Feature Papers in Epigenomes
A topical collection in Epigenomes (ISSN 2075-4655).
Viewed by 13162
Share This Topical Collection
Editors
 Dr. Ivana De la Serna
Dr. Ivana De la Serna
Dr. Ivana De la Serna
E-Mail
Website
Collection Editor
Cancer Biology, College of Medicine and Life Sciences, University of Toledo, Toledo, OH 43614, USA
Interests: SWI/SNF chromatin remodeling enzymes; melanocyte differentiation and regulation of pigmentation; epigenetic regulation of the response to ultraviolet radiation; cancer epigenetics
Special Issues, Collections and Topics in MDPI journals
 Prof. Dr. Che-Kun James Shen
Prof. Dr. Che-Kun James Shen
Prof. Dr. Che-Kun James Shen
E-Mail
Website
Collection Editor
1. The PhD Program for Neural Regenerative Medicine, Taipei Medical University, Taipei 115, Taiwan
2. Institute of Molecular Biology, Academia Sinica, Taipei 115, Taiwan
Interests: gene regulation; chromatin; DNA methylation; neurofunction; transcription
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
This Topical Collection aims to collect high-quality papers on epigenetics and epigenomics. We encourage researchers and experts from various fields within the journal’s scope to contribute papers that highlight the latest developments in their research field. The potential topics of this Special Issue include, but are not limited to, the following:
- Functional epigenomic studies;
- Genome-wide epigenetic status and the regulation of cells or tissues;
- Chromatin modifications and remodeling in diseases;
- Epigenetics in physical diseases;
- Environmental changes in the epigenetic status of cells or tissues;
- The inheritance or fixation of epigenetic characteristics;
- The description of novel methods to study epigenetic regulation;
- Novel tools, protocols, and technologies for epigenetic studies and therapeutics;
- Long noncoding RNA (LncRNA), microRNA (miR), and chromatin crosstalk.
Dr. Ivana De la Serna
Prof. Dr. Che-Kun James Shen
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Epigenomes is an international peer-reviewed open access quarterly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 1500 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- epigenomic
- epigenetic
- chromatin modification
- DNA methylation
- DNA methyltransferases
- cancer epigenetics
- histones
Published Papers (8 papers)
Open AccessArticle
The Role of Different TET Proteins in Cytosine Demethylation Revealed by Mathematical Modeling
by
Karolina Kurasz, Joanna Rzeszowska-Wolny, Ryszard Oliński, Marek Foksiński and Krzysztof Fujarewicz
Viewed by 195
Abstract
In living cells, some reactions can be conducted by more than one enzyme and sometimes it is difficult to establish which enzyme is responsible. Such is the case with proteins from the TET family, capable of converting 5-methyl-2’-deoxycytidine (5-
)
[...] Read more.
In living cells, some reactions can be conducted by more than one enzyme and sometimes it is difficult to establish which enzyme is responsible. Such is the case with proteins from the TET family, capable of converting 5-methyl-2’-deoxycytidine (5-
) in DNA to 5-(hydroxymethyl)-2’-deoxycytidine (5-
) and further to 5-formyl-2’-deoxycytidine (5-
) and 5-carboxy-2’-deoxycytidine (5-
). The estimation of the efficiency of particular TETs in particular oxidative reactions and different cell types is important but experimentally difficult. Here, we propose an approach with mathematical modeling in which methylation and known deoxycytidine modification pathways are presented by 343 possible model versions with assumed different combinations of TET1, 2, and 3 activities in different pathways. Model parameters were calculated on the basis of 5-
, 5-
, 5-
, 5-
, and 5-
levels experimentally assessed in five human cultured cell lines and previously published. Selection of the model versions that give in simulations the best average fit to experimental data suggested that not all TET proteins participate in all modification reactions and that TET3 activity may be especially important in the reaction of 5-
removal.
Full article
►▼
Show Figures
Open AccessArticle
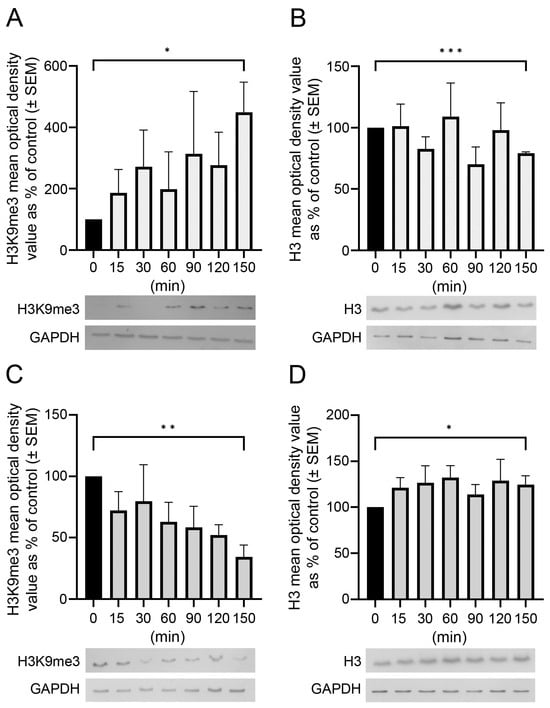
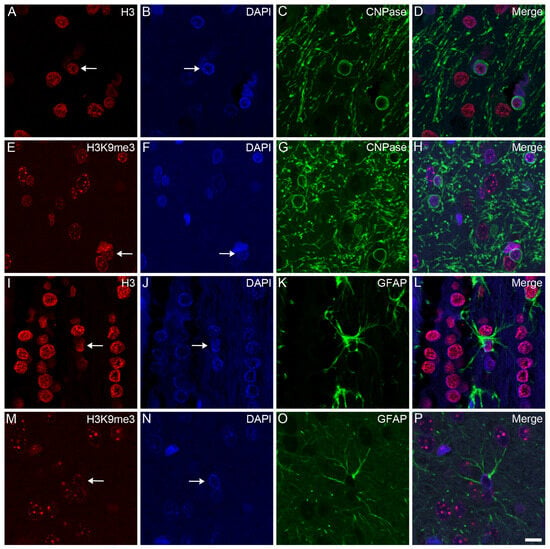
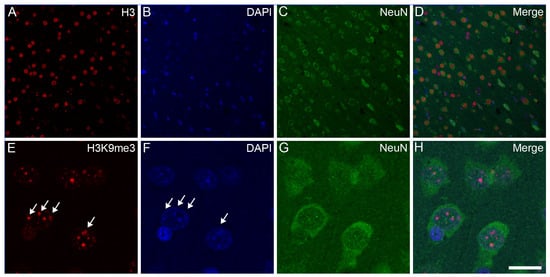
Opposite and Differently Altered Postmortem Changes in H3 and H3K9me3 Patterns in the Rat Frontal Cortex and Hippocampus
by
Karolina Dulka, Noémi Lajkó, Kálmán Nacsa and Karoly Gulya
Viewed by 1113
Abstract
Temporal and spatial epigenetic modifications in the brain occur during ontogenetic development, pathophysiological disorders, and aging. When epigenetic marks, such as histone methylations, in brain autopsies or biopsy samples are studied, it is critical to understand their postmortem/surgical stability. For this study, the
[...] Read more.
Temporal and spatial epigenetic modifications in the brain occur during ontogenetic development, pathophysiological disorders, and aging. When epigenetic marks, such as histone methylations, in brain autopsies or biopsy samples are studied, it is critical to understand their postmortem/surgical stability. For this study, the frontal cortex and hippocampus of adult rats were removed immediately (controls) or after a postmortem delay of 15, 30, 60, 90, 120, or 150 min. The patterns of unmodified H3 and its trimethylated form H3K9me3 were analyzed in frozen samples for Western blot analysis and in formalin-fixed tissues embedded in paraffin for confocal microscopy. We found that both the unmodified H3 and H3K9me3 showed time-dependent but opposite changes and were altered differently in the frontal cortex and hippocampus with respect to postmortem delay. In the frontal cortex, the H3K9me3 marks increased approximately 450% with a slow parallel 20% decrease in the unmodified H3 histones after 150 min. In the hippocampus, the change was opposite, since H3K9me3 marks decreased steadily by approximately 65% after 150 min with a concomitant rapid increase of 20–25% in H3 histones at the same time. Confocal microscopy located H3K9me3 marks in the heterochromatic regions of the nuclei of all major cell types in the control brains: oligodendrocytes, astrocytes, neurons, and microglia. Therefore, epigenetic marks could be affected differently by postmortem delay in different parts of the brain.
Full article
►▼
Show Figures
Open AccessArticle
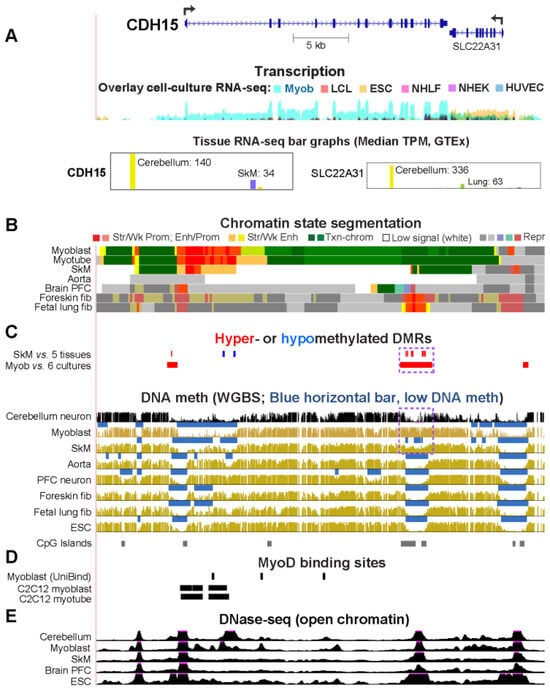
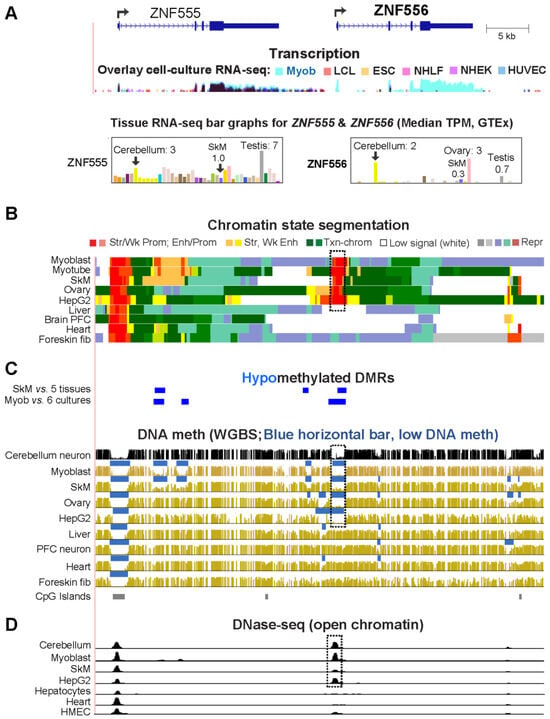
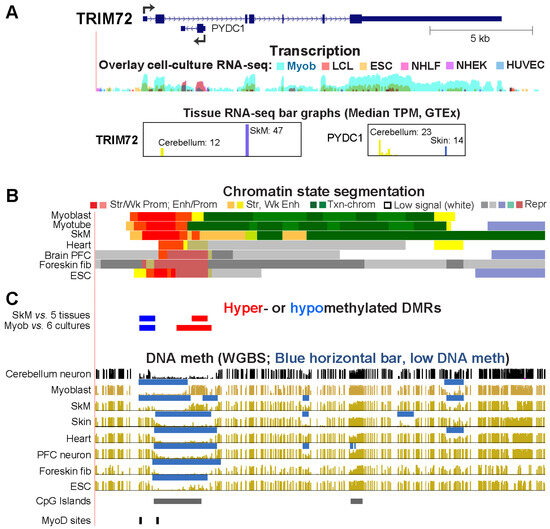
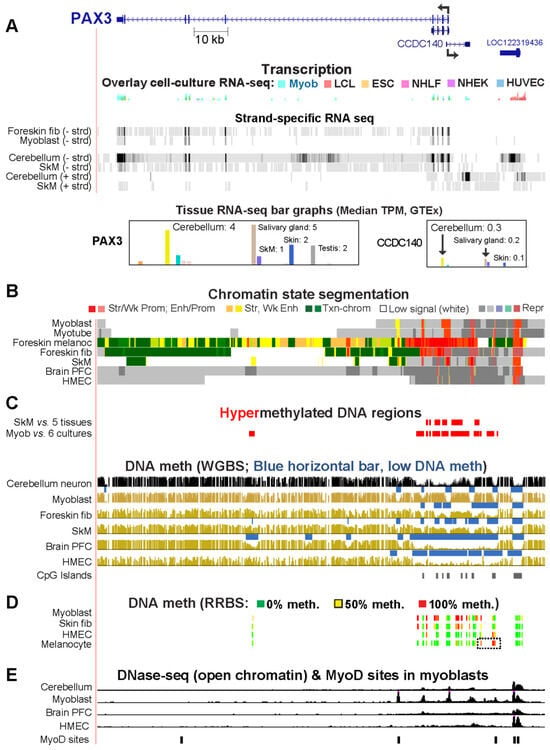
Epigenetics of Genes Preferentially Expressed in Dissimilar Cell Populations: Myoblasts and Cerebellum
by
Melanie Ehrlich, Kenneth C. Ehrlich, Michelle Lacey, Carl Baribault, Sagnik Sen, Pierre-Olivier Estève and Sriharsa Pradhan
Viewed by 1854
Abstract
While studying myoblast methylomes and transcriptomes, we found that
CDH15 had a remarkable preference for expression in both myoblasts and cerebellum. To understand how widespread such a relationship was and its epigenetic and biological correlates, we systematically looked for genes with similar transcription
[...] Read more.
While studying myoblast methylomes and transcriptomes, we found that
CDH15 had a remarkable preference for expression in both myoblasts and cerebellum. To understand how widespread such a relationship was and its epigenetic and biological correlates, we systematically looked for genes with similar transcription profiles and analyzed their DNA methylation and chromatin state and accessibility profiles in many different cell populations. Twenty genes were expressed preferentially in myoblasts and cerebellum (Myob/Cbl genes). Some shared DNA hypo- or hypermethylated regions in myoblasts and cerebellum. Particularly striking was
ZNF556, whose promoter is hypomethylated in expressing cells but highly methylated in the many cell populations that do not express the gene. In reporter gene assays, we demonstrated that its promoter’s activity is methylation sensitive. The atypical epigenetics of
ZNF556 may have originated from its promoter’s hypomethylation and selective activation in sperm progenitors and oocytes. Five of the Myob/Cbl genes (
KCNJ12,
ST8SIA5,
ZIC1,
VAX2, and
EN2) have much higher RNA levels in cerebellum than in myoblasts and displayed myoblast-specific hypermethylation upstream and/or downstream of their promoters that may downmodulate expression. Differential DNA methylation was associated with alternative promoter usage for Myob/Cbl genes
MCF2L,
DOK7,
CNPY1, and
ANK1. Myob/Cbl genes
PAX3,
LBX1,
ZNF556,
ZIC1,
EN2, and
VAX2 encode sequence-specific transcription factors, which likely help drive the myoblast and cerebellum specificity of other Myob/Cbl genes. This study extends our understanding of epigenetic/transcription associations related to differentiation and may help elucidate relationships between epigenetic signatures and muscular dystrophies or cerebellar-linked neuropathologies.
Full article
►▼
Show Figures
Open AccessArticle
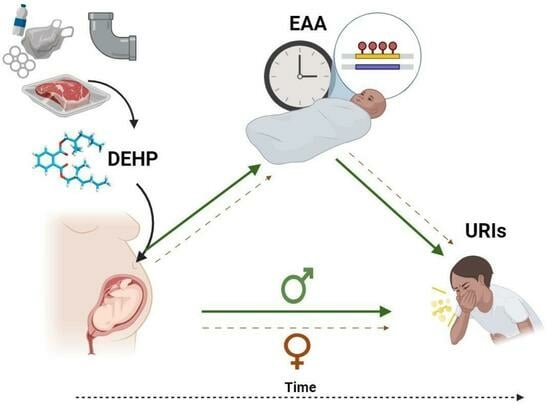
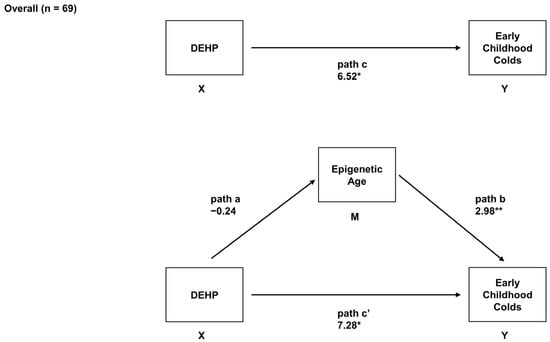
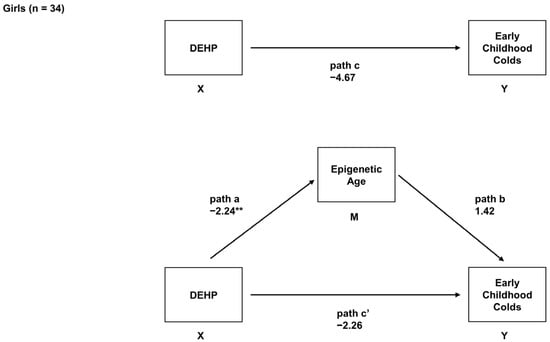
Sex-Specific Associations between Prenatal Exposure to Di(2-ethylhexyl) Phthalate, Epigenetic Age Acceleration, and Susceptibility to Early Childhood Upper Respiratory Infections
by
Sarah M. Merrill, Nicole Letourneau, Gerald F. Giesbrecht, Karlie Edwards, Julia L. MacIsaac, Jonathan W. Martin, Amy M. MacDonald, David W. Kinniburgh, Michael S. Kobor, Deborah Dewey, Gillian England-Mason and The APrON Study Team
Viewed by 1841
Abstract
Di(2-ethylhexyl) phthalate (DEHP) is a common plasticizer that can affect immune system development and susceptibility to infection. Aging processes (measured as epigenetic age acceleration (EAA)) may mediate the immune-related effects of prenatal exposure to DEHP. This study’s objective was to examine associations between
[...] Read more.
Di(2-ethylhexyl) phthalate (DEHP) is a common plasticizer that can affect immune system development and susceptibility to infection. Aging processes (measured as epigenetic age acceleration (EAA)) may mediate the immune-related effects of prenatal exposure to DEHP. This study’s objective was to examine associations between prenatal DEHP exposure, EAA at three months of age, and the number of upper respiratory infections (URIs) from 12 to 18 months of age using a sample of 69 maternal–child pairs from a Canadian pregnancy cohort. Blood DNA methylation data were generated using the Infinium HumanMethylation450 BeadChip; EAA was estimated using Horvath’s pan-tissue clock. Robust regressions examined overall and sex-specific associations. Higher prenatal DEHP exposure (
B = 6.52,
95% CI = 1.22, 11.81) and increased EAA (
B = 2.98,
95% CI = 1.64, 4.32) independently predicted more URIs. In sex-specific analyses, some similar effects were noted for boys, and EAA mediated the association between prenatal DEHP exposure and URIs. In girls, higher prenatal DEHP exposure was associated with decreased EAA, and no mediation was noted. Higher prenatal DEHP exposure may be associated with increased susceptibility to early childhood URIs, particularly in boys, and aging biomarkers such as EAA may be a biological mechanism. Larger cohort studies examining the potential developmental immunotoxicity of phthalates are needed.
Full article
►▼
Show Figures
Open AccessArticle
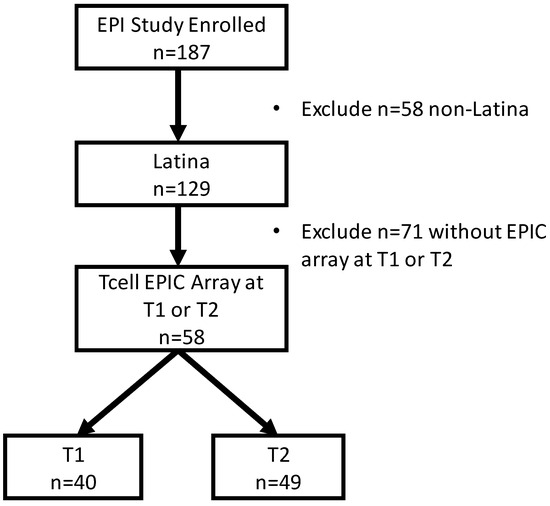
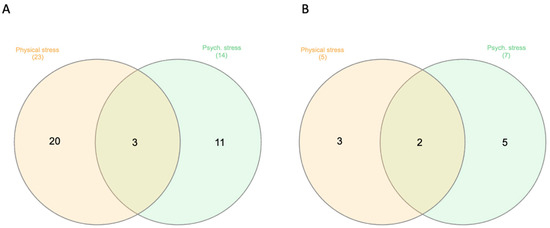
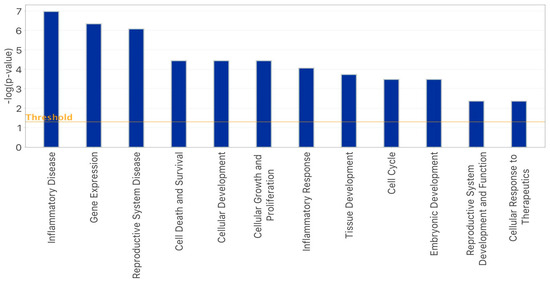
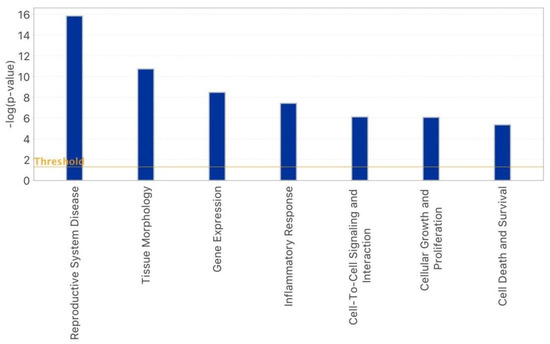
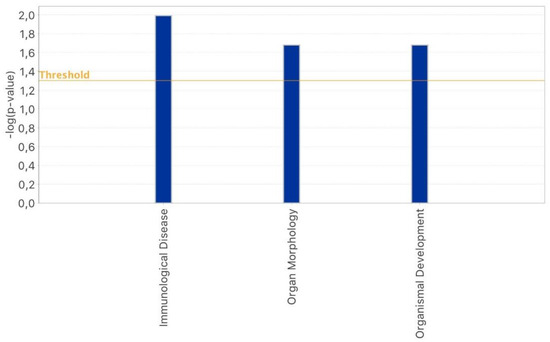
Stress and DNA Methylation of Blood Leukocytes among Pregnant Latina Women
by
Veronica Barcelona, Sameera Abuaish, Seonjoo Lee, Sarah Harkins, Ashlie Butler, Benjamin Tycko, Andrea A. Baccarelli, Kate Walsh and Catherine E. Monk
Viewed by 1781
Abstract
Latinas experience physical and psychological stressors in pregnancy leading to increased morbidity and higher risk for adverse birth outcomes. Epigenetic changes, including DNA methylation (DNAm), have been proposed as markers to create more refined risk stratification, yet few of these studies have examined
[...] Read more.
Latinas experience physical and psychological stressors in pregnancy leading to increased morbidity and higher risk for adverse birth outcomes. Epigenetic changes, including DNA methylation (DNAm), have been proposed as markers to create more refined risk stratification, yet few of these studies have examined these changes in Latinas. We conducted a secondary analysis of stored blood leukocytes of Latina women (n = 58) enrolled in a larger National Institutes of Health funded R01 project (2011–2016). We examined DNAm on eight candidate stress genes to compare physically and psychologically stressed participants to healthy (low stress) participants. We found unique CpGs that were differentially methylated in stressed women early- and mid-pregnancy compared to the healthy group, though none remained significant after FDR correction. Both physical and psychological stress were associated with hypomethylation at two consecutive CpG sites on NR3C1 in early pregnancy and one CpG site on NR3C1 in mid-pregnancy before adjustment. Stress was also associated with hypomethylation at two CpG sites on FKBP5 in early and mid-pregnancy but were no longer significant after FDR adjustment. Though we did not find statistically significant differences in DNAm during pregnancy between stressed and healthy women in this sample, signals were consistent with previous findings. Future work in larger samples should further examine the associations between stress and DNAm in pregnancy as this mechanism may explain underlying perinatal health inequities.
Full article
►▼
Show Figures
Open AccessArticle
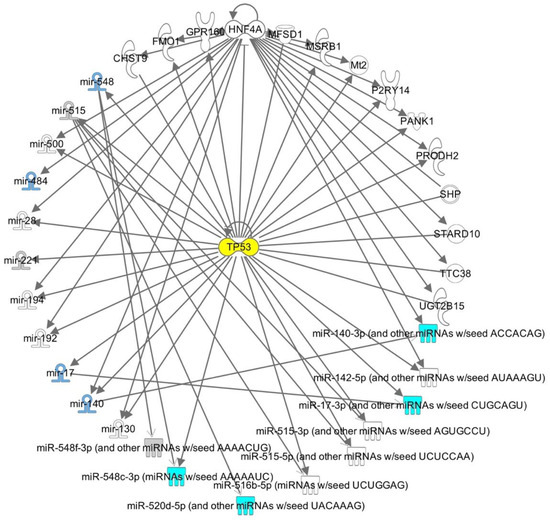
Maternal MicroRNA Profile Changes When LH Is Added to the Ovarian Stimulation Protocol: A Pilot Study
by
Fani Konstantinidou, Martina Placidi, Giovanna Di Emidio, Liborio Stuppia, Carla Tatone, Valentina Gatta and Paolo Giovanni Artini
Viewed by 1570
Abstract
While the use of follicle-stimulating hormone (FSH) in ovarian stimulation for in vitro fertilization (IVF) is an established practice, the use of luteinizing hormone (LH) remains debatable. MicroRNAs (miRNAs) are short, endogenous, non-coding transcripts that control a variety of cellular functions, such as
[...] Read more.
While the use of follicle-stimulating hormone (FSH) in ovarian stimulation for in vitro fertilization (IVF) is an established practice, the use of luteinizing hormone (LH) remains debatable. MicroRNAs (miRNAs) are short, endogenous, non-coding transcripts that control a variety of cellular functions, such as gonadotrophin production and follicular development. The goal of this pilot study was to investigate whether the employment of recombinant LH (rLH) in ovarian stimulation protocols results in changes in the miRNA profiles in human oocytes. Patients were divided into two groups: seven received recombinant FSH (rFSH, 225 IU), and six received rFSH (150 IU) plus rLH (75 IU). MiRNA predesigned panels and real-time PCR technology were used to analyze the oocytes retrieved from the follicular ovarian retrieval. Among the miRNAs evaluated, a series of them evidenced upregulation or downregulation in their expression in the FSH plus LH group compared to the FSH group. Considering the results obtained from the functional and network analysis, the different maternal miRNA profiles in the two groups revealed a differential modulation of pathways involved in numerous biological functions. Overall, based on the pathways associated with most of these maternal miRNAs, the presence of LH may result in a different modulation of pathways regulating survival under the control of a Tp53-related mechanism. Interestingly, among the miRNAs differentially expressed in oocytes of the two groups, we have found miRNAs already investigated at ovarian, follicular, oocyte, and embryonic levels: hsa-miR-484, hsa-miR-222, hsa-miR-520d-5p, hsa-miRNA-17, hsa-miR-548, and hsa-miR-140. Thus, investigation into the role of these miRNAs in oocyte molecular pathways may help determine how LH affects oocyte competence and eventually leads to the clinical improvement of IVF.
Full article
►▼
Show Figures
Open AccessReview
The ErbB Signaling Network and Its Potential Role in Endometrial Cancer
by
Georgios Androutsopoulos, Ioanna Styliara, Evgenia Zarogianni, Nadia Lazurko, George Valasoulis, Georgios Michail and Georgios Adonakis
Cited by 3 | Viewed by 1816
Abstract
Endometrial cancer (EC) is the second most common malignancy of the female reproductive system worldwide. The updated EC classification emphasizes the significant role of various signaling pathways such as PIK3CA-PIK3R1-PTEN and RTK/RAS/β-catenin in EC pathogenesis. Some of these pathways are part of the
[...] Read more.
Endometrial cancer (EC) is the second most common malignancy of the female reproductive system worldwide. The updated EC classification emphasizes the significant role of various signaling pathways such as PIK3CA-PIK3R1-PTEN and RTK/RAS/β-catenin in EC pathogenesis. Some of these pathways are part of the EGF system signaling network, which becomes hyperactivated by various mechanisms and participates in cancer pathogenesis. In EC, the expression of ErbB receptors is significantly different, compared with the premenopausal and postmenopausal endometrium, mainly because of the increased transcriptional activity of ErbB encoding genes in EC cells. Moreover, there are some differences in ErbB-2 receptor profile among EC subgroups that could be explained by the alterations in pathophysiology and clinical behavior of various EC histologic subtypes. The fact that ErbB-2 receptor expression is more common in aggressive EC histologic subtypes (papillary serous and clear cell) could indicate a future role of ErbB-targeted therapies in well-defined EC subgroups with overexpression of ErbB receptors.
Full article
►▼
Show Figures
Open AccessArticle
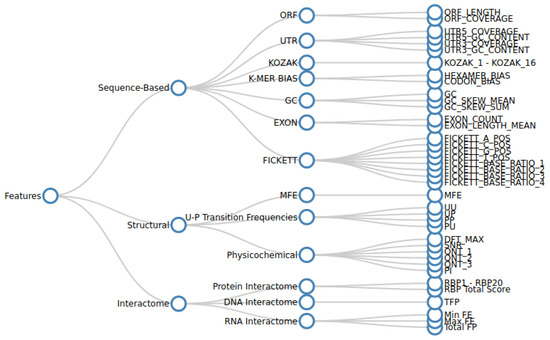
Linc2function: A Comprehensive Pipeline and Webserver for Long Non-Coding RNA (lncRNA) Identification and Functional Predictions Using Deep Learning Approaches
by
Yashpal Ramakrishnaiah, Adam P. Morris, Jasbir Dhaliwal, Melcy Philip, Levin Kuhlmann and Sonika Tyagi
Cited by 1 | Viewed by 1971
Abstract
Long non-coding RNAs (lncRNAs), comprising a significant portion of the human transcriptome, serve as vital regulators of cellular processes and potential disease biomarkers. However, the function of most lncRNAs remains unknown, and furthermore, existing approaches have focused on gene-level investigation. Our work emphasizes
[...] Read more.
Long non-coding RNAs (lncRNAs), comprising a significant portion of the human transcriptome, serve as vital regulators of cellular processes and potential disease biomarkers. However, the function of most lncRNAs remains unknown, and furthermore, existing approaches have focused on gene-level investigation. Our work emphasizes the importance of transcript-level annotation to uncover the roles of specific transcript isoforms. We propose that understanding the mechanisms of lncRNA in pathological processes requires solving their structural motifs and interactomes. A complete lncRNA annotation first involves discriminating them from their coding counterparts and then predicting their functional motifs and target bio-molecules. Current in silico methods mainly perform primary-sequence-based discrimination using a reference model, limiting their comprehensiveness and generalizability. We demonstrate that integrating secondary structure and interactome information, in addition to using transcript sequence, enables a comprehensive functional annotation. Annotating lncRNA for newly sequenced species is challenging due to inconsistencies in functional annotations, specialized computational techniques, limited accessibility to source code, and the shortcomings of reference-based methods for cross-species predictions. To address these challenges, we developed a pipeline for identifying and annotating transcript sequences at the isoform level. We demonstrate the effectiveness of the pipeline by comprehensively annotating the lncRNA associated with two specific disease groups. The source code of our pipeline is available under the MIT licensefor local use by researchers to make new predictions using the pre-trained models or to re-train models on new sequence datasets. Non-technical users can access the pipeline through a web server setup.
Full article
►▼
Show Figures






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}