
Spectroscopic Identification of Hydrogen Bond Vibrations and Quasi-Isostructural Polymorphism in N-Salicylideneaniline

 , , , and
, , , and
Abstract
: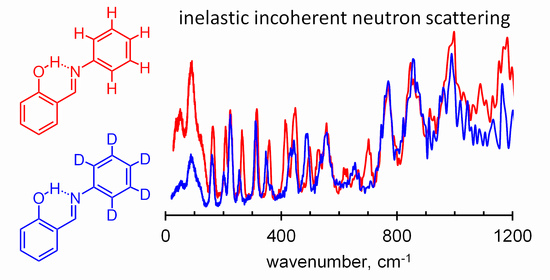
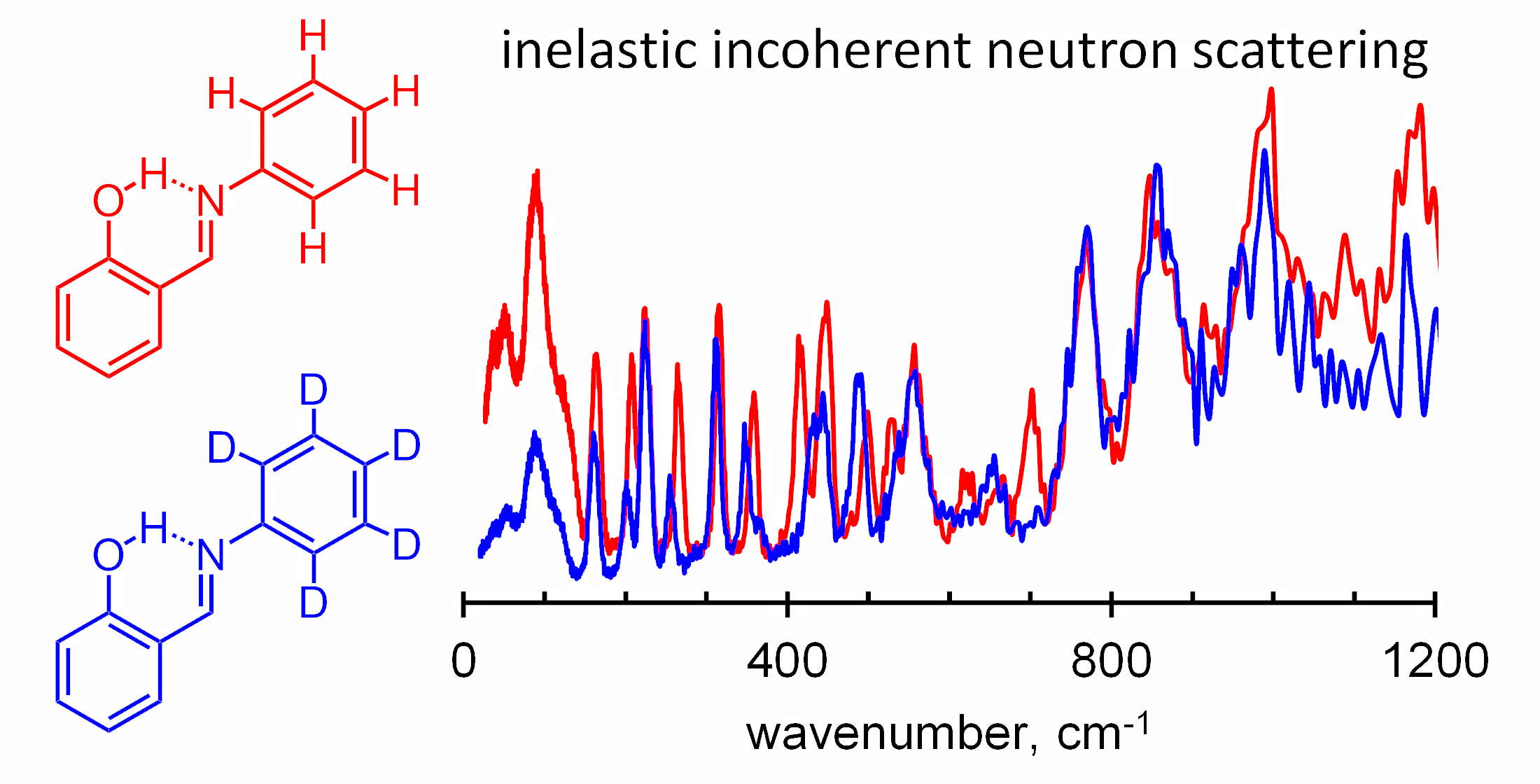{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
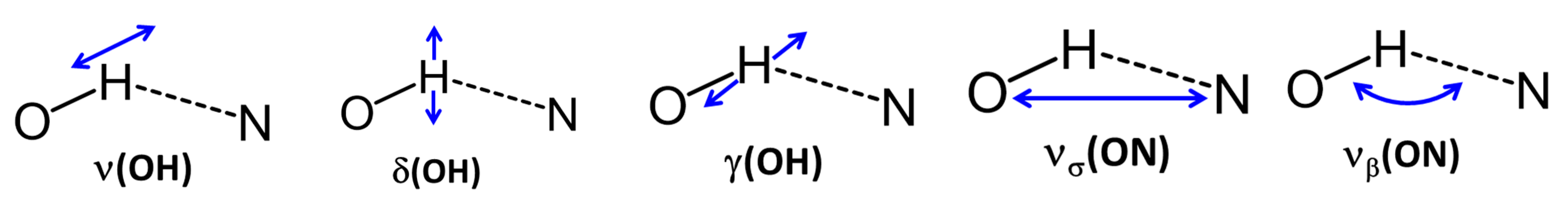{kind=link}
1. Introduction
2. Results and Discussion
2.1. Assignment of Hydrogen Bonding Vibrations
2.2. Spectral Manifestations of Polymorphism
2.3. X-ray Powder Diffraction (XPD) Study of Polymorphism in SA
3. Materials and Methods
3.1. Compounds and Deuteration
3.2. Infrared, Raman and IINS Measurements
3.3. X-ray Powder Diffraction
3.4. Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schiff, H. Mittheilungen aus dem Universitätslaboratorium in Pisa Eine neue Reihe organischer Basen. Justus Liebigs Ann. Chem. 1864, 131, 118–119. [Google Scholar] [CrossRef] [Green Version]
- Tidwell, T.T. Hugo (Ugo) Schiff, Schiff Bases, and a Century of β-Lactam Synthesis. Angew. Chem. 2008, 47, 1016–1020. [Google Scholar] [CrossRef]
- Anselrnino, O. Isomere Schiff’sche Basen. Ber. Dtsch. Chem. Ges. 1905, 38, 3989–3997. [Google Scholar] [CrossRef] [Green Version]
- Carletta, A.; Dubois, J.; Tilborg, A.; Wouters, J. Solid-state investigation on a new dimorphic substituted N-salicylidene compound: Insights into its thermochromic behaviour. CrystEngComm 2015, 17, 3509–3518. [Google Scholar] [CrossRef]
- Ossowicz, P.; Janus, E.; Szady-Chelmieniecka, A.; Rozwadowski, Z. Influence of modification of the amino acids ionic liquids on their physico-chemical properties: Ionic liquids versus ionic liquids-supported Schiff bases. J. Mol. Liq. 2016, 224, 211–218. [Google Scholar] [CrossRef]
- Liu, H.; Ye, K.; Zhang, Z.; Zhang, H. An Organic Crystal with High Elasticity at an Ultra-Low Temperature (77 K) and Shape ability at High Temperatures. Angew. Chem. Int. Ed. 2019, 58, 19081–19086. [Google Scholar] [CrossRef]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef]
- Kawato, T.; Koyama, H.; Kanatomi, H.; Shigemizu, H. Difference in the Rate of Photo-induced Unimolecular Motion of Chiral Salicylideneamines in the Chiral Crystal Environments. Chem. Lett. 1997, 26, 401–402. [Google Scholar] [CrossRef]
- Koshima, H.; Matsuo, R.; Matsudomi, M.; Uemura, Y.; Shiro, M. Light-Driven Bending Crystals of Salicylidenephenylethylamines in Enantiomeric and Racemate Forms. Cryst. Growth Des. 2013, 13, 4330–4337. [Google Scholar] [CrossRef]
- Takanabe, A.; Tanaka, M.; Johmoto, K.; Uekusa, H.; Mori, T.; Koshima, H.; Asahi, T. Optical Activity and Optical Anisotropy in Photomechanical Crystals of Chiral Salicylidenephenylethylamines. J. Am. Chem. Soc. 2016, 45, 15066–15077. [Google Scholar] [CrossRef]
- Centore, R.; Jazbinsek, M.; Tuzi, A.; Roviello, A.; Capobianco, A.; Peluso, A. A series of compounds forming polar crystals and showing single-crystal-to-single-crystal transitions between polar phases. CrystEngComm 2012, 14, 2645–2653. [Google Scholar] [CrossRef]
- Takanabe, A.; Katsufuji, T.; Johmoto, K.; Uekusa, H.; Shiro, M.; Koshima, H.; Asahi, T. Reversible Single-Crystal-to-Single-Crystal Phase Transition of Chiral Salicylidenephenylethylamine. Crystals 2017, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.D.; Schmidt, G.M. Photochromy and thermochromy of anils. J. Phys. Chem. 1962, 66, 2442–2446. [Google Scholar] [CrossRef]
- Riddle, J.A.; Bollinger, J.C.; Lee, D. Escape from a Nonporous Solid: Mechanically Coupled Biconcave Molecules. Angew. Chem. Int. Ed. 2005, 44, 6689–6693. [Google Scholar] [CrossRef]
- Jia, Y.; Li, J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem. Rev. 2015, 115, 1597–1621. [Google Scholar] [CrossRef]
- Kandappa, S.K.; Valloli, L.K.; Ahuja, S.; Parthiban, J.; Sivaguru, J. Taming the excited state reactivity of imines—from non-radiative decay to aza Paternó–Büchi reaction. Chem. Soc. Rev. 2021, 50, 1617–1641. [Google Scholar] [CrossRef] [PubMed]
- Muriel, W.A.; Botero-Cadavid, J.F.; Cardenas, C.; Rodriguez-Cordoba, W. A theoretical study of the photodynamics of salicylidene-2-anthrylamine in acetonitrile solution. Phys. Chem. Chem. Phys. 2018, 20, 29399–29411. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, E.; Cintas, P.; Light, M.E.; Palacios, J.C. Electronic effects in tautomeric equilibria: The case of chiral imines from d-glucamine and 2-hydroxyacetophenones. Org. Biomol. Chem. 2019, 17, 10209–10222. [Google Scholar] [CrossRef]
- Filarowski, A.; Lopatkova, M.; Lipkowski, P.; Van der Auwerar, M.; Leen, V.; Dehaen, W. Solvatochromism of BODIPY-Schiff base Dye. J. Phys. Chem. B 2015, 119, 2576–2584. [Google Scholar] [CrossRef]
- Cohen, M.D.; Schmidt, G.M.J.; Flavian, S. Topochemistry. Part VI. Experiments on photochromy and thermochromy of crystalline anils of salicylaldehydes. J. Chem. Soc. 1964, 2041–2051. [Google Scholar] [CrossRef]
- Andes, R.V.; Manikowski, D.M. Photochromism of Salicylidene Aniline. Appl. Opt. 1968, 7, 1179–1183. [Google Scholar] [CrossRef]
- Destro, R.; Gavezzotti, A.; Simonetta, M. Salicylideneaniline. Acta Crystallogr. 1978, 34, 2867–2869. [Google Scholar] [CrossRef]
- Hadjoudis, E.; Vittorakis, M.; Moustakali-Mavridis, I. Photochromism of organic compounds in the crystal state. Tetrahedron 1987, 43, 1345–1360. [Google Scholar] [CrossRef]
- Ogawa, K.; Kasahara, Y.; Ohtani, Y.; Harada, J. Crystal Structure Change for the Thermochromy of N-Salicylideneanilines. The First Observation by X-ray Diffraction. J. Am. Chem. Soc. 1998, 120, 7107–7108. [Google Scholar]
- Gao, A.-H.; Wang, M.-S. Nonadiabatic ab initio molecular dynamics study of photoisomerization in N-salicilydenemethylfurylamine (SMFA). J. Chem. Phys. 2017, 146, 124312. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.S.M.; Mal, S.; Singh, P. Photochromism in Anils—A Review. Open Chem. J. 2015, 2, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Arod, F.; Pattison, P.; Schenk, K.J.; Chapuis, G. Polymorphism in N-Salicylideneaniline Reconsidered. Cryst. Growth Des. 2007, 7, 1679–1685. [Google Scholar] [CrossRef]
- Arod, F.; Gardon, M.; Pattison, P.; Chapuis, G. The α2-polymorph of salicylideneaniline. Acta Crystallogr. 2005, 61, o317–o320. [Google Scholar] [CrossRef] [Green Version]
- Grzegorzek, J.; Filarowski, A.; Mielke, Z. The photoinduced isomerization and its implication in the photo-dynamical processes in two simple Schiff bases isolated in solid argon. Phys. Chem. Chem. Phys. 2011, 13, 16596–16605. [Google Scholar] [CrossRef]
- Grzegorzek, J.; Mielke, Z.; Filarowski, A. C=N–N=C conformational isomers of 2’-hydroxyacetophenone azine: FTIR matrix isolation and DFT study. J. Mol. Struct. 2010, 976, 371–376. [Google Scholar] [CrossRef]
- Sıdır, İ.; Gülseven Sıdır, Y.; Góbi, S.; Berber, H.; Fausto, R. Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol. Molecules 2021, 26, 2814. [Google Scholar] [CrossRef] [PubMed]
- Majerz, I.; Pawlukojć, A.; Sobczyk, L.; Dziembowska, T.; Grech, E.; Szady-Chełmieniecka, A. The infrared, Raman and inelastic neutron scattering studies on 5-nitro-N-salicylideneethylamine. J. Mol. Struct. 2000, 552, 243–247. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Lipkowski, P.; Pawlukojć, A. Inelastic neutron scattering and vibrational spectra of 2-(N-methyl-α-iminoethyl)-phenol and 2-(N-methyliminoethyl)-phenol: Experimental and theoretical approach. J. Mol. Struct. 2008, 880, 97–108. [Google Scholar] [CrossRef]
- Moosavi-Tekyeh, Z.; Dastani, N. Intramolecular hydrogen bonding in N-salicylideneaniline: FT-IR spectrum and quantum chemical calculations. J. Mol. Struct. 2015, 1102, 314–322. [Google Scholar] [CrossRef]
- Pająk, J.; Maes, G.; De Borggraeve, W.M.; Boens, N.; Filarowski, A. Matrix-isolation FT-IR and theoretical investigation of the vibrational properties of the sterically hindered ortho-hydroxy acylaromatic Schiff bases. J. Mol. Struct. 2007, 844–845, 83–93. [Google Scholar] [CrossRef]
- Dziembowska, T.; Szafran, M.; Katrusiak, A.; Rozwadowski, Z. Crystal structure of and solvent effect on tautomeric equilibrium in Schiff base derived from 2-hydroxy-1-naphthaldehyde and methylamine studied by X-ray diffraction, DFT, NMR and IR methods. J. Mol. Struct. 2009, 929, 32–42. [Google Scholar] [CrossRef]
- Sobczyk, L.; Obrzud, M.; Filarowski, A. H/D Isotope Effects in Hydrogen Bonded Systems. Molecules 2013, 18, 4467–4476. [Google Scholar] [CrossRef] [Green Version]
- Kwocz, A.; Panek, J.J.; Jezierska, A.; Hetmańczyk, Ł.; Pawlukojć, A.; Kochel, A.; Lipkowski, P.; Filarowski, A. A molecular roundabout: Triple cycle-arranged hydrogen bonds in light of experiment and theory. New J. Chem. 2018, 42, 19467–19477. [Google Scholar] [CrossRef]
- Zundel, G. Easily Polarizable Hydrogen Bonds—Their Interactions with the Environment—IR Continuum and Anomalous Large Conductivity. In The Hydrogen Bond: Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland: Amsterdam, The Netherland, 1976; Volume 2, pp. 683–766. [Google Scholar]
- Shigorin, D.N. Vodorodnaya Svyaz’ (Hydrogen Bond); Sokolov, N.D., Chulanovsky, A.D., Eds.; Nauka: Moscow, Russia, 1964; p. 195. [Google Scholar]
- Hadži, D.; Bratos, S. Vibrational spectroscopy of the hydrogen bond. In The Hydrogen Bond: Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland: Amsterdam, The Netherlands, 1976; Volume 2, pp. 565–611. [Google Scholar]
- Tsubomura, H. Nature of the Hydrogen Bond. III. The Measurement of the Infrared Absorption Intensities of Free and Hydrogen-Bonded OH Bands. Theory of the Increase of the Intensity Due to the Hydrogen Bond. J. Chem. Soc. 1956, 24, 927. [Google Scholar] [CrossRef]
- Hadzi, D. Absorption spectra and structure of some solid hydroxyazo-compounds. J. Chem. Soc. 1956, 2143–2150. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A. Intergrated intensity of νs(OH) absorption bands in bent hydrogen bonds in ortho-dialkylaminomethyl phenols. Vib. Spectrosc. 1996, 12, 15–24. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A. Specificity of the intramolecular hydrogen bond. The differences in spectroscopic characteristics of the intermolecular and intramolecular H-bonds. Vib. Spectrosc. 1998, 17, 123–131. [Google Scholar] [CrossRef]
- Takasuka, M.; Matsui, Y. Experimental Observations and CN D0/2 Calculations for Hydroxy Stretching Frequency Shifts, Intensities, and Hydrogen Bond Energies of Intramolecular Hydrogen Bonds in ortho-Substituted Phenols. J. Chem. Soc. Perkin II 1979, 1743–1750. [Google Scholar] [CrossRef]
- Nguyen, Y.H.; Lampkin, B.J.; Venkatesh, A.; Ellern, A.; Rossini, A.J.; VanVeller, B. Open-Resonance-Assisted Hydrogen Bonds and Competing Quasiaromaticity. J. Org. Chem. 2018, 83, 9850–9857. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Hetmańczyk, Ł.; Szklarz, P.; Kwocz, A.; Wierzejewska, M.; Pagacz-Kostrzewa, M.; Melnikov, M.Y.; Tolstoy, P.M.; Filarowski, A. Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions. Molecules 2021, 26, 3109. [Google Scholar] [CrossRef]
- Piękoś, P.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Pozharskii, A.F.; Antonov, A.S.; Tolstoy, P.M.; Filarowski, A. Symmetry/asymmetry of the NHN hydrogen bond in protonated 1,8-bis(dimethylamino)naphthalene. Symmetry 2020, 12, 1924. [Google Scholar] [CrossRef]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]
- Prohens, R.; Barbas, R.; Font-Bardia, M. Morphotropism and “Quasi-Isostructurality” in the Three High Z′ Concomitant Polymorphs of Efinaconazole. Cryst. Grow. Des. 2020, 20, 4238−4242. [Google Scholar] [CrossRef]
- Kálmán, A.; Párkányi, L.; Argay, G. Classification of the isostructurality of organic molecules in the crystalline state. Acta Cryst. 1993, 49, 1039–1049. [Google Scholar] [CrossRef]
- Dey, D.; Thomas, S.P.; Spackman, M.A.; Chopra, D. Quasi-isostructural polymorphism’ in molecular crystals: Inputs from interaction hierarchy and energy frameworks. Chem. Commun. 2016, 52, 2141–2144. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Lima, J.A.; Freire, P.T.C.; Melo, F.E.A.; Havenith, R.W.A.; Filho, J.M.; Broer, R.; Eckert, J.; Bordallo, H.N. Molecular flexibility and structural instabilities in crystalline L-methionine. Biophys. Chem. 2013, 180–181, 76–85. [Google Scholar] [CrossRef]
- De Souza, J.M.; Freire, P.T.C.; Argyriou, D.N.; Stride, J.A.; Barthòs, M.; Kalceff, W.; Bordallo, H.N. Raman and Neutron Scattering Study of Partially Deuterated l-Alanine: Evidence of a Solid-Solid Phase Transition. ChemPhysChem 2009, 10, 3337–3343. [Google Scholar] [CrossRef]
- Hetmańczyk, J.; Hetmańczyk, Ł.; Bilski, P.; Kozak, A. Complex water dynamics in crystalline [Ca(H2O)2](ReO4)2, studied by the vibrational spectroscopy and proton magnetic resonance. J. Mol. Struct. 2020, 1205, 127610. [Google Scholar] [CrossRef]
- Szostak, E.; Hetmańczyk, J.; Migdał-Mikuli, A. Inelastic and elastic neutron scattering studies of the vibrational and reorientational dynamics, crystal structure and solid–solid phase transition in [Mn(OS(CH3)2)6](ClO4)2 supported by theoretical (DFT) calculations. Spectrochim. Acta A 2015, 145, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Hetmańczyk, J.; Hetmańczyk, Ł.; Nowicka-Scheibe, J.; Pawlukojć, A.; Maurin, J.K.; Schilf, W. Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid. Materials 2021, 14, 3292. [Google Scholar] [CrossRef] [PubMed]
- Shapenova, D.S.; Shiryaev, A.A.; Bolte, M.; Kukułka, M.; Szczepanik, D.W.; Hooper, J.; Babashkina, M.G.; Mahmoudi, G.; Mitoraj, M.P.; Safin, D.A. Resonance Assisted Hydrogen Bonding Phenomenon Unveiled from Both Experiment and Theory—An Example of New Family of Ethyl N-salicylideneglycinate Dyes. Chem. Eur. J. 2020, 26, 12987–12995. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Babashkina, M.G.; Filinchuk, Y.; Rotaru, A.; Jureschi, C.; Mitoraj, M.P.; Hooper, J.; Brela, M.; Garcia, Y. Polymorphism driven optical properties of an anil dye. CrystEngComm 2016, 18, 7249–7259. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Van Alsenoy, C. GAR2PED; University of Antwerp: Antwerpen, Belgium, 1995. [Google Scholar]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V.R.; Zicovich-Wilson, C.M. CRYSTAL: A computational tool for the ab initio study of the electronic properties of crystals. Z. Kristallogr. 2005, 220, 571–573. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL09 User’s Manual; University of Torino: Torino, Italy, 2009. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Vener, M.V. Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds. Symmetry 2021, 13, 425. [Google Scholar] [CrossRef]
- Vener, M.V.; Sauer, J. Environmental effects on vibrational proton dynamics in H5O2+: DFT study on crystalline H5O2+ClO4−. Phys. Chem. Chem. Phys. 2005, 7, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Mitev, P.D.; Eriksson, A.; Hermansson, K. H-bond and electric field correlations for water in highly hydrated crystals. Int. J. Quantum Chem. 2016, 116, 67–80. [Google Scholar] [CrossRef]
- Červinka, C.; Fulem, M. Cohesive properties of the crystalline phases of twenty proteinogenic α-aminoacids from first-principles calculations. Phys. Chem. Chem. Phys. 2019, 21, 18501–18515. [Google Scholar] [CrossRef] [PubMed]
- Beran, G.J.O. Modeling Polymorphic Molecular Crystals with Electronic Structure Theory. Chem. Rev. 2016, 116, 5567–5613. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Manin, A.N.; Voronin, A.P.; Churakov, A.V.; Perlovich, G.L.; Vener, M.V. Weak Interactions Cause Packing Polymorphism in Pharmaceutical Two-Component crystals. The case study of the Salicylamide Cocrystal. Cryst. Growth Des. 2017, 17, 1425–1437. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hetmańczyk, Ł.; Goremychkin, E.A.; Waliszewski, J.; Vener, M.V.; Lipkowski, P.; Tolstoy, P.M.; Filarowski, A. Spectroscopic Identification of Hydrogen Bond Vibrations and Quasi-Isostructural Polymorphism in N-Salicylideneaniline. Molecules 2021, 26, 5043. https://doi.org/10.3390/molecules26165043
Hetmańczyk Ł, Goremychkin EA, Waliszewski J, Vener MV, Lipkowski P, Tolstoy PM, Filarowski A. Spectroscopic Identification of Hydrogen Bond Vibrations and Quasi-Isostructural Polymorphism in N-Salicylideneaniline. Molecules. 2021; 26(16):5043. https://doi.org/10.3390/molecules26165043
Chicago/Turabian StyleHetmańczyk, Łukasz, Eugene A. Goremychkin, Janusz Waliszewski, Mikhail V. Vener, Paweł Lipkowski, Peter M. Tolstoy, and Aleksander Filarowski. 2021. "Spectroscopic Identification of Hydrogen Bond Vibrations and Quasi-Isostructural Polymorphism in N-Salicylideneaniline" Molecules 26, no. 16: 5043. https://doi.org/10.3390/molecules26165043