G.; Kállay, M.; Gauss, J.; Valeev, E.F.; Flowers, B.A.; Vázquez, J.; Stanton, J.F. HEAT: High accuracy extrapolated ab. initio. thermochemistry. J. Chem. Phys. 2004, 121, 11599. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Fradera, X.; Poater, J.; Simon, S.; Duran, M. Electron-pairing analysis from localization and delocalization indices in the framework of atoms-in-molecules theory. Theor. Chem. Acc. 2002, 108, 214–224. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of Hydrogen-Bond Interaction Potentials from the Electron Density: Integration of Noncovalent Interaction Regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narth, C.; Maroun, Z.; Boto, C.R.; Bonnet, M.L.; Piquemal, J.P.; Contreras-Garcia, J. A Complete NCI Perspective from new bonds to reactivity. In Applications of Topological Methods in Molecular Chemistry; Springer: Cham, Switzerland, 2016; Volume 22, pp. 491–527. [Google Scholar]
- Wilson, E.B.; Decius, J.C.; Cross, P.G. Molecular Vibrations: The Theory of Infrared and Raman Vibratioal Spectra; McGraw-Hill Book Co.: New York, NY, USA, 1955; Dover reissue, Courier Corporation 1980. [Google Scholar]
- Konkoli, J.Z.; Cremer, D. A new way of analyzing vibrational spectra. I. Derivation of adiabatic internal modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Kraka, E.; Zou, W.; Tao, Y. Decoding chemical information from vibrational spectroscopy data: Local vibrational mode theory. WIREs Comput. Mol. Sci. 2020, 34, 1480. [Google Scholar] [CrossRef]
- Freindorf, M.; Kraka, E.; Cremer, D. A comprehensive analysis of hydrogen bond interactions based on local vibrational modes. Int. J. Quantum Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E. Cremer D Local vibrational modes of the water dimer—Comparison of theory and experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local vibrational modes of the formic acid dimer—The strength of the double H-bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01. 2009. Available online: https://gaussian.com (accessed on 29 April 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, A.03. 2016. Available online: https://gaussian.com (accessed on 29 April 2021).
- Keith, T.A.; Todd, A.; Keith, T.K. Gristmill Software. AIMAll (Version 19.10.12). 2019. Available online: http://aim.tkgristmill.com (accessed on 29 April 2021).
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-Garcia, J. NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions. J. Chem. Theory Comput. 2020, 16, 4150–4158. Available online: http://www.lct.jussieu.fr/pagesperso/contrera/index-nci.html and https://github.com/juliacontrerasgarcia/nciplot (accessed on 29 April 2021). [CrossRef]
- Zou, W.; Tao, Y.; Freindorf, M.; Makos, M.; Verma, N.; Kraka, E. LMODEA2020; Southern Methodist University: Dallas, TX, USA, 2020. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No H Bonding | CH…O or CH…N | OH…O, OH…N, NH…O |
|---|---|---|
| THREE (+62) EIGHT (+101) | SIX (+43) SEVEN (+38) NINE (+91) TEN (61) | ONE (4.0) TWO (0.0) FOUR (+42) FIVE (+38) ELEVEN (+48) |
| Species | Interaction | ρ | ∇2ρ | H | V | Q(H) | Order | DI |
|---|---|---|---|---|---|---|---|---|
| ONE | C=O…HN | 0.0241 | 0.0931 | 0.00640 | −0.0177 | 0.4636 | 0.164 | 0.0642 |
| TWO | -OH…O= | 0.0615 | 0.1158 | −0.01860 | −0.0661 | 0.3273 | 0.221 | 0.1254 |
| THREE | NCI | 0.0110 | 0.0564 | −0.0080 | 0.5876 | |||
| FOUR | HO…HN | 0.0218 | 0.0974 | 0.00390 | −0.0167 | 0.4485 | 0.158 | 0.0541 |
| FIVE | −OH…NH2 | 0.0324 | 0.0870 | −0.00220 | −0.0262 | 0.3661 | 0.180 | 0.0831 |
| SIX | CH…O= | 0.0202 | 0.0782 | 0.00300 | −0.0135 | 0.1164 | 0.155 | 0.0715 |
| SEVEN | CH…NH2 | 0.0140 | 0.0517 | 0.00230 | −0.0084 | 0.0604 | 0.138 | 0.0531 |
| EIGHT | NCI | 0.0133 | 0.0560 | −0.0081 | 0.5978 | |||
| NINE | CH…OH | 0.0114 | 0.0532 | 0.00260 | −0.0081 | 0.0140 | 0.129 | 0.0384 |
| TEN | CH…N | 0.0137 | 0.0511 | 0.00220 | −0.0083 | 0.0434 | 0.142 | 0.0276 |
| ELEVEN | =O…HN | 0.0391 | 0.1192 | −0.00260 | −0.0350 | 0.5022 | 0.191 | 0.1003 |
| Species | NH2 Stretches | Double Bond Stretches | OH Stretches | ||
|---|---|---|---|---|---|
| ONE | 3725a | 3566s | 1790 1 | 1757 2 | None |
| TWO | 3777a | 3630s | 1725 | 1710 | 3087 |
| THREE | 3747a | 3609s | 1782 | 1748 | 3836 |
| FOUR | 3777a | 3626s | 1773 | 1736 | 3853 |
| FIVE | 3659s | 3537s | 1780 | 1704 | 3571 |
| SIX | 3766a | 3622s | 1771 | 1742 | 3842 |
| SEVEN | 3726a | 3591s | 1731 | 1772 | 3850 |
| EIGHT | 3679a | 3559s | 1763 | 1714 | 3807 |
| NINE | 3673a | 3554s | 1712 | 1750 | 3803 |
| TEN | 3726a | 3591s | 1772 | 1731 | 3850 |
| ELEVEN | 3655a | 3347s | 1685 | 1742 | 3818 |
| Species | CH3 Stretches4 | CCC Bend5 | CO Stretch | ||
| ONE | 3208s | 3157a | 3076b | 236 | None |
| TWO | 3198s | 3164a | 3084b | 246 | 1356 |
| THREE | 3202s | 3155a | 3074b | 178 | 1385 |
| FOUR | 3198a | 3143s | 3063b | 226 | 1347 |
| FIVE | 3203s | 3162a | 3084b | 226 | 1317 |
| SIX | 3213s | 3135a | 3062b | 226 | 1348, 1267 |
| SEVEN | 3213s | 3132a | 3063b | 193 | 1351, 1273 |
| EIGHT | 3202s | 3155a | 3074b | 178 | 1467 |
| NINE | 3205s | 3175a | 3089b | 207 | 1435 |
| TEN | 3214a | 3132s | 3063b | 194 | 1273 |
| ELEVEN | 3204s | 3161a | 3079b | 242 | 1367 |
| Species | C=O | C=O′ | NH | NH′ | C-H | C-H′ | C-H″ | H…O |
|---|---|---|---|---|---|---|---|---|
| ONE | 11.7 1700 | 11.7 1699 | 7.4 3654 | 7.0 3564 | 5.5 3181 | 5.3 3116 | 5.3 3112 | 0.161 537 |
| Species | C=O | C-O | NH | NH′ | C-H | C-H′ | C-H″ | O-H | X…Y 1 |
|---|---|---|---|---|---|---|---|---|---|
| TWO | 9.7 1552 | 6.5 1273 | 7.4 3651 | 7.0 3670 | 5.5 3169 | 5.4 3130 | 5.4 3130 | 4.3 2801 | 0.323 760 |
| THREE | 11.7 1701 | 6.2 1237 | 7.3 3641 | 7.3 3630 | 5.5 3174 | 5.3 3103 | 5.3 3103 | 8.2 3829 | NCI |
| FOUR | 11.4 1678 | 5.6 1177 | 7.6 3666 | 7.6 3661 | 5.5 3178 | 5.3 3099 | 5.3 3099 | 8.3 3848 | 0.150 764 |
| FIVE | 11.8 1708 | 6.0 1214 | 7.1 3578 | 7.0 3560 | 5.5 3176 | 5.4 3129 | 5.4 3127 | 6.7 3475 | 0.201 712 |
| SIX | 11.3 1671 | 5.8 1202 | 7.4 3657 | 7.4 3644 | 5.6 3186 | 5.3 3100 | 5.3 3100 | 8.2 3836 | 0.142 732 |
| SEVEN | 11.6 1694 | 5.8 1195 | 7.2 3616 | 7.3 3631 | 5.6 3191 | 5.3 3098 | 5.3 3098 | 8.2 3844 | 0.109 612 |
| EIGHT | 11.6 1694 | 5.8 1253 | 7.0 3557 | 7.2 3610 | 5.5 3176 | 5.3 3113 | 5.3 3113 | 8.0 3757 | NCI |
| NINE | 11.2 1666 | 6.0 1219 | 7.0 3553 | 7.2 3606 | 5.5 3172 | 5.4 3127 | 5.4 3127 | 8.0 3789 | 0.093 711 |
| TEN | 11.2 1664 | 5.8 1197 | 6.8 3511 | 6.8 3598 | 5.2 3176 | 5.0 3022 | 5.2 2995 | 7.8 3728 | 0.115 688 |
| ELEVEN | 9.9 1566 | 6.0 1219 | 7.2 3594 | 5.7 3212 | 5.2 3176 | 5.2 3076 | 5.2 3076 | 8.1 3806 | 0.231 804 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altun, Z.; Bleda, E.A.; Trindle, C. Focal Point Evaluation of Energies for Tautomers and Isomers for 3-hydroxy-2-butenamide: Evaluation of Competing Internal Hydrogen Bonds of Types -OH…O=, -OH…N, -NH…O=, and CH…X (X=O and N). Molecules 2021, 26, 2623. https://doi.org/10.3390/molecules26092623
Altun Z, Bleda EA, Trindle C. Focal Point Evaluation of Energies for Tautomers and Isomers for 3-hydroxy-2-butenamide: Evaluation of Competing Internal Hydrogen Bonds of Types -OH…O=, -OH…N, -NH…O=, and CH…X (X=O and N). Molecules. 2021; 26(9):2623. https://doi.org/10.3390/molecules26092623
Chicago/Turabian StyleAltun, Zikri, Erdi Ata Bleda, and Carl Trindle. 2021. "Focal Point Evaluation of Energies for Tautomers and Isomers for 3-hydroxy-2-butenamide: Evaluation of Competing Internal Hydrogen Bonds of Types -OH…O=, -OH…N, -NH…O=, and CH…X (X=O and N)" Molecules 26, no. 9: 2623. https://doi.org/10.3390/molecules26092623