
Current Approaches to Epigenetic Therapy
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Pharmacotherapeutic Approach for Epigenome Modulation
Oncometabolites and Metabolic Rewiring
3. Innovative Molecular and Genetic Approaches to the Modulation of Epigenetic Regulation
3.1. CRISPR/Cas9
3.2. Non-Coding RNAs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Deichmann, U. Epigenetics: The origins and evolution of a fashionable topic. Dev. Biol. 2016, 416, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, D. DNA methyltransferase-1 in acute myeloid leukaemia: Beyond the maintenance of DNA methylation. Ann. Med. 2022, 54, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Fiñana, C.; Gómez-Molina, N.; Alonso-Moreno, S.; Belver, L. Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia. Cancers 2022, 14, 1335. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Wang, Y.; Huang, G.; Li, X.; Xie, Z.; Zhou, Z. Insights into the Role of DNA Methylation in Immune Cell Development and Autoimmune Disease. Front. Cell Dev. Biol. 2021, 9, 757318. [Google Scholar] [CrossRef]
- Sharma, V.; Wright, K.L.; Epling-Burnette, P.K.; Reuther, G.W. Metabolic Vulnerabilities and Epigenetic Dysregulation in Myeloproliferative Neoplasms. Front. Immunol. 2020, 11, 604142. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug. Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Daher-Reyes, G.S.; Merchan, B.M.; Yee, K.W.L. Guadecitabine (SGI-110): An investigational drug for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin. Investig. Drugs. 2019, 28, 835–849. [Google Scholar] [CrossRef]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Sun, G.; Jiang, W.; He, X.; Shi, Y.; Ma, F.; Liu, P. Histone deacetylases inhibitor chidamide synergizes with humanized PD1 antibody to enhance T-cell chemokine expression and augment Ifn-γ response in NK-T cell lymphoma. eBioMedicine 2023, 87, 104420. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Johnston, S.K.; Mrugala, M.M.; Beard, B.C.; Guyman, L.A.; Baldock, A.L.; Bridge, C.A.; Hawkins-Daarud, A.; Gori, J.L.; Born, D.E.; et al. Gene therapy enhances chemotherapy tolerance and efficacy in glioblastoma patients. J. Clin. Investig. 2014, 124, 4082–4092. [Google Scholar] [CrossRef]
- Ruan, J.; Moskowitz, A.J.; Mehta-Shah, N.; Sokol, L.; Chen, Z.; Rahim, R.; Song, W.; Van Besien, K.; Horwitz, S.M.; Rutherford, S.C. Multi-Center Phase II Study of Oral Azacitidine (CC-486) Plus CHOP As Initial Treatment for Peripheral T-Cell Lymphoma (PTCL). Blood 2020, 136 (Suppl. S1), 33–34. [Google Scholar] [CrossRef]
- Montesinos, P.P.; Roboz, G.J.; Bulabois, C.E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Roboz, G.; Walsh, K.; Kantarjian, H.; Ritchie, E.; Kropf, P.; O’Connell, C.; Tibes, R.; Lunin, S.; Rosenblat, T.; et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: Phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet. Haematol. 2019, 6, e317–e327. [Google Scholar] [CrossRef]
- Sébert, M.; Renneville, A.; Bally, C.; Peterlin, P.; Beyne-Rauzy, O.; Legros, L.; Gourin, M.P.; Sanhes, L.; Wattel, E.; Gyan, E.; et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica 2019, 104, 1565–1571. [Google Scholar] [CrossRef]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef] [PubMed]
- Navarro Mendivil, A.F.; Gutierrez, S.; Bullock, R.; Buesa, C. 1806P Final safety and efficacy data from CLEPSIDRA trial in 2L ED-SCLC. Ann. Oncol. 2020, 31, S1044. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Erickson, S.W.; Han, R.; Teicher, B.A.; Smith, M.A.; Roth, M.; Gorlick, R.; Houghton, P.J. In vivo evaluation of the lysine-specific demethylase (KDM1A/LSD1) inhibitor SP-2577 (Seclidemstat) against pediatric sarcoma preclinical models: A report from the Pediatric Preclinical Testing Consortium (PPTC). Pediatr. Blood Cancer 2021, 68, e29304. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.; Northrop, W.; Ellison, G.; Sabapathy, T.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Statins Do Not Directly Inhibit the Activity of Major Epigenetic Modifying Enzymes. Cancers 2019, 11, 516. [Google Scholar] [CrossRef]
- Lauria, A.; Mannino, S.; Gentile, C.; Mannino, G.; Martorana, A.; Peri, D. DRUDIT: Web-based DRUgs DIscovery Tools to design small molecules as modulators of biological targets. Bioinformatics 2020, 36, 1562–1569. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Letouzé, E.; Gimenez-Roqueplo, A.P.; Favier, J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Qin, Y. Connections between metabolism and epigenetics: Mechanisms and novel anti-cancer strategy. Front. Pharmacol. 2022, 13, 935536. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M. Wild-type IDH1 inhibition enhances chemotherapy response in melanoma. J. Exp. Clin. Cancer Res. 2022, 41, 283. [Google Scholar] [CrossRef]
- Fan, D.; Yue, Q.; Chen, J.; Wang, C.; Yu, R.; Jin, Z.; Yin, S.; Wang, Q.; Chen, L.; Liao, X.; et al. Reprogramming the immunosuppressive microenvironment of IDH1 wild-type glioblastoma by blocking Wnt signaling between microglia and cancer cells. Oncoimmunology 2021, 10, 1932061. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X. Recent advances of IDH1 mutant inhibitor in cancer therapy. Front. Pharmacol. 2022, 3, 982424. [Google Scholar] [CrossRef]
- Issa, G.C.; DiNardo, C.D. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021, 11, 107. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kornepati, A.V.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bloj, B.; Moses, C.; Sgro, A.; Plani-Lam, J.; Arooj, M.; Duffy, C.; Thiruvengadam, S.; Sorolla, A.; Rashwan, R.; Mancera, R.L.; et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget 2016, 7, 60535–60554. [Google Scholar] [CrossRef]
- Balboa, D.; Weltner, J.; Eurola, S.; Trokovic, R.; Wartiovaara, K.; Otonkoski, T. Conditionally Stabilized dCas9 Activator for Controlling Gene Expression in Human Cell Reprogramming and Differentiation. Stem Cell Rep. 2015, 5, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Pham, T.L.H.; Choi, S. Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells. Int. J. Mol. Sci. 2021, 22, 397. [Google Scholar] [CrossRef]
- Elbasani, E.; Falasco, F.; Gramolelli, S.; Nurminen, V.; Günther, T.; Weltner, J.; Balboa, D.; Grundhoff, A.; Otonkoski, T.; Ojala, P.M. Kaposi’s Sarcoma-Associated Herpesvirus Reactivation by Targeting of a dCas9-Based Transcription Activator to the ORF50 Promoter. Viruses 2020, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Villamizar, O.; Waters, S.A.; Scott, T.; Saayman, S.; Grepo, N.; Urak, R.; Davis, A.; Jaffe, A.; Morris, K.V. Targeted Activation of Cystic Fibrosis Transmembrane Conductance Regulator. Mol. Ther. 2019, 27, 1737–1748. [Google Scholar] [CrossRef]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Sajwan, S.; Mannervik, M. Gene activation by dCas9-CBP and the SAM system differ in target preference. Sci. Rep. 2019, 9, 18104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Sun, Y.; Xiao, R.; Wai, K.; Ahmad, M.J.; Khan, F.A.; Zhou, H.; Li, Z.; Zhang, Y.; Zhou, A.; et al. Porcine antiviral activity is increased by CRISPRa-SAM system. Biosci. Rep. 2019, 39, BSR20191496. [Google Scholar] [CrossRef]
- Kunii, A.; Hara, Y.; Takenaga, M.; Hattori, N.; Fukazawa, T.; Ushijima, T.; Yamamoto, T.; Sakuma, T. Three-Component Repurposed Technology for Enhanced Expression: Highly Accumulable Transcriptional Activators via Branched Tag Arrays. CRISPR J. 2018, 1, 337–347. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442. [Google Scholar] [CrossRef]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin 2019, 12, 26. [Google Scholar] [CrossRef]
- Alerasool, N.; Segal, D.; Lee, H.; Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods 2020, 17, 1093–1096. [Google Scholar] [CrossRef]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Gjaltema, R.A.F.; Goubert, D.; Huisman, C.; Pilar García Tobilla, C.D.; Koncz, M.; Jellema, P.G.; Wu, D.; Brouwer, U.; Kiss, A.; Verschure, P.J.; et al. KRAB-Induced Heterochromatin Effectively Silences PLOD2 Gene Expression in Somatic Cells and Is Resilient to TGFβ1 Activation. Int. J. Mol. Sci. 2020, 21, 3634. [Google Scholar] [CrossRef]
- Das, S.; Chadwick, B.P. CRISPR mediated targeting of DUX4 distal regulatory element represses DUX4 target genes dysregulated in Facioscapulohumeral muscular dystrophy. Sci. Rep. 2021, 11, 12598. [Google Scholar] [CrossRef]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Tomkova, M.; Combs, J.A.; Tilley, E.K.; Segal, D.J. Determinants of heritable gene silencing for KRAB-dCas9 + DNMT3 and Ezh2-dCas9 + DNMT3 hit-and-run epigenome editing. Nucleic Acids Res. 2022, 50, 3239–3253. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, M.; Liu, X.; Song, S.; Wang, L.; Yang, X.; Song, Y. Construction and validation of the CRISPR/dCas9-EZH2 system for targeted H3K27Me3 modification. Biochem. Biophys. Res. Commun. 2019, 511, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regula tion. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]
- Duke, C.G.; Bach, S.V.; Revanna, J.S.; Sultan, F.A.; Southern, N.T.; Davis, M.N.; Carullo, N.V.N.; Bauman, A.J.; Phillips, R.A., 3rd; Day, J.J. An Improved CRISPR/dCas9 Interference Tool for Neuronal Gene Suppression. Front. Genome Ed. 2020, 2, 9. [Google Scholar] [CrossRef]
- Perillo, B.; Tramontano, A.; Pezone, A.; Migliaccio, A. LSD1: More than demethylation of histone lysine residues. Exp. Mol. Med. 2020, 52, 1936–1947. [Google Scholar] [CrossRef]
- Haldeman, J.M.; Conway, A.E.; Arlotto, M.E.; Slentz, D.H.; Muoio, D.M.; Becker, T.C.; Newgard, C.B. Creation of versatile cloning platforms for transgene expression and dCas9-based epigenome editing. Nucleic Acids Res. 2019, 47, e23. [Google Scholar] [CrossRef]
- Ngeow, J.; Eng, C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb. Perspect. Med. 2020, 10, a036087. [Google Scholar] [CrossRef]
- Wang, H.; Guo, R.; Du, Z.; Bai, L.; Li, L.; Cui, J.; Li, W.; Hoffman, A.R.; Hu, J.F. Epigenetic Targeting of Granulin in Hepatoma Cells by Synthetic CRISPR dCas9 Epi-suppressors. Mol. Ther. Nucleic Acids 2018, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol. Ther. Methods Clin. Dev. 2020, 20, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Colasante, G.; Lignani, G.; Brusco, S.; Di Berardino, C.; Carpenter, J.; Giannelli, S.; Valassina, N.; Bido, S.; Ricci, R.; Castoldi, V.; et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther. 2020, 28, 235–253. [Google Scholar] [CrossRef]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Wang, S.E.; Jiang, Y.h. Potential of Epigenetic Therapy for Prader-Willi Syndrome. Trends Pharmacol. Sci. 2019, 40, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, B.S.; Stafrace, Y.; Calleja-Agius, J. Silver-Russell Syndrome: A Review. Neonatal Netw. 2017, 36, 206–212. [Google Scholar] [CrossRef]
- Butler, M.G. Imprinting disorders in humans: A review. Curr. Opin. Pediatr. 2020, 32, 719–729. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Webber, A.L.; Brunkow, M.E.; Tilghman, S.M. Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes Dev. 1993, 7, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, K.D.; Saam, J.R.; Ingram, R.S.; Tilghman, S.M.; Bartolomei, M.S. A paternal-specific methylation imprint marks the alleles of the mouse H19 gene. Nat. Genet. 1995, 9, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Horii, T.; Morita, S.; Hino, S.; Kimura, M.; Hino, Y.; Kogo, H.; Nakao, M.; Hatada, I. Successful generation of epigenetic disease model mice by targeted demethylation of the epigenome. Genome Biol. 2020, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of fragile x syndrome. Annu. Rev. Genom. Hum. Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Contractor, A.; Klyachko, V.A.; Portera-Cailliau, C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 2015, 87, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef]
- Ricci, R.; Colasante, G. CRISPR/dCas9 as a Therapeutic Approach for Neurodevelopmental Disorders: Innovations and Limitations Compared to Traditional Strategies. Dev. Neurosci. 2021, 43, 253–261. [Google Scholar] [CrossRef]
- Weiss, B.; Davidkova, G.; Zhou, L.W. Antisense RNA gene therapy for studying and modulating biological processes. Cell. Mol. Life Sci. 1999, 55, 334–358. [Google Scholar] [CrossRef]
- Saw, P.E.; Song, E.W. siRNA therapeutics: A clinical reality. Sci. China Life Sci. 2020, 63, 485–500. [Google Scholar] [CrossRef]
- Mollocana-Lara, E.C.; Ni, M.; Agathos, S.N.; Gonzales-Zubiate, F.A. The infinite possibilities of RNA therapeutics. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab063. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, X.Y.; Lu, A.; Wang, X.Y.; Jiang, L.X.; Wang, J.C. Non-viral vectors for RNA delivery. J. Control. Release 2022, 342, 241. [Google Scholar] [CrossRef]
- Czauderna, F.; Fechtner, M.; Dames, S.; Aygün, H.; Klippel, A.; Pronk, G.J.; Giese, K.; Kaufmann, J. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003, 31, 2705–2716. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Foster, D.J.; Brown, C.R.; Shaikh, S.; Trapp, C.; Schlegel, M.K.; Qian, K.; Sehgal, A.; Rajeev, K.G.; Jadhav, V.; Manoharan, M.; et al. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018, 26, 708–717. [Google Scholar] [CrossRef]
- Shen, W.; De Hoyos, C.L.; Sun, H.; Vickers, T.A.; Liang, X.H.; Crooke, S.T. Acute hepatotoxicity of 2′ fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 2018, 46, 2204–2217. [Google Scholar] [CrossRef]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): Highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Yan, Y.; Kos, P.; Chen, X.; Siegwart, D.J. PEI fluorination reduces toxicity and promotes liver-targeted siRNA delivery. Drug Deliv. Transl. Res. 2021, 11, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Sevilla, I.; Artiga, Á.; Mitchell, S.G.; De Matteis, L.; de la Fuente, J.M. Natural Polysaccharides for siRNA Delivery: Nanocarriers Based on Chitosan, Hyaluronic Acid, and Their Derivatives. Molecules 2019, 24, 2570. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. NAR Breakthrough Article Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of Small Interfering RNA at the Organ and Cellular Levels after Lipid Nanoparticle-mediated Delivery. J. Histochem. Cytochem. 2011, 59, 727. [Google Scholar] [CrossRef]
- Sioud, M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 2005, 348, 1079–1090. [Google Scholar] [CrossRef]
- Sledz, C.A.; Holko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003, 5, 834–839. [Google Scholar] [CrossRef]
- Marques, J.T.; Devosse, T.; Wang, D.; Zamanian-Daryoush, M.; Serbinowski, P.; Hartmann, R.; Fujita, T.; Behlke, M.A.; Williams, B.R. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 2006, 24, 559–565. [Google Scholar] [CrossRef]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef]
- Forsbach, A.; Nemorin, J.G.; Montino, C.; Müller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B.; et al. Identification of RNA Sequence Motifs Stimulating Sequence-Specific TLR8-Dependent Immune Responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Cao, H.; Zhu, Y.; Zheng, J.; Zhou, H. Extraordinary GU-rich single-strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect. 2013, 15, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Yan, F.; Tu, J.; Yang, X.; Xing, Y.; Chen, Z. 5′-Triphosphate-siRNA activates RIG-I-dependent type i interferon production and enhances inhibition of hepatitis B virus replication in HepG2.2.15 cells. Eur. J. Pharmacol. 2013, 721, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Coleman, L.G.; Zou, J.; Crews, F.T. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J. Neuroinflammation 2017, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Naeli, P.; Winter, T.; Hackett, A.P.; Alboushi, L.; Jafarnejad, S.M. The intricate balance between microRNA-induced mRNA decay and translational repression. FEBS J. 2023, 290, 2508–2524. [Google Scholar] [CrossRef]
- Gangopadhyay, S.; Gore, K.R. Advances in siRNA therapeutics and synergistic effect on siRNA activity using emerging dual ribose modifications. RNA Biol. 2022, 19, 452–467. [Google Scholar] [CrossRef]
- Herrera-Carrillo, E.; Liu, Y.P.; Berkhout, B. Improving miRNA delivery by optimizing mirna expression cassettes in diverse virus vectors. Hum. Gene. Ther. Methods 2017, 28, 177–190. [Google Scholar] [CrossRef]
- Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Petrek, H.; Yu, A.M. MicroRNAs in non-small cell lung cancer: Gene regulation, impact on cancer cellular processes, and therapeutic potential. Pharmacol. Res. Perspect. 2019, 7, e00528. [Google Scholar] [CrossRef]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. MicroRNA therapeutics in cancer—An emerging concept. eBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Circular RNAs as Therapeutic Agents and Targets. Front. Physiol. 2018, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Breuer, J.; Rossbach, O. Production and Purification of Artificial Circular RNA Sponges for Application in Molecular Biology and Medicine. Methods Protoc. 2020, 3, 42. [Google Scholar] [CrossRef]
- Rama, A.R.; Quiñonero, F.; Mesas, C.; Melguizo, C.; Prados, J. Synthetic Circular miR-21 Sponge as Tool for Lung Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 2963. [Google Scholar] [CrossRef]
- Li, D.; Zhang, J.; Li, J. Role of miRNA sponges in hepatocellular carcinoma. Clin. Chim. Acta 2020, 500, 10–19. [Google Scholar] [CrossRef]
- Jost, I.; Shalamova, L.A.; Gerresheim, G.K.; Niepmann, M.; Bindereif, A.; Rossbach, O. Functional sequestration of microRNA-122 from Hepatitis C Virus by circular RNA sponges. RNA Biol. 2018, 15, 1032–1039. [Google Scholar] [CrossRef]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Wesselhoeft, R.A.; Kowalski, P.S.; Parker-Hale, F.C.; Huang, Y.; Bisaria, N.; Anderson, D.G. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell. 2019, 74, 508. [Google Scholar] [CrossRef]
- Schreiner, S.; Didio, A.; Hung, L.H.; Bindereif, A. Design and application of circular RNAs with protein-sponge function. Nucleic Acids Res. 2020, 48, 12326–12335. [Google Scholar] [CrossRef]
- Abe, N.; Matsumoto, K.; Nishihara, M.; Nakano, Y.; Shibata, A.; Maruyama, H.; Shuto, S.; Matsuda, A.; Yoshida, M.; Ito, Y.; et al. Rolling Circle Translation of Circular RNA in Living Human Cells. Sci. Rep. 2015, 5, 16435. [Google Scholar] [CrossRef] [PubMed]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A microRNA-29 mimic (Remlarsen) represses extracellular matrix expression and fibroplasia in the skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Lindow, M.; Kauppinen, S. Discovering the first microrna-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; van Doorn, L.J.; van der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef]



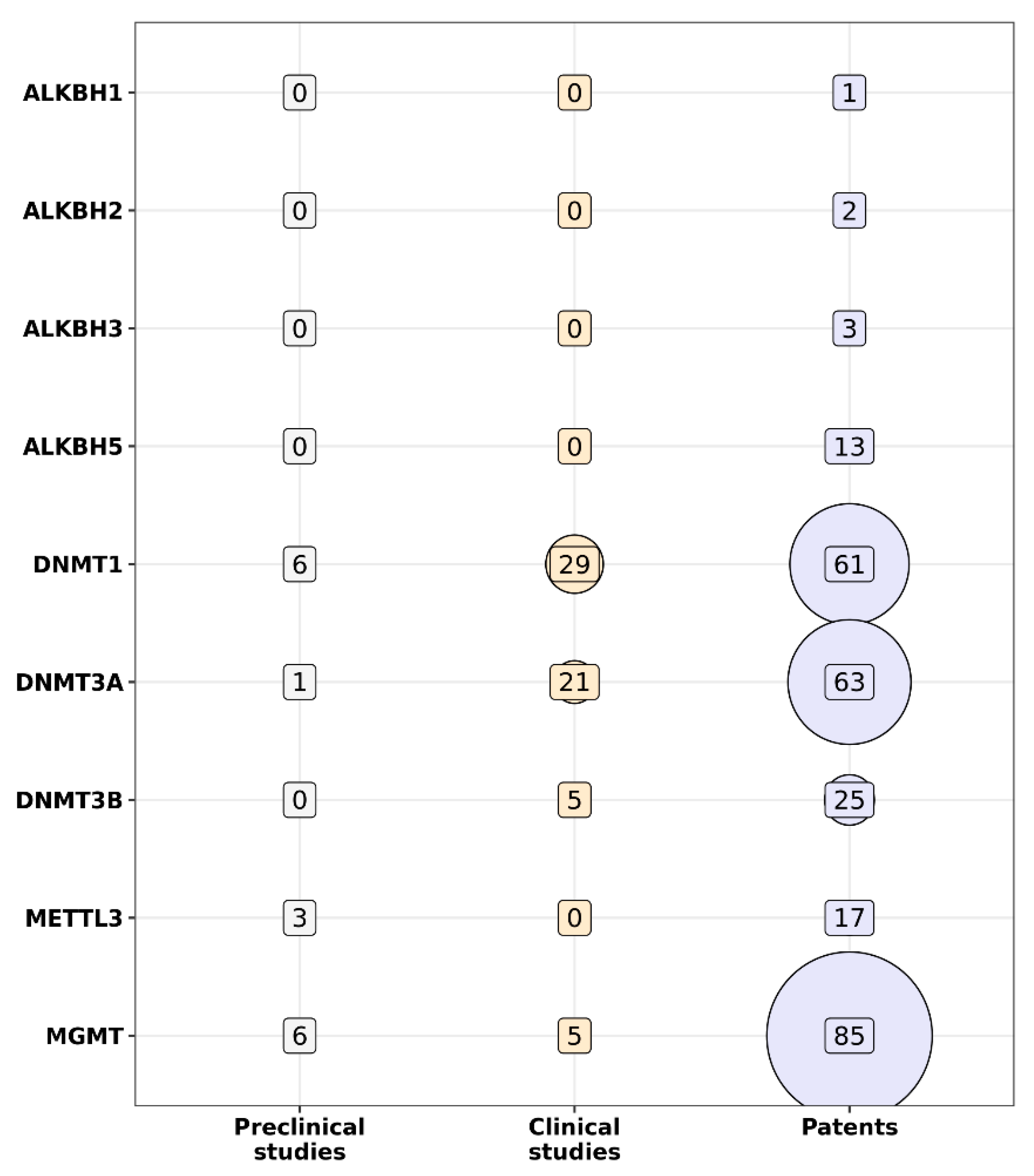{kind=link}
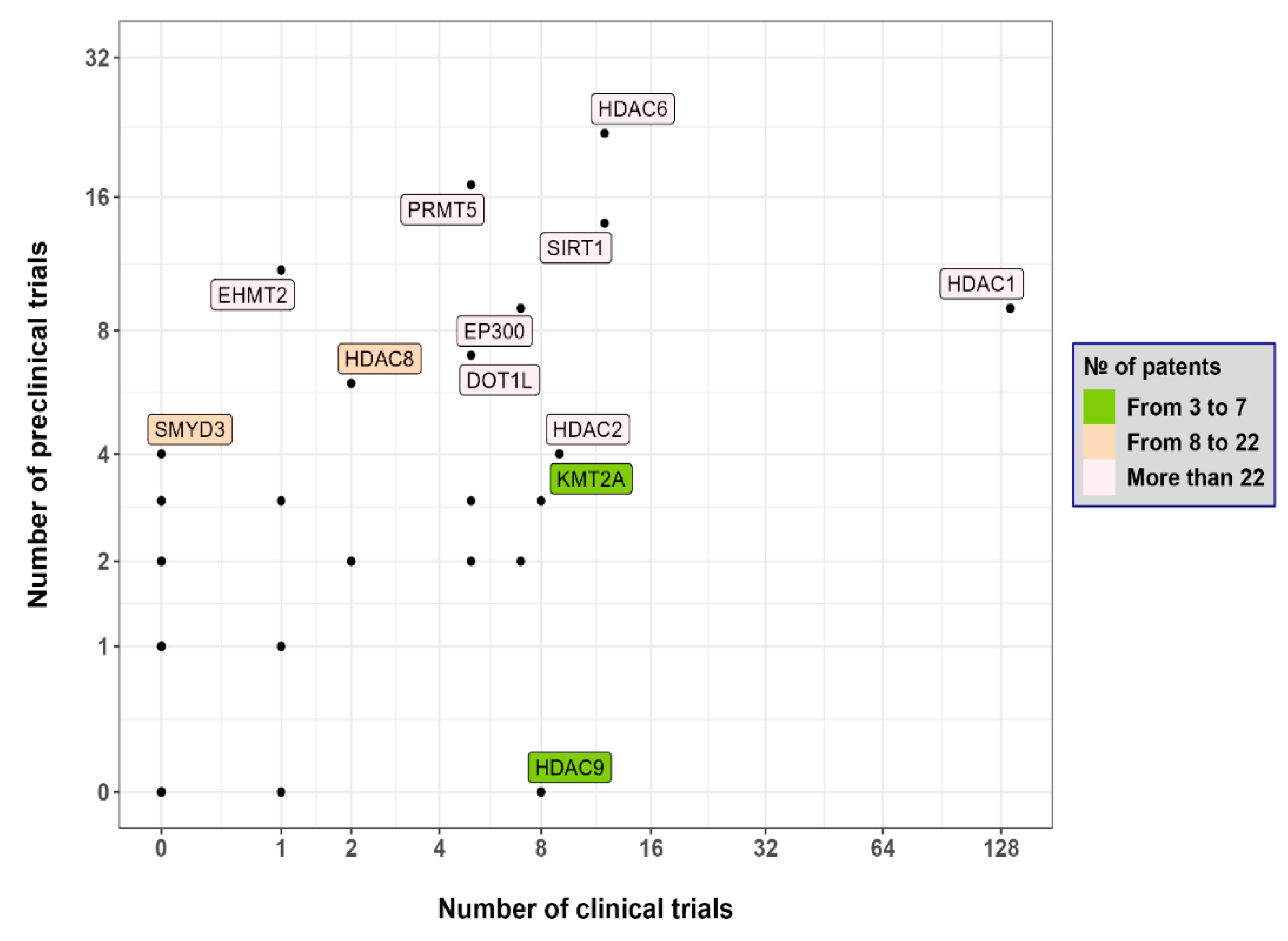{kind=link}
| Protein Name | Drug | ID Trial (www.ClinicalTrials.gov, Accessed on 16 September 2023) | Phase | Disease |
|---|---|---|---|---|
| DNA methyltransferase DNMT | Guadecitabine, SGI-110 | NCT03206047 | I/II | Platinum-Resistant Fallopian Tube Carcinoma, Platinum-Resistant Ovarian Carcinoma, Platinum-Resistant Primary Peritoneal Carcinoma, etc. |
| NCT01261312 | I/II | Myelodysplastic Syndromes (MDS), Acute myeloid leukemia (AML), Chronic Myelomonocytic Leukemia (CMML) | ||
| NCT02197676 | II | Myelodysplastic Syndromes (MDS), | ||
| Decitabine | NCT02472145 | II/III | Acute myeloid leukemia (AML) | |
| NCT04051996 | II | Acute myeloid leukemia (AML) | ||
| Azacitidine, CC-486 | NCT03542266 | II | Peripheral T-cell lymphoma (PTCL) | |
| Histone acetyltransferase EP300 | Inobrodib, CCS1477 | NCT03568656 | I/II | Metastatic Castration-Resistant Prostate Cancer, Metastatic Breast Cancer, Non-small Cell Lung Cancer, Advanced Solid Tumors |
| Histone acetyltransferase DOT1L | Pinometostat | NCT03701295, NCT03724084 | I/II | Acute myeloid leukemia (AML) |
| Histone methyltransferase PRMT5 | Pemramethostat, GSK3326595 | NCT04676516 | II | Early stages of breast cancer |
| Histone demethylase KDM1A | Tranylcypramine | NCT02717884 | I/II | Acute myeloid leukemia (AML) Myelodysplastic Syndrome |
| Seclidemstat | NCT03600649 | I/II | Ewing Sarcoma, Myxoid Liposarcoma, Sarcoma, Soft Tissue, Desmoplastic Small Round Cell Tumor, etc. | |
| Iadademstat | NCT05546580 | I | Acute myeloid leukemia (AML) | |
| NCT05420636 | II | Small cell lung cancer (SCLC), Neuroendocrine Carcinoma | ||
| NAD-dependent deacetylase SIRT1 | Selisistat, SEN0014196 | NCT01521585 | II | Huntington’s disease |
| Chidamide, HBI-8000,Tucidinostat, | ChiCTR1800017698 * | IV | Diffuse large B-cell lymphoma (DLBCL) | |
| ChiCTR2000034301 * | N/A | Advanced breast cancer | ||
| ChiCTR-OIC-17011303 * | IV | Peripheral T-cell lymphoma (PTCL) | ||
| NCT03023358 | III | Peripheral T-cell lymphoma (PTCL) | ||
| NCT04674683 | III | Metastatic inoperable melanoma | ||
| NCT04231448 | III | Diffuse large B-cell lymphoma (DLBCL) | ||
| NCT04040491 | IV | Peripheral T-cell lymphoma | ||
| Fimepinostat, CUDC-907 | NCT03002623 | II | Thyroid Neoplasms, Poorly Differentiated and Undifferentiated Thyroid Cancer, Differentiated Thyroid Cancer | |
| Histone deacetylase HDAC | Givinostat | NCT01901432 | I/II | Polycythemia Vera |
| Ricolinostat, ACY-1215 | NCT01997840 | I/II | Multiple myeloma | |
| NCT02856568 | I | Non-Resectable Cholangiocarcinoma, Recurrent Cholangiocarcinoma, Stage III Extrahepatic Bile Duct Cancer, Stage III Intrahepatic Cholangiocarcinoma, etc. | ||
| Quizinostat | NCT01486277 | II | T cell lymphoma | |
| MGCD-0103 | NCT00358982 | II | Hodgkin’s lymphoma | |
| Resminostat, 4SC-201 | NCT00943449 | II | Advanced hepatocellular carcinoma | |
| Entinostat | NCT00866333 | II | Hodgkin’s lymphoma |
| Protein Name | Drug Name | DrugBank ID | Status |
|---|---|---|---|
| HDAC1-3, HDAC6, HDAC8 | Vorinostat | DB02546 | FDA approved |
| HDAC1-3, HDAC6 | Pracinostat | DB05223 | Investigational |
| HDAC1, HDAC2, HDAC4, HDAC6 | Atorvastatin | DB06176 | FDA approved |
| HDAC1–3 | Mocetinostat | DB11830 | Investigational |
| HDAC2 HDAC9 | Valproic acid | DB00313 | FDA approved |
| HDAC10 HDAC6 | Bufexamac | DB13346 | FDA approved |
| HDAC7 HDAC8 | Trichostatin A | DB04297 | Experimental |
| HDAC1 | Abexinostat | DB12565 | Investigational |
| Fingolimod | DB08868 | FDA approved | |
| HDAC2 | Atorvastatin | DB06176 | FDA approved |
| Fluvastatin | DB01095 | FDA approved | |
| Pravastatin | DB00175 | FDA approved | |
| Lovastatin | DB00227 | FDA approved | |
| Simvastatin | DB00641 | FDA approved | |
| HDAC4 | CID 24836810 * | DB08613 | Experimental |
| CID 24836811 * | DB07879 | Experimental | |
| HDAC8 | CID 3994 * | DB02565 | Experimental |
| CID 5287979 * | DB07586 | Experimental | |
| CID 10379137 * | DB07350 | Experimental | |
| CID 449096 * | DB02917 | Experimental | |
| Cumarin 120 | DB08168 | Experimental |
| KEGG Terms * | p-Value | Protein * |
|---|---|---|
| hsa00350: Tyrosine metabolism | 0.0002 | Amine oxidase copper containing 3 (AOC3) |
| Monoamine oxidase A (MAOA) | ||
| Tyrosinase (TYR) | ||
| 4-hydroxyphenylpyruvate dioxygenase (HPD) | ||
| hsa00360: Phenylalanine metabolism | 0.0016 | Amine oxidase copper containing three proteins (AOC3) |
| Monoamine oxidase A (MAOA) | ||
| 4-hydroxyphenylpyruvate dioxygenase (HPD) | ||
| hsa04068: FoxO signaling pathway | 0.0012 | Mitogen-activated protein kinase 10 (MAPK10) |
| Mitogen-activated protein kinase 9 (MAPK9) | ||
| Serine/threonine kinase 4 (STK4) | ||
| Serum/glucocoticoid regulated kinase 1 (SGK1) | ||
| Sirtuin 1 (SIRT1) | ||
| hsa04024: cAMP signaling pathway | 0.0052 | Mitogen-activated protein kinase 10 (MAPK10) |
| Mitogen-activated protein kinase 9 (MAPK9) | ||
| Phosphodiesterase 4D (PDE4D) | ||
| 5-hydroxytryptamine receptor 1A (HTR1A) | ||
| Phosphodiesterase 4A (PDE4A) | ||
| hsa04010: MAPK signaling pathway | 0.0120 | Mitogen-activated protein kinase 10 (MAPK10) |
| Mitogen-activated protein kinase 9 (MAPK9) | ||
| Calcium voltage-gated channel auxiliary subunit alpha2 delta 1 (CACNA2D1) | ||
| Protein tyrosine phosphatase non-receptor type 7 (PTPN7) | ||
| Serine/threonine kinase 4 (STK4) | ||
| hsa04012: ErbB signaling pathway | 0.0392 | Mitogen-activated protein kinase 10 (MAPK10) |
| Mitogen-activated protein kinase 9 (MAPK9) | ||
| ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1) | ||
| hsa04014: Ras signaling pathway | 0.0472 | Mitogen-activated protein kinase 10 (MAPK10) |
| Mitogen-activated protein kinase 9 (MAPK9) | ||
| ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1) | ||
| Serine/threonine kinase 4 (STK4) |
| ID * | Phase | Cancer Type | Oncometabolite | Drug Combination |
|---|---|---|---|---|
| NCT03449901 | II | Soft tissue sarcoma | Arginine | ADI-PEG20, gemcitabine, docetaxel |
| NCT04776889 | IV | Prostate cancer (metastatic) | Cholesterol | Rosuvastatin |
| NCT04862260 | Early Phase I | Pancreatic cancer | Cholesterol | FOLFIRINOX, ezetimibe, atorvastatin, evolocumab |
| NCT04164901 | III | Glioma | 2-hydroxyglutarate | Vorasidenib |
| NCT03173248 | III | Acute myeloid leukemia | 2-hydroxyglutarate | Ivosidenib, azacitidine |
| Commercial Name | Substance | Clinical Trial No. | Target | Progress |
|---|---|---|---|---|
| ONPATTRO | patisiran | NCT01617967 NCT02510261 NCT04201418 NCT03997383 NCT05040373 | Transthyretin (TTR) | FDA approved, long-term studies, pregnancy safety studies |
| LEQVIO | Inclisiran [92] | NCT05362903, NCT04929249 NCT05682378 NCT03159416 NCT05399992 | Proprotein convertase subtilisin/kexin type 9 (PCSK9) | FDA approved, long-term study, combination therapy effectiveness study, extension trials |
| OXLUMO | Lumasiran | NCT04152200 NCT03905694 NCT03350451 NCT04982393 | Hydroxyacid oxidase (OA1) | FDA approved, observational study, extension trials |
| GIVLAARI | givosiran | NCT04883905 NCT02452372 | aminolevulinic acid synthase 1 (ALAS1) | FDA approved, combination therapy |
| - | Cemdisiran | NCT02352493 NCT05070858 NCT04601844 NCT05744921 NCT05133531 | Complement 5 | Phase I/II completed, Combination therapy trials |
| Anti-EpHa2 siRNA | Anti-EpHa2 siRNA | NCT01591356 | ephrin type-A receptor 2 (EpHa2) | Phase I estimated in 2024 |
| CAS3/SS3 | Anti-CpG-STAT3 siRNA | NCT04995536 | TLR9 receptor and signal transducer and activator of transcription 3 (STAT3) | Phase I estimated in 2024 |
| NBF-006 | Anti-KRAS siRNA | NCT03819387 | KRAS proto-oncogene (KRAS) | Phase I estimated in 2024 |
| ALN-KHK | antiKHK siRNA | NCT05761301 | ketohexokinase (KHK) | Phase I/II estimated in 2025 |
| OLX10212 | Asymmetric siRNA | NCT05643118 | Pathways upstream of VEGF (vascular endothelial growth factor) | Phase I estimated in 2024 |
| ADX-038 | Anti-PK siRNA | NCT05876312 | Prekallikrein (PK), | Phase I estimated in 2025 |
| SRN-001 | Anti-AREG siRNA | NCT05984992 | Amphiregulin (AREG) | Phase I estimated in 2024 |
| AOC 1020 | Anti-DUX4 siRNA | NCT05747924 | Double homeobox 4(DUX4) | Phase I estimated in 2025 |
| AGX148/PH-762 | Anti PD-1 siRNA | NCT05902520 | siRNA Modulation of PD-1 | Phase I estimated in 2026 |
| Commercial Name | Substance | Clinical Trial No. | Progress |
|---|---|---|---|
| INT-1B3 | miR-193a-3p mimic | NCT04675996 | Phase I estimated in 2024 |
| MRX34 | miR-34a mimic | NCT01829971 NCT03033329 | Terminated with adverse effects in 2017 Phase I completed in 2017 |
| MRG-201 | Remlarsen [132] | NCT03601052 NCT02603224 | Phase 2 completed in 2020 |
| SPC3649 | Miravirsen [133,134] | NCT00979927 NCT00688012 NCT01200420 | Phase 2 discontinued in 2021 |
| MRG-106 | Cobomarsen | NCT03837457 NCT02580552 NCT03713320 | Phase 2 terminated in 2020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griazeva, E.D.; Fedoseeva, D.M.; Radion, E.I.; Ershov, P.V.; Meshkov, I.O.; Semyanihina, A.V.; Makarova, A.S.; Makarov, V.V.; Yudin, V.S.; Keskinov, A.A.; et al. Current Approaches to Epigenetic Therapy. Epigenomes 2023, 7, 23. https://doi.org/10.3390/epigenomes7040023
Griazeva ED, Fedoseeva DM, Radion EI, Ershov PV, Meshkov IO, Semyanihina AV, Makarova AS, Makarov VV, Yudin VS, Keskinov AA, et al. Current Approaches to Epigenetic Therapy. Epigenomes. 2023; 7(4):23. https://doi.org/10.3390/epigenomes7040023
Chicago/Turabian StyleGriazeva, Ekaterina D., Daria M. Fedoseeva, Elizaveta I. Radion, Pavel V. Ershov, Ivan O. Meshkov, Alexandra V. Semyanihina, Anna S. Makarova, Valentin V. Makarov, Vladimir S. Yudin, Anton A. Keskinov, and et al. 2023. "Current Approaches to Epigenetic Therapy" Epigenomes 7, no. 4: 23. https://doi.org/10.3390/epigenomes7040023