
Linc2function: A Comprehensive Pipeline and Webserver for Long Non-Coding RNA (lncRNA) Identification and Functional Predictions Using Deep Learning Approaches

 , ,
, ,
Abstract
:1. Introduction
1.1. Structure to Function
1.2. Interactome
1.3. Computational Identification and Annotation of lncRNA
2. Methods
2.1. Rule-Based Machine Learning Approach (ANN Models)
2.1.1. Data
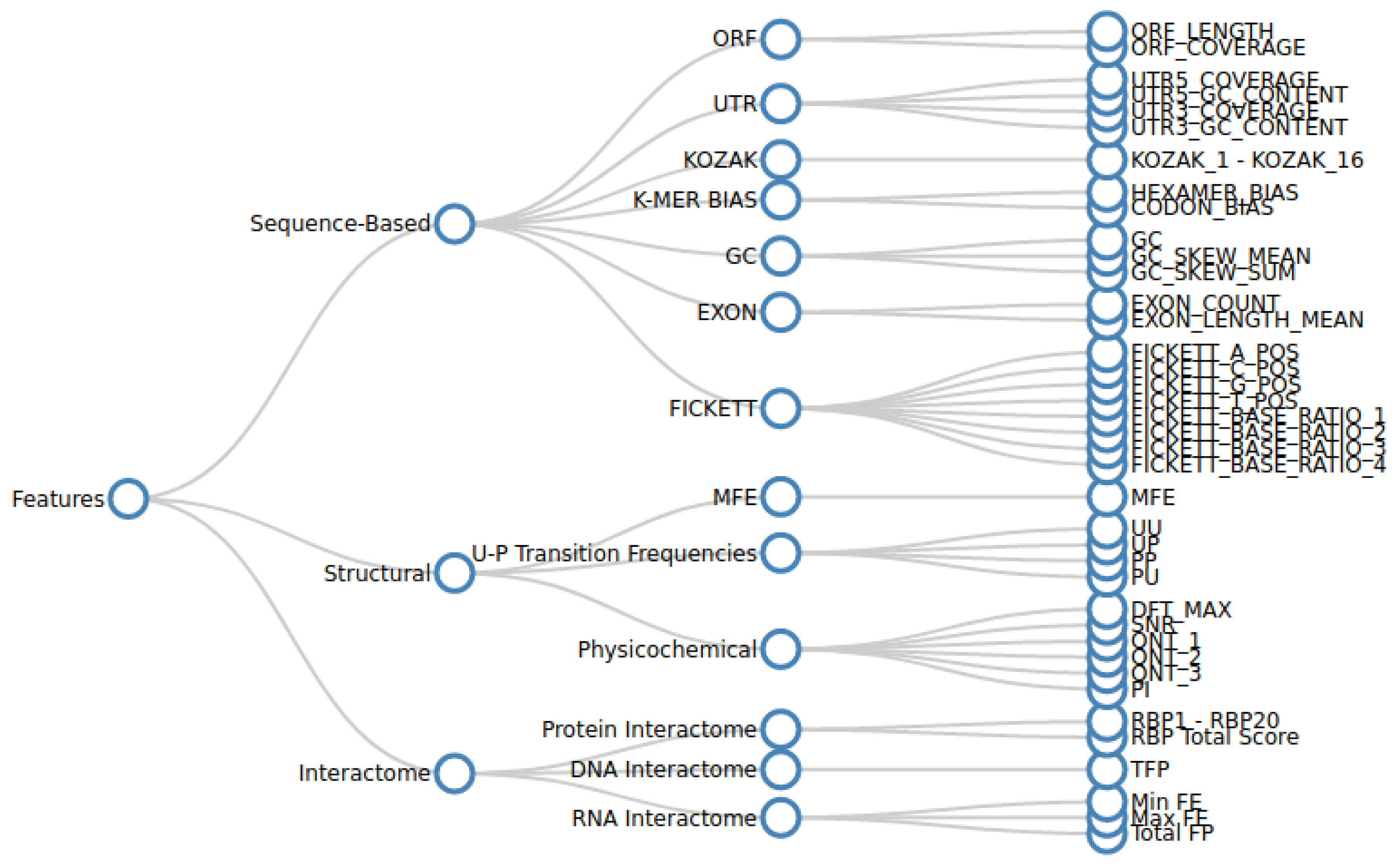
2.1.2. Feature Extraction
2.1.3. Model Training and Performance Evaluation
2.2. Deep Learning-Based Approach (LRN2 Model)
2.2.1. Modification of Existing Model and Recoding
2.2.2. Model and Training and Performance Evaluation
2.3. Benchmarking and Comparative Analysis of Models
2.3.1. Comparison of ANN with Other Rule-Based Approaches
2.3.2. lncRNA Identification Using Different Models
2.3.3. Cross-Species Comparison
2.4. lncRNA Annotation
3. Results and Discussion
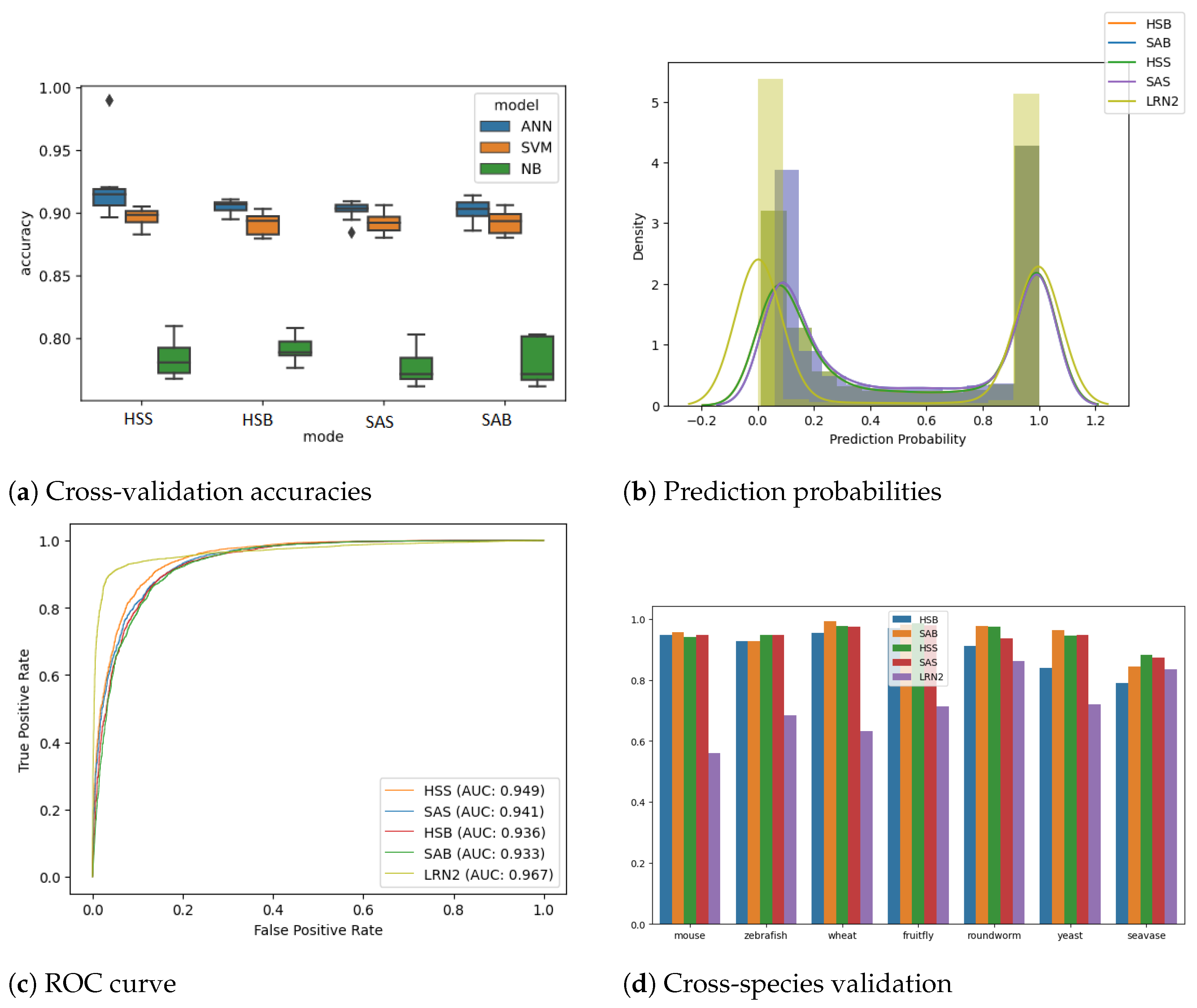
3.1. ANN Outperforms Other Rule-Based Models
3.2. ANNand RNN Models
3.3. Species Agnostic Models Perform Better at Cross-Species Analysis
3.4. Our Pipeline Is Accessible via a User-Friendly Webserver Interface
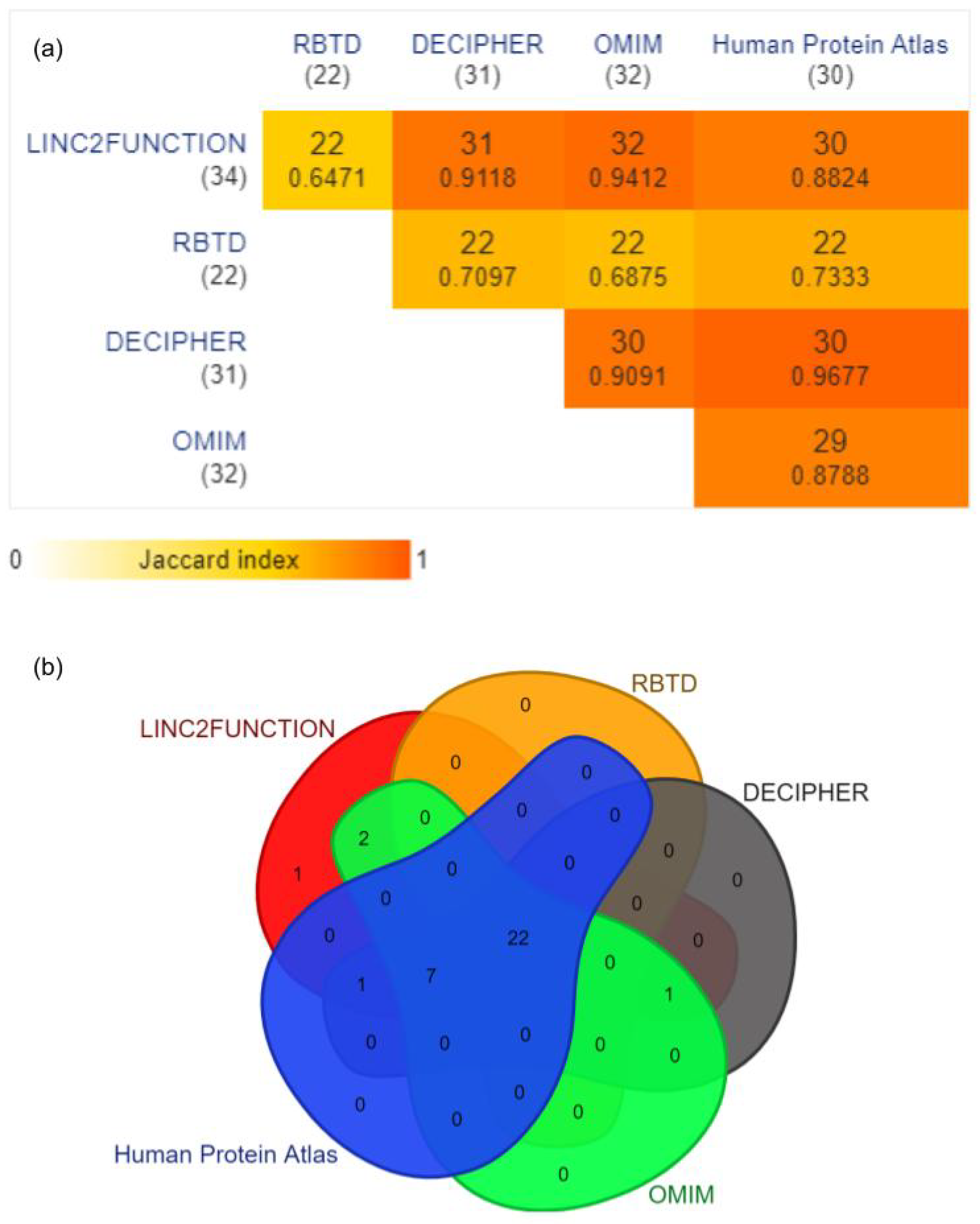
3.5. Application of linc2function Pipeline for Comprehensive Disease Associated lncRNA Annotations
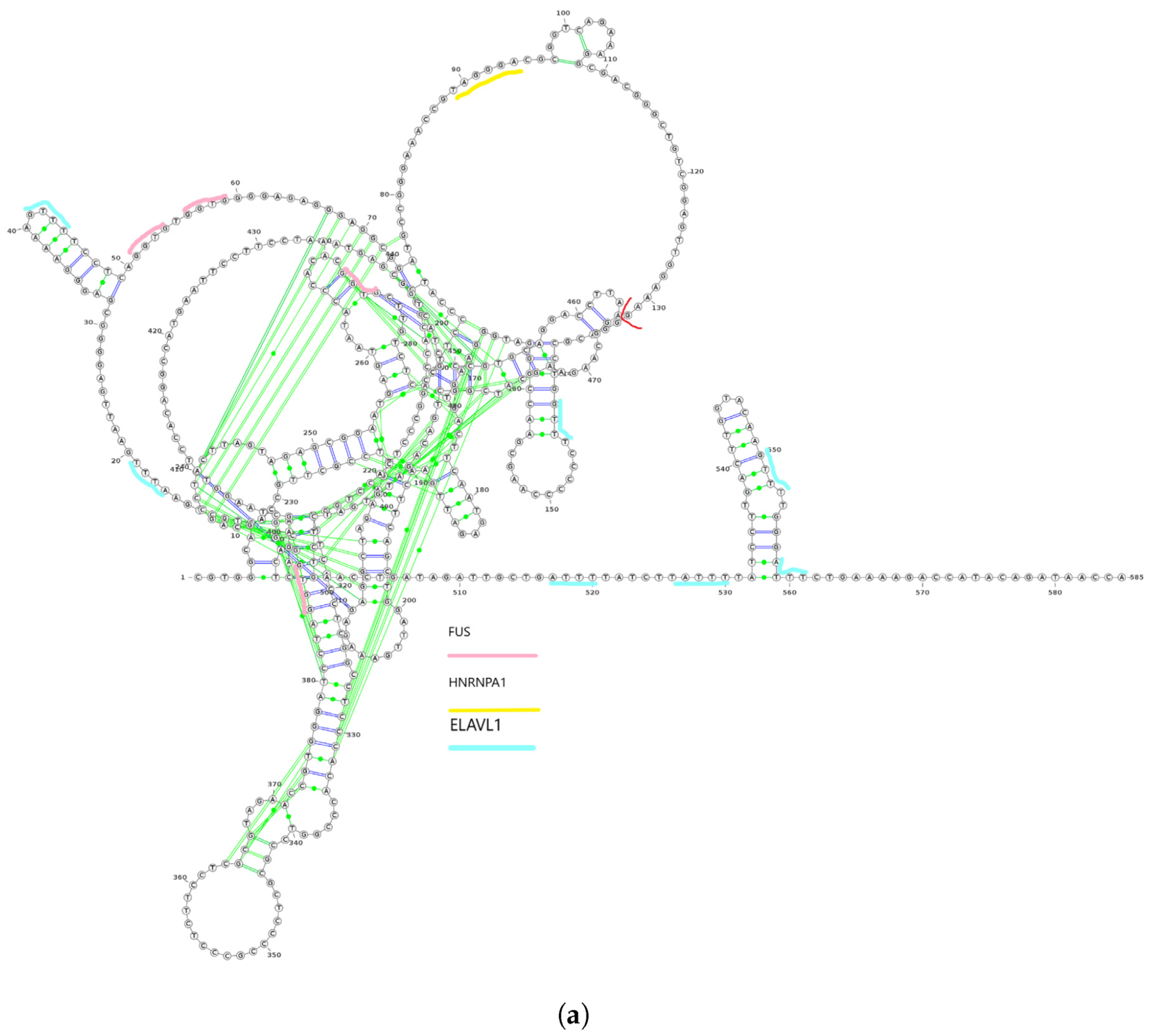
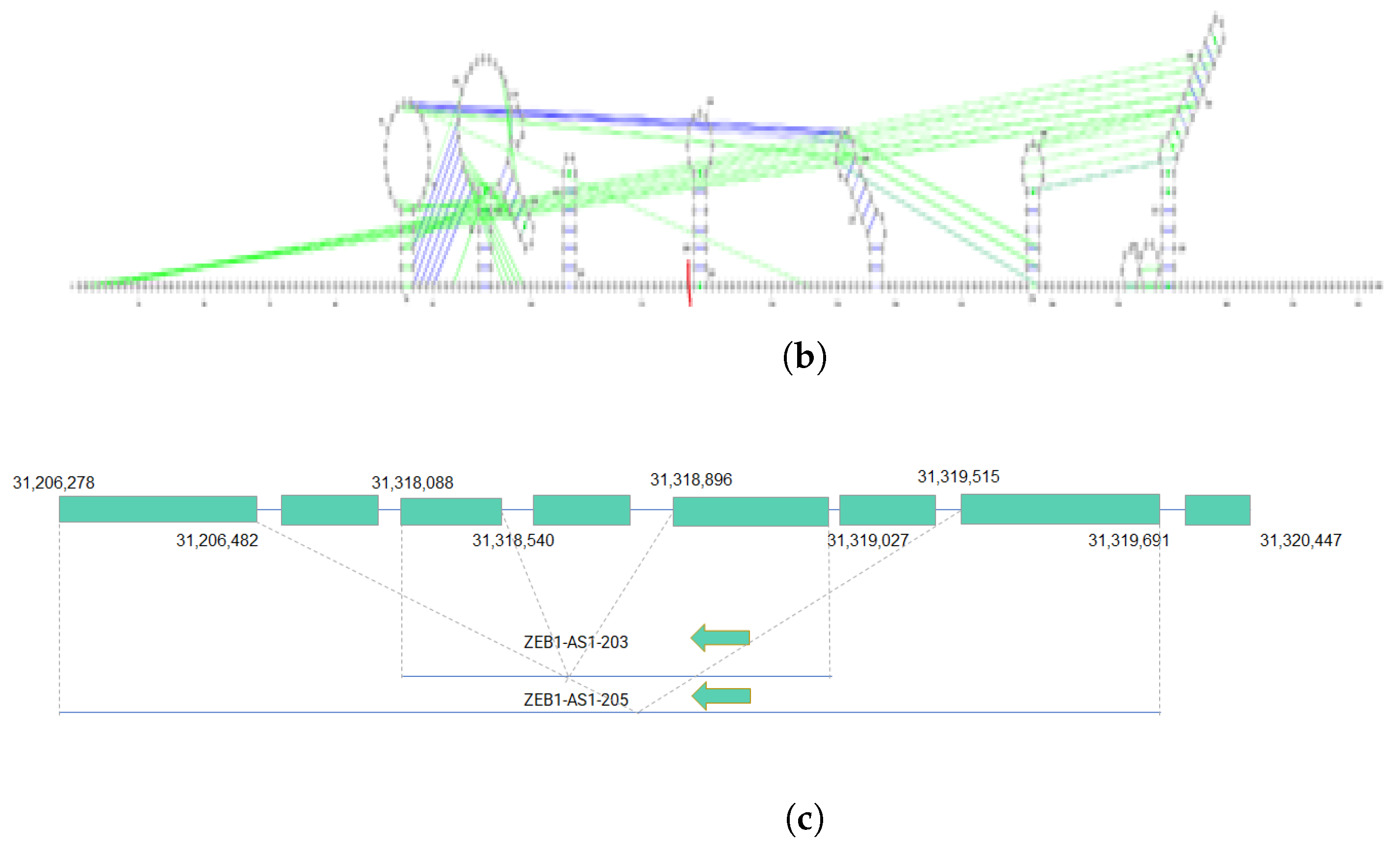
3.5.1. Case Study 1: Isoform-Level Annotation Reveals Functional Diveristy of Transcript Variants
Summary
3.5.2. Case Study 2: Predicting Interactome Associated with Cancer
RNA Interactome Validation
Summary
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, H.; Osak, M.; Bogu, G.K.; Stanton, L.W.; Johnson, R.; Lipovich, L. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 2010, 16, 1478–1487. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Kashi, K.; Henderson, L.; Bonetti, A.; Carninci, P. Discovery and functional analysis of lncRNAs: Methodologies to investigate an uncharacterized transcriptome. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 3–15. [Google Scholar] [CrossRef]
- van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most “Dark Matter” Transcripts Are Associated with Known Genes. PLoS Biol. 2010, 8, e1000371. [Google Scholar] [CrossRef]
- Agrawal, S.; Alam, T.; Koido, M.; Kulakovskiy, I.V.; Severin, J.; Abugessaisa, I.; Buyan, A.; Dostie, J.; Itoh, M.; Kondo, N.; et al. Functional annotation of human long noncoding RNAs using chromatin conformation data. bioRxiv 2021. bioRxiv:2021.01.13.426305. [Google Scholar] [CrossRef]
- Ramakrishnaiah, Y.; Kuhlmann, L.; Tyagi, S. Towards a comprehensive pipeline to identify and functionally annotate long noncoding RNA (lncRNA). Comput. Biol. Med. 2020, 127, 104028. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2012, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2018, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Acuña, S.; Aoki, J.; Floeter-Winter, L.; Muxel, S. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H. Long non-coding RNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2016, 18, 558–576. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef]
- de Goede, O.M.; Nachun, D.C.; Ferraro, N.M.; Gloudemans, M.J.; Rao, A.S.; Smail, C.; Eulalio, T.Y.; Aguet, F.; Ng, B.; Xu, J.; et al. Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease. Cell 2021, 184, 2633–2648.e19. [Google Scholar] [CrossRef]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Orom, U.A.; Derrien, T.; Guigo, R.; Shiekhattar, R. Long Noncoding RNAs as Enhancers of Gene Expression. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs: Sequence, structure, function. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-coding RNA Structure and Function: Is There a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Clements, J.; Eddy, S.R. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2016, 14, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.C.; Pyle, A.M.; Somarowthu, S. Phylogenetic Analysis with Improved Parameters Reveals Conservation in lncRNA Structures. J. Mol. Biol. 2019, 431, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E. RNA structure prediction using positive and negative evolutionary information. PLoS Comput. Biol. 2020, 16, e1008387. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Xi, Y.; Sung, S. Modular function of long noncoding RNA, COLDAIR, in the vernalization response. PLoS Genet. 2017, 13, e1006939. [Google Scholar] [CrossRef] [PubMed]
- Buske, F.A.; Mattick, J.S.; Bailey, T.L. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011, 8, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S. RNA–protein complexes. Curr. Opin. Struct. Biol. 1999, 9, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hudson, W.H.; Ortlund, E.A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014, 15, 749–760. [Google Scholar] [CrossRef]
- Philip, M.; Chen, T.; Tyagi, S. A Survey of Current Resources to Study lncRNA–protein Interactions. Non-Coding RNA 2021, 7, 33. [Google Scholar] [CrossRef]
- Szcześniak, M.W.; Makałowska, I. lncRNA-RNA Interactions across the Human Transcriptome. PLoS ONE 2016, 11, e0150353. [Google Scholar] [CrossRef]
- Tyagi, S.; Vaz, C.; Gupta, V.; Bhatia, R.; Maheshwari, S.; Srinivasan, A.; Bhattacharya, A. CID-miRNA: A web server for prediction of novel miRNA precursors in human genome. Biochem. Biophys. Res. Commun. 2008, 372, 831–834. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Kato, M.; Carninci, P. Genome-Wide Technologies to Study RNA–Chromatin Interactions. Non-Coding RNA 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2015, 17, 47–62. [Google Scholar] [CrossRef]
- Krchňáková, Z.; Thakur, P.K.; Krausová, M.; Bieberstein, N.; Haberman, N.; Müller-McNicoll, M.; Staněk, D. Splicing of long non-coding RNAs primarily depends on polypyrimidine tract and 5′ splice-site sequences due to weak interactions with SR proteins. Nucleic Acids Res. 2018, 47, 911–928. [Google Scholar] [CrossRef]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Lee, B.; Kwon, S.; Yoon, S. LncRNAnet: Long non-coding RNA identification using deep learning. Bioinformatics 2018, 34, 3889–3897. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019, 10, 5407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, Y.; Kwoh, C.K. Deep learning based DNA:RNA triplex-forming potential prediction. BMC Bioinform. 2020, 21, 522. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.B.; Kazan, H.; Zuberi, K.; Morris, Q.; Hughes, T.R. RBPDB: A database of RNA-binding specificities. Nucleic Acids Res. 2010, 39, D301–D308. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Hamada, M. RIblast: An ultrafast RNA–RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, K.; Yu, Y.; Le, N.Q.K.; Chua, M.C.H. Prediction of anticancer peptides based on an ensemble model of deep learning and machine learning using ordinal positional encoding. Brief. Bioinform. 2023, 24, bbac630. [Google Scholar] [CrossRef]
- Kha, Q.H.; Le, V.H.; Hung, T.N.K.; Nguyen, N.T.K.; Le, N.Q.K. Development and Validation of an Explainable Machine Learning-Based Prediction Model for Drug-Food Interactions from Chemical Structures. Sensors 2023, 23, 3962. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhao, H.; Yu, J.; Guo, C.; Dou, X.; Song, F.; Hu, G.; Cao, Z.; Qu, Y.; Yang, Y.; et al. EVLncRNAs: A manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2017, 46, D100–D105. [Google Scholar] [CrossRef] [PubMed]
- Amand, J.; Fehlmann, T.; Backes, C.; Keller, A. DynaVenn: Web-based computation of the most significant overlap between ordered sets. BMC Bioinform. 2019, 20, 743. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Year | ML Model | SB | Con | Str | Exp | Int |
|---|---|---|---|---|---|---|---|
| CPC | 2007 | SVM | y | y | n | n | n |
| CPAT | 2013 | LR | y | n | n | n | n |
| CNCI | 2013 | SVM | y | n | n | n | n |
| PLEK | 2014 | SVM | y | n | n | n | n |
| lncRScanSVM | 2015 | SVM | y | y | n | n | n |
| lncRNAID | 2015 | RF | y | y | n | n | n |
| lncRNAMFDL | 2015 | ANN | y | y | y | n | n |
| lncScore | 2016 | LR | y | y | n | n | n |
| DeepLNC | 2016 | ANN | y | n | n | n | n |
| CPC2 | 2017 | SVM | y | n | y | n | n |
| COME | 2017 | RF | y | y | n | y | n |
| Longdist | 2017 | SVM | y | n | n | n | n |
| FEElnc | 2017 | RF | y | n | n | n | n |
| LncADeep | 2018 | DL | y | y | n | n | n |
| LncRNAnet | 2018 | CNN, RNN | y | n | n | n | n |
| EVlncRNApred | 2019 | SVM | y | y | y | y | y |
| LncFinder | 2019 | LR, SVM, RF, ELM, ANN | y | n | y | n | n |
| linc2function | 2023 | ANN, CNN, RNN | y | n | y | n | y |
| Model | Precision | Recall | Specificity | F1 | AUROC |
|---|---|---|---|---|---|
| Linc2function HSS | 0.8771 | 0.8852 | 0.8760 | 0.8812 | 0.949 |
| Linc2function SAS | 0.8717 | 0.8625 | 0.8730 | 0.8671 | 0.941 |
| Linc2function HSB | 0.8592 | 0.8815 | 0.8555 | 0.8702 | 0.936 |
| Linc2function SAB | 0.8591 | 0.8702 | 0.8572 | 0.8646 | 0.933 |
| Linc2function LRN2 | 0.9412 | 0.9160 | 0.9428 | 0.9284 | 0.967 |
| LncRNAnet * | 0.9750 | 0.8962 | 0.9770 | 0.9340 | 0.977 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramakrishnaiah, Y.; Morris, A.P.; Dhaliwal, J.; Philip, M.; Kuhlmann, L.; Tyagi, S. Linc2function: A Comprehensive Pipeline and Webserver for Long Non-Coding RNA (lncRNA) Identification and Functional Predictions Using Deep Learning Approaches. Epigenomes 2023, 7, 22. https://doi.org/10.3390/epigenomes7030022
Ramakrishnaiah Y, Morris AP, Dhaliwal J, Philip M, Kuhlmann L, Tyagi S. Linc2function: A Comprehensive Pipeline and Webserver for Long Non-Coding RNA (lncRNA) Identification and Functional Predictions Using Deep Learning Approaches. Epigenomes. 2023; 7(3):22. https://doi.org/10.3390/epigenomes7030022
Chicago/Turabian StyleRamakrishnaiah, Yashpal, Adam P. Morris, Jasbir Dhaliwal, Melcy Philip, Levin Kuhlmann, and Sonika Tyagi. 2023. "Linc2function: A Comprehensive Pipeline and Webserver for Long Non-Coding RNA (lncRNA) Identification and Functional Predictions Using Deep Learning Approaches" Epigenomes 7, no. 3: 22. https://doi.org/10.3390/epigenomes7030022